
Comparative Genomic Analysis of Antimicrobial-Resistant Escherichia coli from South American Camelids in Central Germany


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain Selection
2.2. Whole-Genome Sequencing
2.3. Closure of the Gap in the Plasmid p20E0407A
3. Results
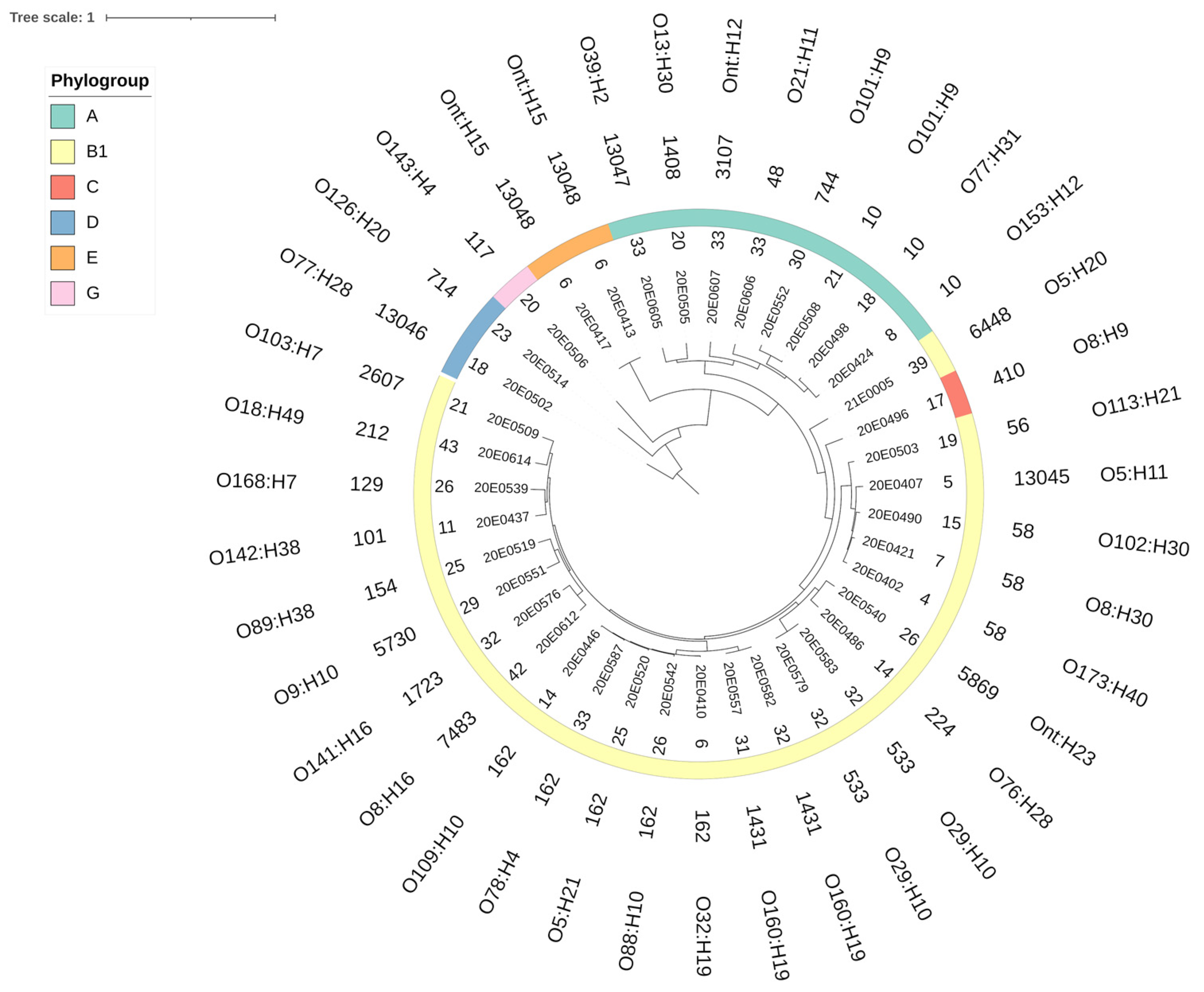
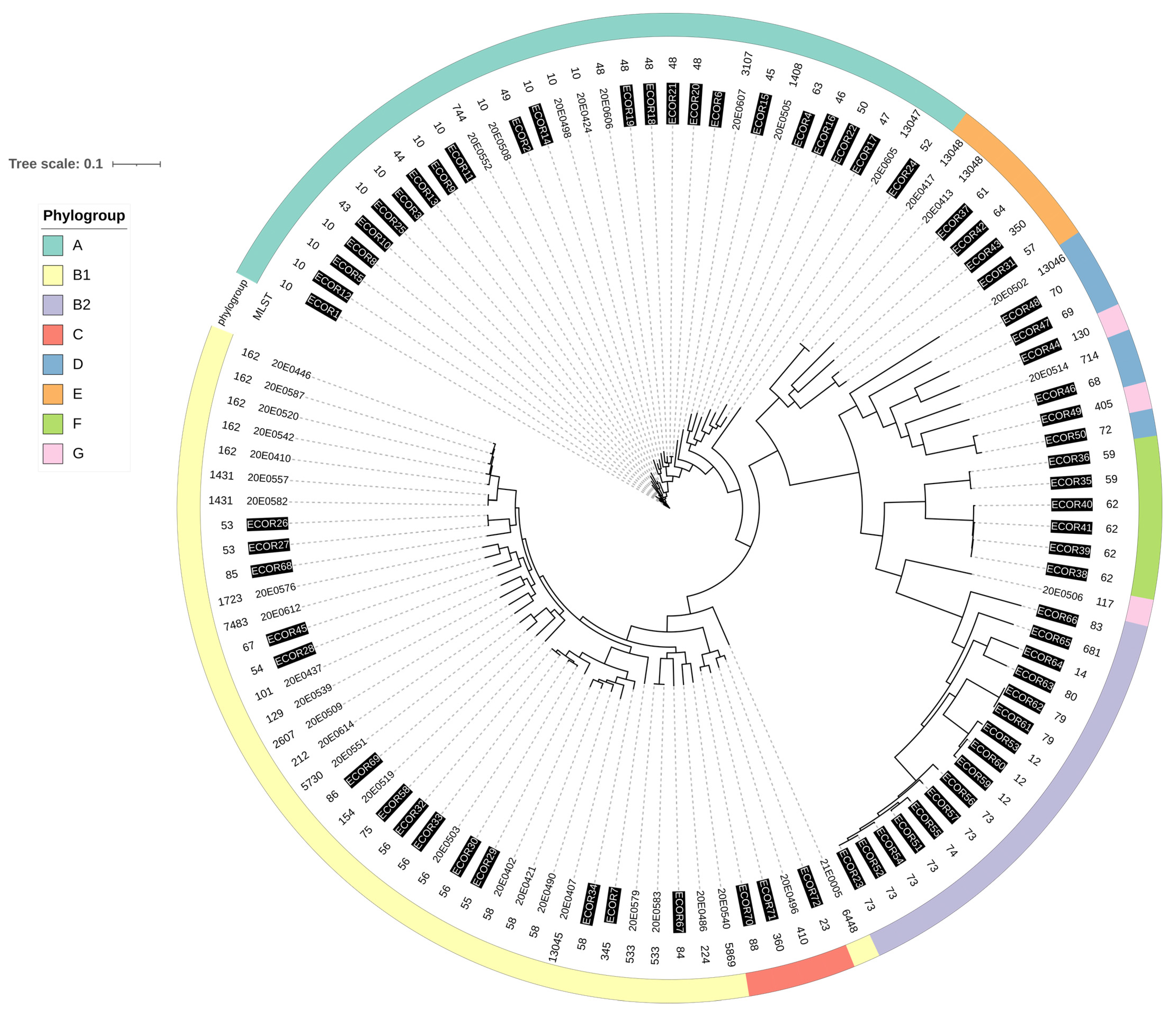
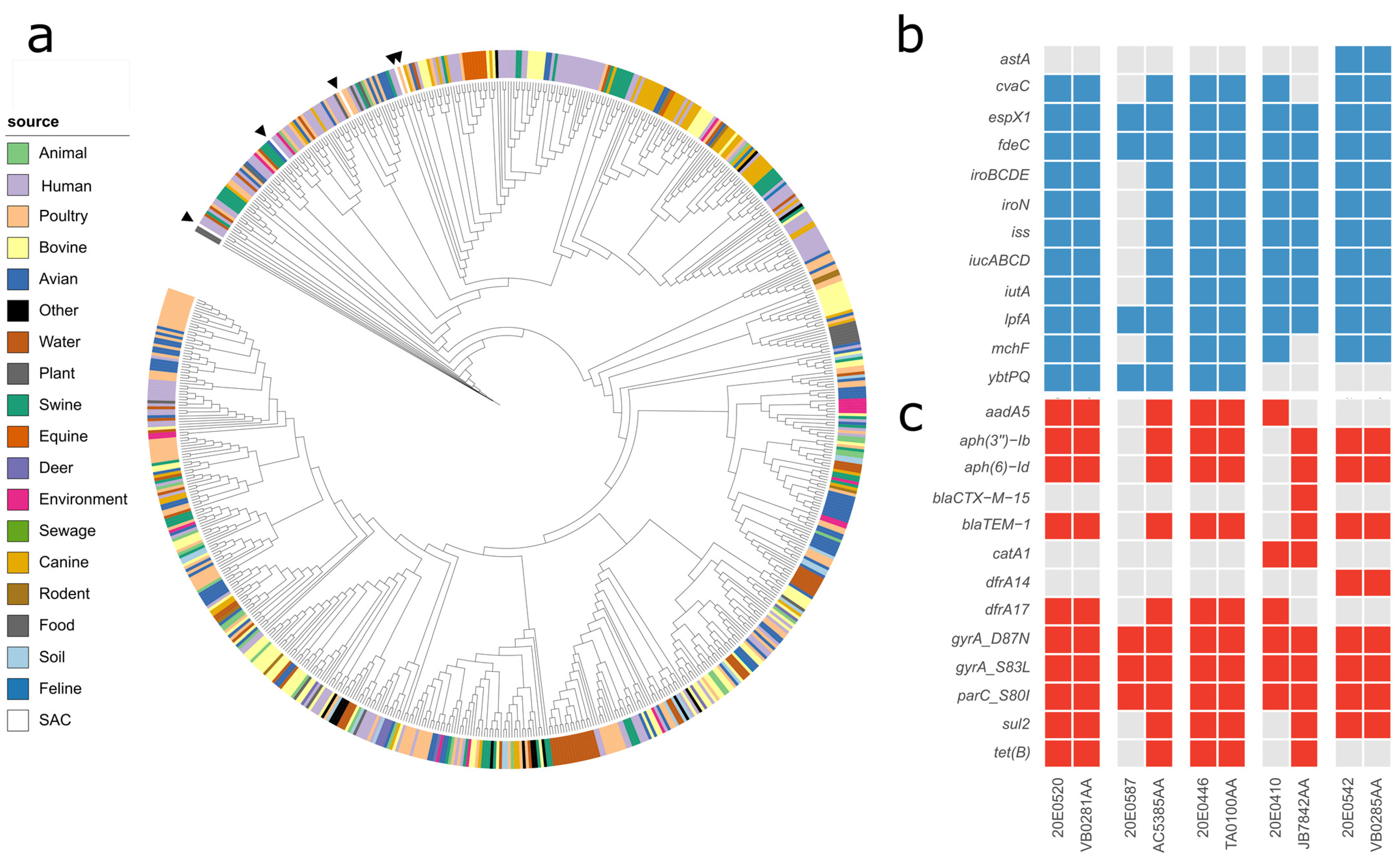
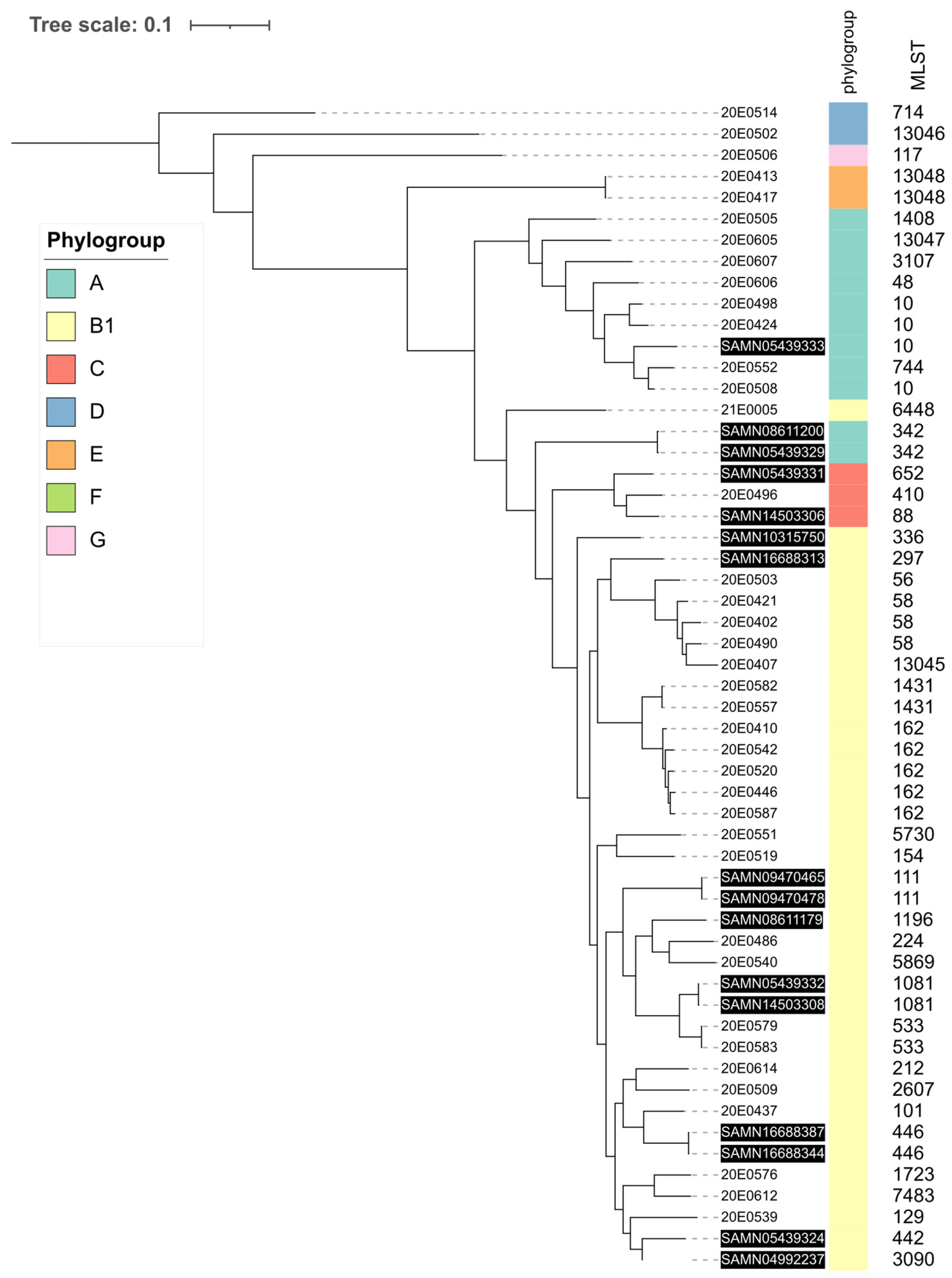
3.1. Phylogenetic Analysis
3.2. Virulence Factors
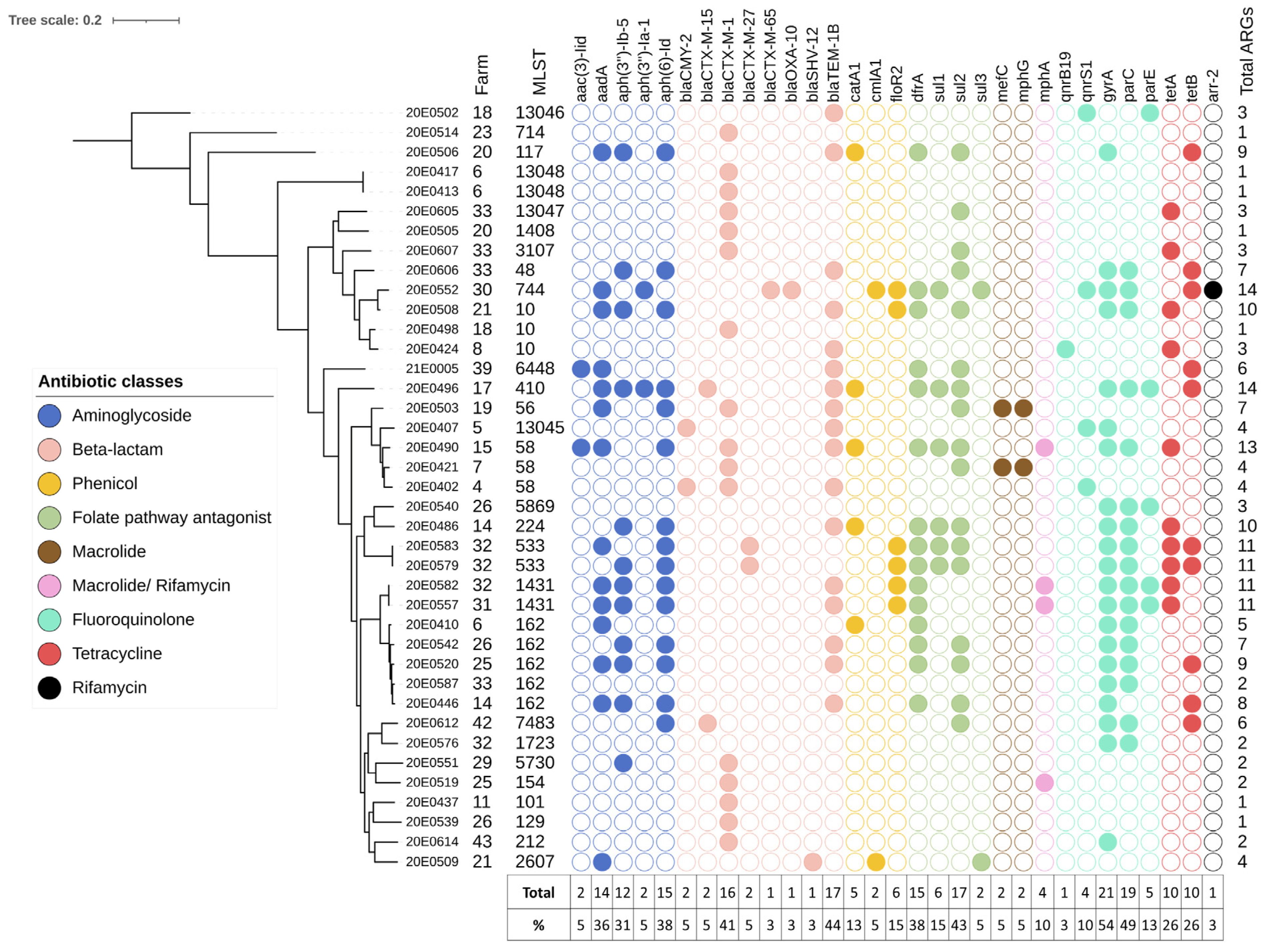
3.3. Antimicrobial Resistance
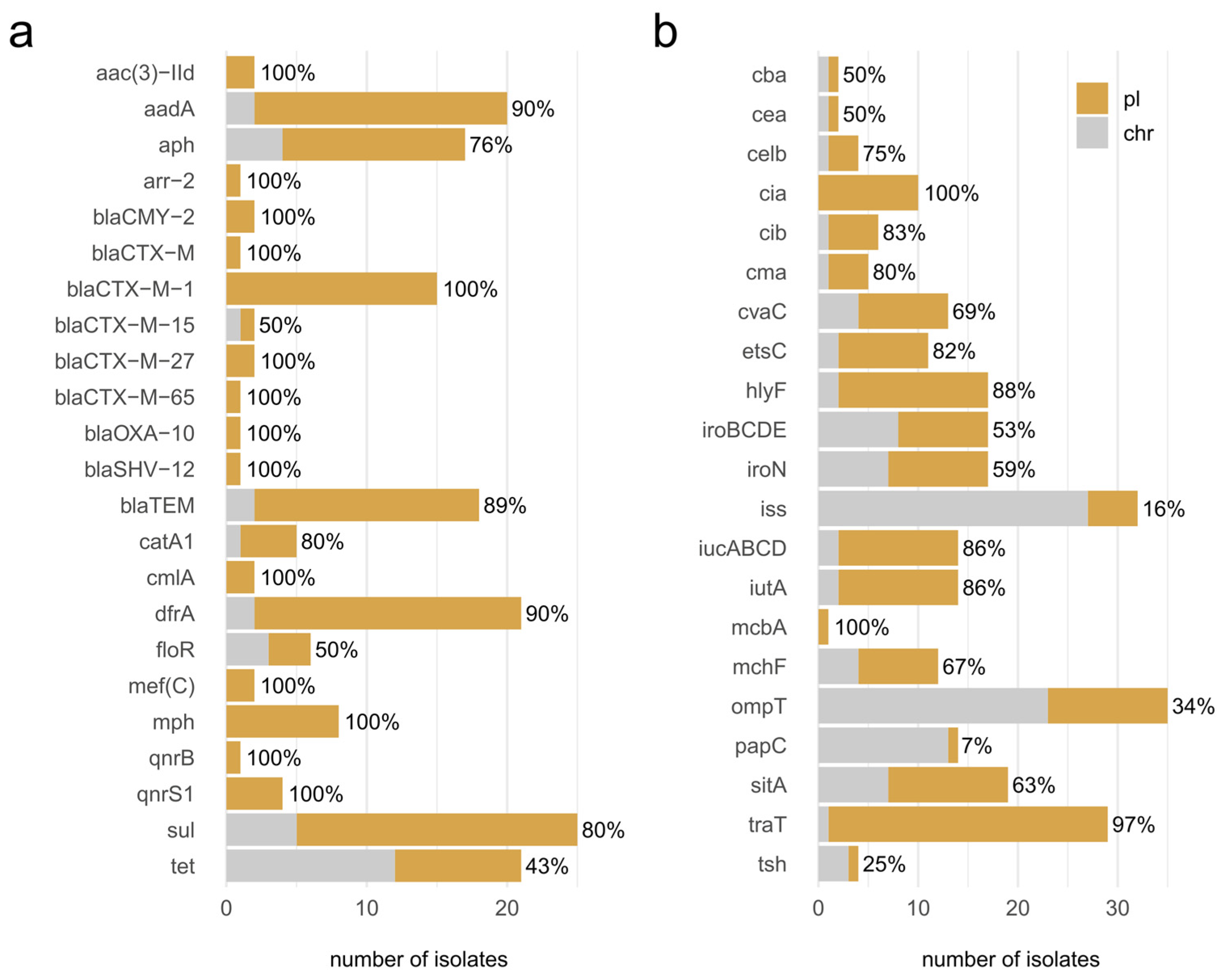
3.4. Plasmids
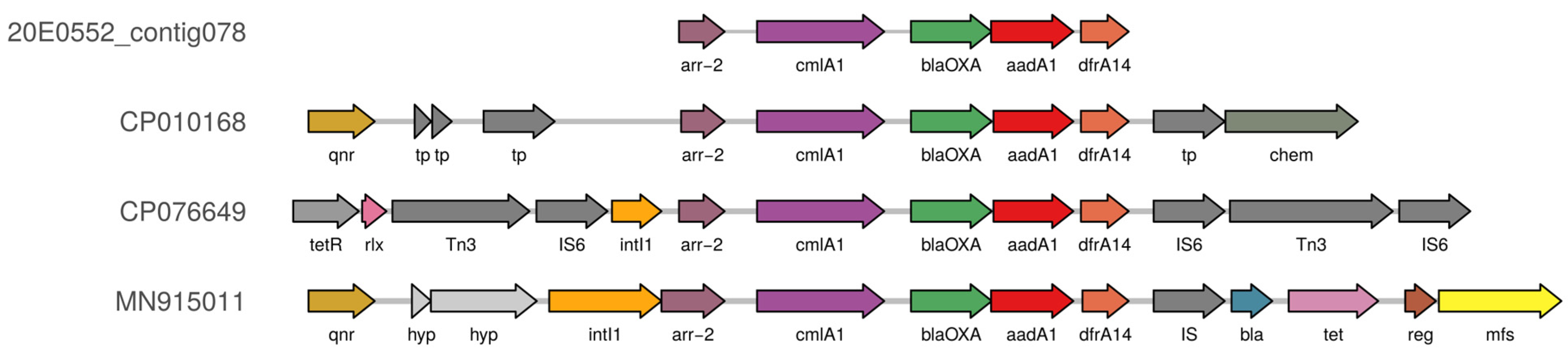
3.5. Co-Localization of AMR Genes and Virulence Factors on Plasmids
3.6. Comparison of ST162 Isolates with Enterobase Isolates
3.7. Comparison with Other Alpaca and Llama Isolates
4. Discussion
4.1. The E. coli Strains Isolated from the Central German SAC Are Phylogenetically Diverse
4.2. The Load of Virulence Factors Indicates a Low Pathogenic Potential
4.3. The AMR Genes Are Unevenly Distributed between Strains
4.4. The AMR Genotypes Correlate Well with the Phenotypes of the Strains
4.5. Conserved Plasmids Are Identified despite Apparent Plasmid Heterogeneity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zarrin, M.; Riveros, J.L.; Ahmadpour, A.; De Almeida, A.M.; Konuspayeva, G.; Vargas-Bello-Perez, E.; Faye, B.; Hernandez-Castellano, L.E. Camelids: New players in the international animal production context. Trop. Anim. Health Prod. 2020, 52, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Fowler, M.E. Camelids are not ruminants. In Zoo and Wild Animal Medicine; Elsevier Inc.: Amsterdam, The Netherlands, 2009; pp. 375–385. [Google Scholar]
- Neubert, S.; von Altrock, A.; Wendt, M.; Wagener, M.G. Llama and Alpaca management in Germany—Results of an online survey among owners on farm structure, health problems and self-reflection. Animals 2021, 11, 102. [Google Scholar] [CrossRef]
- González-Santamarina, B.; Schnee, C.; Köhler, H.; Weber, M.; Methner, U.; Seyboldt, C.; Berens, C.; Menge, C. Survey on shedding of selected pathogenic, zoonotic or antimicrobial resistant bacteria by South American camelids in Central Germany. Berl. Münchener Tierärztliche Wochenschr. 2022, 135, 1–16. [Google Scholar] [CrossRef]
- Halsby, K.; Twomey, D.F.; Featherstone, C.; Foster, A.; Walsh, A.; Hewitt, K.; Morgan, D. Zoonotic diseases in South American camelids in England and Wales. Epidemiol. Infect. 2017, 145, 1037–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Antimicrobial Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 14 January 2021).
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Lechner, I.; Freivogel, C.; Stark, K.D.C.; Visschers, V.H.M. Exposure pathways to antimicrobial resistance at the human-animal interface—A qualitative comparison of Swiss expert and consumer opinions. Front. Public Health 2020, 8, 345. [Google Scholar] [CrossRef]
- EFSA; ECDC. The European Union Summary Report on Antimicrobial Resistance in zoonotic and indicator bacteria from humans, animals and food in 2017/2018. EFSA J. 2020, 18, 166. [Google Scholar] [CrossRef] [Green Version]
- Hesp, A.; Veldman, K.; Van der Goot, J.; Mevius, D.; Van Schaik, G. Monitoring antimicrobial resistance trends in commensal Escherichia coli from livestock, The Netherlands, 1998 to 2016. Eurosurveillance 2019, 24, 1800438. [Google Scholar] [CrossRef] [Green Version]
- Aerts, M.; Battisti, A.; Hendriksen, R.; Kempf, I.; Teale, C.; Tenhagen, B.A.; Veldman, K.; Wasyl, D.; Guerra, B.; Liébana, E.; et al. Technical specifications on harmonised monitoring of antimicrobial resistance in zoonotic and indicator bacteria from food-producing animals and food. EFSA J. 2019, 17, e05709. [Google Scholar] [CrossRef] [Green Version]
- Barrios-Arpi, M.; Siever, M.C.; Villacaqui-Ayllon, E. Susceptibilidad antibiótica de cepas de Escherichia coli en crías de alpaca con y sin diarrea. Rev. Investig. Vet. Perú 2016, 27, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Luna, E.L.; Maturrano, H.L.; Rivera, G.H.; Zanabria, H.V.; Rosadio, A.R. Genotipificación, evaluación toxigénica in vitro y sensibilidad a antibióticos de cepas de Escherichia coli aisladas de casos diarreicos y fatales en alpacas neonatas. Rev. Investig. Vet. Perú 2012, 23, 280–288. [Google Scholar] [CrossRef] [Green Version]
- Gestrich, A.; Bedenice, D.; Ceresia, M.; Zaghloul, I. Pharmacokinetics of intravenous gentamicin in healthy young-adult compared to aged alpacas. J. Vet. Pharmacol. Ther. 2018, 41, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Denamur, E.; Clermont, O.; Bonacorsi, S.; Gordon, D. The population genetics of pathogenic Escherichia coli. Nat. Rev. Microbiol. 2021, 19, 37–54. [Google Scholar] [CrossRef]
- Maturrano, L.; Aleman, M.; Carhuaricra, D.; Maximiliano, J.; Siuce, J.; Luna, L.; Rosadio, R. Draft genome sequences of enterohemorrhagic and enteropathogenic Escherichia coli strains isolated from alpacas in Peru. Genome Announc. 2018, 6, e01391-7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangiredla, J.; Mammel, M.K.; Barnaba, T.J.; Tartera, C.; Gebru, S.T.; Patel, I.R.; Leonard, S.R.; Kotewicz, M.L.; Lampel, K.A.; Elkins, C.A.; et al. Species-wide collection of Escherichia coli isolates for examination of genomic diversity. Genome Announc. 2017, 5, e01321-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacher, D.W.; Mammel, M.K.; Gangiredla, J.; Gebru, S.T.; Barnaba, T.J.; Majowicz, S.A.; Dudley, E.G. Draft genome sequences of isolates of diverse host origin from the E. coli reference center at Penn State University. Microbiol. Resour. Announc. 2020, 9, e01005-20. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.F.; Mohamed, K.; Fan, Y.; Agama Study, G.; Achtman, M. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020, 30, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Caméléna, F.; Birgy, A.; Smail, Y.; Courroux, C.; Mariani-Kurkdjian, P.; Le Hello, S.; Bonacorsi, S.; Bidet, P. Rapid and simple universal Escherichia coli genotyping method based on multiple-locus variable-number tandem-repeat analysis using single-tube multiplex PCR and standard gel electrophoresis. Appl. Environ. Microbiol. 2019, 85, e02812-18. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.H.; Hsu, C.H.; McDermott, P.F.; et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 2020, 64, e00483-19. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Hasman, H.; Kaas, R.S.; Seyfarth, A.M.; Agersø, Y.; Lund, O.; Larsen, M.V.; Aarestrup, F.M. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J. Antimicrob. Chemother. 2013, 68, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.F.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.Y.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.D.; Jin, Q.; Chen, L.H.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Tetzschner, A.M.M.; Johnson, J.R.; Johnston, B.D.; Lund, O.; Scheutz, F. In silico genotyping of Escherichia coli isolates for extraintestinal virulence genes by use of whole-genome sequencing data. J. Clin. Microbiol. 2020, 58, e01269-20. [Google Scholar] [CrossRef]
- Johnson, J.R.; Murray, A.C.; Gajewski, A.; Sullivan, M.; Snippes, P.; Kuskowski, M.A.; Smith, K.E. Isolation and molecular characterization of nalidixic acid-resistant extraintestinal pathogenic Escherichia coli from retail chicken products. Antimicrob. Agents Chemother. 2003, 47, 2161–2168. [Google Scholar] [CrossRef] [Green Version]
- Spurbeck, R.R.; Dinh, P.C.; Walk, S.T.; Stapleton, A.E.; Hooton, T.M.; Nolan, L.K.; Kim, K.S.; Johnson, J.R.; Mobley, H.L.T. Escherichia coli isolates that carry vat, fyuA, chuA, and yfcV efficiently colonize the urinary tract. Infect. Immun. 2012, 80, 4115–4122. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.R.; Porter, S.; Johnston, B.; Kuskowski, M.A.; Spurbeck, R.R.; Mobley, H.L.T.; Williamson, D.A. Host characteristics and bacterial traits predict experimental virulence for Escherichia coli bloodstream isolates from patients with urosepsis. Open Forum Infect. Dis. 2015, 2, ovf083. [Google Scholar] [CrossRef]
- Wirth, T.; Falush, D.; Lan, R.; Colles, F.; Mensa, P.; Wieler, L.H.; Karch, H.; Reeves, P.R.; Maiden, M.C.; Ochman, H.; et al. Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 2006, 60, 1136–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, P.; Aarestrup, F.M.; Lund, O. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinform. 2018, 19, 307. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Tetzschner, A.M.M.; Iguchi, A.; Aarestrup, F.M.; Scheutz, F. Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 2015, 53, 2410–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaper, J.B. Pathogenic Escherichia coli. Int. J. Med. Microbiol. 2005, 295, 355–356. [Google Scholar] [CrossRef]
- DebRoy, C.; Fratamico, P.M.; Yan, X.; Baranzoni, G.; Liu, Y.; Needleman, D.S.; Tebbs, R.; O’Connell, C.D.; Allred, A.; Swimley, M.; et al. Comparison of O-antigen gene clusters of all O-serogroups of Escherichia coli and proposal for adopting a new nomenclature for O-typing. PLoS ONE 2016, 11, e0147434. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Furevi, A.; Perepelov, A.V.; Guo, X.; Cao, H.; Wang, Q.; Reeves, P.R.; Knirel, Y.A.; Wang, L.; Widmalm, G. Structure and genetics of Escherichia coli O antigens. FEMS Microbiol. Rev. 2020, 44, 655–683. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, C.; Roberts, I.S. Structure, assembly and regulation of expression of capsules in Escherichia coli. Mol. Microbiol. 1999, 31, 1307–1319. [Google Scholar] [CrossRef]
- Schwengers, O.; Barth, P.; Falgenhauer, L.; Hain, T.; Chakraborty, T.; Goesmann, A. Platon: Identification and characterization of bacterial plasmid contigs in short-read draft assemblies exploiting protein sequence-based replicon distribution scores. Microb. Genom. 2020, 6, mgen000398. [Google Scholar] [CrossRef]
- Beghain, J.; Bridier-Nahmias, A.; Le Nagard, H.; Denamur, E.; Clermont, O. ClermonTyping: An easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 2018, 4, mgen000192. [Google Scholar] [CrossRef]
- Clermont, O.; Dixit, O.V.A.; Vangchhia, B.; Condamine, B.; Dion, S.; Bridier-Nahmias, A.; Denamur, E.; Gordon, D. Characterization and rapid identification of phylogroup G in Escherichia coli, a lineage with high virulence and antibiotic resistance potential. Environ. Microbiol. 2019, 21, 3107–3117. [Google Scholar] [CrossRef]
- Tenaillon, O.; Skurnik, D.; Picard, B.; Denamur, E. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 2010, 8, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Selander, R.K. Standard reference strains of Escherichia coli from natural populations. J. Bacteriol. 1984, 157, 690–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, I.R.; Gangiredla, J.; Mammel, M.K.; Lampel, K.A.; Elkins, C.A.; Lacher, D.W. Draft genome sequences of the Escherichia coli reference (ECOR) collection. Microbiol. Resour. Announc. 2018, 7, e01133-18. [Google Scholar] [CrossRef] [Green Version]
- Croxen, M.A.; Law, R.J.; Scholz, R.; Keeney, K.M.; Wlodarska, M.; Finlay, B.B. Recent advances in understanding enteric pathogenic Escherichia coli. Clin. Microbiol. Rev. 2013, 26, 822–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, L.W. Distinguishing Pathovars from Nonpathovars: Escherichia coli. Microbiol. Spectrum. 2020, 8, AME-0014-2020. [Google Scholar] [CrossRef] [PubMed]
- Morgado, S.; Fonseca, E.; Vicente, A.C. Genomic epidemiology of rifampicin ADP-ribosyltransferase (Arr) in the Bacteria domain. Sci. Rep. 2021, 11, 19775. [Google Scholar] [CrossRef]
- Nonaka, L.; Maruyama, F.; Miyamoto, M.; Miyakoshi, M.; Kurokawa, K.; Masuda, M. Novel conjugative transferable multiple drug resistance plasmid pAQU1 from Photobacterium damselae subsp damselae isolated from marine aquaculture environment. Microbes Environ. 2012, 27, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Bizot, E.; Cointe, A.; Bidet, P.; Mariani-Kurkdjian, P.; Hobson, C.A.; Courroux, C.; Liguori, S.; Bridier-Nahmias, A.; Magnan, M.; Merimèche, M.; et al. Azithromycin resistance in Shiga toxin-producing Escherichia coli in France between 2004 and 2020 and detection of mef(C)-mph(G) genes. Antimicrob. Agents Chemother. 2022, 66, e0194921. [Google Scholar] [CrossRef]
- Seiffert, S.N.; Carattoli, A.; Schwendener, S.; Collaud, A.; Endimiani, A.; Perreten, V. Plasmids carrying blaCMY-2/4 in Escherichia coli from poultry, poultry meat, and humans belong to a novel IncK subgroup designated IncK2. Front. Microbiol. 2017, 8, 407. [Google Scholar] [CrossRef] [Green Version]
- Carattoli, A. Resistance plasmid families in Enterobacteriaceae. Antimicrob. Agents Chemother. 2009, 53, 2227–2238. [Google Scholar] [CrossRef] [Green Version]
- Alonso, C.A.; Michael, G.B.; Li, J.; Somalo, S.; Simon, C.; Wang, Y.; Kaspar, H.; Kadlec, K.; Torres, C.; Schwarz, S. Analysis of blaSHV-12-carrying Escherichia coli clones and plasmids from human, animal and food sources. J. Antimicrob. Chemother. 2017, 72, 1589–1596. [Google Scholar] [CrossRef]
- Touzain, F.; Le Devendec, L.; De Boisseson, C.; Baron, S.; Jouy, E.; Perrin-Guyomard, A.; Blanchard, Y.; Kempf, I. Characterization of plasmids harboring blaCTX-M and blaCMY genes in E. coli from French broilers. PLoS ONE 2018, 13, e0188768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagdasarian, M.; Bailone, A.; Bagdasarian, M.M.; Manning, P.A.; Lurz, R.; Timmis, K.N.; Devoret, R. An inhibitor of SOS induction, specified by a plasmid locus in Escherichia coli. Proc. Natl. Acad. Sci. USA 1986, 83, 5723–5726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anjum, M.F.; Schmitt, H.; Borjesson, S.; Berendonk, T.U.; on behalf of the WAWES network. The potential of using E. coli as an indicator for the surveillance of antimicrobial resistance (AMR) in the environment. Curr. Opin. Microbiol. 2021, 64, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Perrin, A.; De Sousa, J.A.M.; Vangchhia, B.; Burn, S.; O’Brien, C.L.; Denamur, E.; Gordon, D.; Rocha, E.P. Phylogenetic background and habitat drive the genetic diversification of Escherichia coli. PLoS Genet. 2020, 16, e1008866. [Google Scholar] [CrossRef] [PubMed]
- Isler, M.; Wissmann, R.; Morach, M.; Zurfluh, K.; Stephan, R.; Nüesch-Inderbinen, M. Animal petting zoos as sources of Shiga toxin-producing Escherichia coli, Salmonella and extended-spectrum β-lactamase (ESBL)-producing Enterobacteriaceae. Zoonoses Public Health 2021, 68, 79–87. [Google Scholar] [CrossRef]
- Fuentes-Castillo, D.; Esposito, F.; Cardoso, B.; Dalazen, G.; Moura, Q.; Fuga, B.; Fontana, H.; Cerdeira, L.; Dropa, M.; Rottmann, J.; et al. Genomic data reveal international lineages of critical priority Escherichia coli harbouring wide resistome in Andean condors (Vultur gryphus Linnaeus, 1758). Mol. Ecol. 2020, 29, 1919–1935. [Google Scholar] [CrossRef]
- Matamoros, S.; Van Hattem, J.M.; Arcilla, M.S.; Willemse, N.; Melles, D.C.; Penders, J.; Vinh, T.N.; Hoa, N.T.; Bootsma, M.C.J.; Van Genderen, P.J.; et al. Global phylogenetic analysis of Escherichia coli and plasmids carrying the mcr-1 gene indicates bacterial diversity but plasmid restriction. Sci. Rep. 2017, 7, 15364. [Google Scholar] [CrossRef] [Green Version]
- Köhler, C.-D.; Dobrindt, U. What defines extraintestinal pathogenic Escherichia coli? Int. J. Med. Microbiol. 2011, 301, 642–647. [Google Scholar] [CrossRef]
- Manges, A.R.; Geum, H.M.; Guo, A.; Edens, T.J.; Fibke, C.D.; Pitout, J.D.D. Global extraintestinal pathogenic Escherichia coli (ExPEC) lineages. Clin. Microbiol. Rev. 2019, 32, e00135-18. [Google Scholar] [CrossRef]
- Reid, C.J.; Cummins, M.L.; Borjesson, S.; Brouwer, M.S.M.; Hasman, H.; Hammerum, A.M.; Roer, L.; Hess, S.; Berendonk, T.; Nesporova, K.; et al. A role for ColV plasmids in the evolution of pathogenic Escherichia coli ST58. Nat. Commun. 2022, 13, 683. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Stegger, M.; Aziz, M.; Johnson, T.J.; Waits, K.; Nordstrom, L.; Gauld, L.; Weaver, B.; Rolland, D.; Statham, S.; et al. Escherichia coli ST131-H22 as a foodborne uropathogen. mBio 2018, 9, e00470-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, S.; De Almeida, A.; Carniel, E. The Yersinia high-pathogenicity island is present in different members of the family Enterobacteriaceae. FEMS Microbiol. Lett. 2000, 183, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Bélanger, L.; Garenaux, A.; Harel, J.; Boulianne, M.; Nadeau, E.; Dozois, C.M. Escherichia coli from animal reservoirs as a potential source of human extraintestinal pathogenic E. coli. FEMS Immunol. Med. Microbiol. 2011, 62, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carhuapoma Dela Cruz, V.; Valencia Mamani, N.; Huamán Gonzales, T.; Paucar Chanca, R.; Hilario Lizana, E.; Huere Peña, J.L. Resistencia antibiótica de Salmonella spp., Escherichia coli aisladas de alpacas (Vicugna pacus) con y sin diarrea. GRANJA Rev. Cienc. Vida 2020, 31, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Niehaus, A.J.; Anderson, D.E. Tooth root abscesses in llamas and alpacas: 123 cases (1994–2005). J. Am. Vet. Med. Assoc. 2007, 231, 284–289. [Google Scholar] [CrossRef]
- Guerrero-Olmos, K.; Baez, J.; Valenzuela, N.; Gahona, J.; Del Campo, R.; Silva, J. Molecular characterization and antibiotic resistance of Enterococcus species from gut microbiota of Chilean Altiplano camelids. Infect. Ecol. Epidemiol. 2014, 4, 24714. [Google Scholar] [CrossRef] [Green Version]
- Schauer, B.; Krametter-Frotscher, R.; Knauer, F.; Ehricht, R.; Monecke, S.; Fessler, A.T.; Schwarz, S.; Grunert, T.; Spergser, J.; Loncaric, I. Diversity of methicillin-resistant Staphylococcus aureus (MRSA) isolated from Austrian ruminants and New World camelids. Vet. Microbiol. 2018, 215, 77–82. [Google Scholar] [CrossRef]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, I.; Golparian, D.; Dillon, J.-A.R.; Johansson, A.; Ohnishi, M.; Sethi, S.; Chen, S.-C.; Nakayama, S.-I.; Sundqvist, M.; Bala, M.; et al. Characterisation of blaTEM genes and types of β-lactamase plasmids in Neisseria gonorrhoeae—The prevalent and conserved blaTEM-135 has not recently evolved and existed in the Toronto plasmid from the origin. BMC Infect. Dis. 2014, 14, 454. [Google Scholar] [CrossRef] [Green Version]
- Ewers, C.; Bethe, A.; Semmler, T.; Guenther, S.; Wieler, L.H. Extended-spectrum β-lactamase-producing and AmpC-producing Escherichia coli from livestock and companion animals, and their putative impact on public health: A global perspective. Clin. Microbiol. Infect. 2012, 18, 646–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.; Yang, Y.; Li, F.; Li, X.; Liu, H.; Fazilani, S.A.; Guo, W.; Xu, G.; Zhang, X. The prevalence and mechanism of fluoroquinolone resistance in Escherichia coli isolated from swine farms in China. BMC Vet. Res. 2020, 16, 258. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C.; Jacoby, G.A. Mechanisms of drug resistance: Quinolone resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 12–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayer, S.S.; Casanova-Higes, A.; Paladino, E.; Elnekave, E.; Nault, A.; Johnson, T.; Bender, J.; Perez, A.; Alvarez, J. Global distribution of fluoroquinolone and colistin resistance and associated resistance markers in Escherichia coli of swine origin—A systematic review and meta-analysis. Front. Microbiol. 2022, 13, 834793. [Google Scholar] [CrossRef]
- Billane, K.; Harrison, E.; Cameron, D.; Brockhurst, M.A. Why do plasmids manipulate the expression of bacterial phenotypes? Philos. Trans. R. Soc. B 2022, 377, 20200461. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Beltrán, J.; DelaFuente, J.; León-Sampedro, R.; MacLean, R.C.; San Millán, Á. Beyond horizontal gene transfer: The role of plasmids in bacterial evolution. Nat. Rev. Microbiol. 2021, 19, 347–359. [Google Scholar] [CrossRef]
- Carattoli, A.; Villa, L.; Fortini, D.; García-Fernández, A. Contemporary IncI1 plasmids involved in the transmission and spread of antimicrobial resistance in Enterobacteriaceae. Plasmid 2021, 118, 102392. [Google Scholar] [CrossRef] [Green Version]
- Duggett, N.; AbuOun, M.; Randall, L.; Horton, R.; Lemma, F.; Rogers, J.; Crook, D.; Teale, C.; Anjum, M.F. The importance of using whole genome sequencing and extended spectrum β-lactamase selective media when monitoring antimicrobial resistance. Sci. Rep. 2020, 10, 19880. [Google Scholar] [CrossRef]
- Baron, S.; Le Devendec, L.; Lucas, P.; Larvor, E.; Jove, T.; Kempf, I. Characterisation of plasmids harbouring extended-spectrum cephalosporin resistance genes in Escherichia coli from French rivers. Vet. Microbiol. 2020, 243, 108619. [Google Scholar] [CrossRef]
- Homeier-Bachmann, T.; Schütz, A.K.; Dreyer, S.; Glanz, J.; Schaufler, K.; Conraths, F.J. Genomic analysis of ESBL-producing E. coli in wildlife from North-Eastern Germany. Antibiotics 2022, 11, 123. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Santamarina, B.; Weber, M.; Menge, C.; Berens, C. Comparative Genomic Analysis of Antimicrobial-Resistant Escherichia coli from South American Camelids in Central Germany. Microorganisms 2022, 10, 1697. https://doi.org/10.3390/microorganisms10091697
González-Santamarina B, Weber M, Menge C, Berens C. Comparative Genomic Analysis of Antimicrobial-Resistant Escherichia coli from South American Camelids in Central Germany. Microorganisms. 2022; 10(9):1697. https://doi.org/10.3390/microorganisms10091697
Chicago/Turabian StyleGonzález-Santamarina, Belén, Michael Weber, Christian Menge, and Christian Berens. 2022. "Comparative Genomic Analysis of Antimicrobial-Resistant Escherichia coli from South American Camelids in Central Germany" Microorganisms 10, no. 9: 1697. https://doi.org/10.3390/microorganisms10091697