
Computational Identification of Potential Multitarget Inhibitors of Nipah Virus by Molecular Docking and Molecular Dynamics


1
Virology Discovery Unit and Bioinformatics Centre, CSIR-Institute of Microbial Technology, Council of Scientific and Industrial Research, Chandigarh 160036, India
2
Academy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Microorganisms 2022, 10(6), 1181; https://doi.org/10.3390/microorganisms10061181
Submission received: 24 March 2022
/
Revised: 28 April 2022
/
Accepted: 11 May 2022
/
Published: 9 June 2022
(This article belongs to the Special Issue Henipaviruses: Emergence, Pathogenesis, and Control Measures)
Abstract
:Nipah virus (NiV) is a recently emerged paramyxovirus that causes severe encephalitis and respiratory diseases in humans. Despite the severe pathogenicity of this virus and its pandemic potential, not even a single type of molecular therapeutics has been approved for human use. Considering the role of NiV attachment glycoprotein G (NiV-G), fusion glycoprotein (NiV-F), and nucleoprotein (NiV-N) in virus replication and spread, these are the most attractive targets for anti-NiV drug discovery. Therefore, to prospect for potential multitarget chemical/phytochemical inhibitor(s) against NiV, a sequential molecular docking and molecular-dynamics-based approach was implemented by simultaneously targeting NiV-G, NiV-F, and NiV-N. Information on potential NiV inhibitors was compiled from the literature, and their 3D structures were drawn manually, while the information and 3D structures of phytochemicals were retrieved from the established structural databases. Molecules were docked against NiV-G (PDB ID:2VSM), NiV-F (PDB ID:5EVM), and NiV-N (PDB ID:4CO6) and then prioritized based on (1) strong protein-binding affinity, (2) interactions with critically important binding-site residues, (3) ADME and pharmacokinetic properties, and (4) structural stability within the binding site. The molecules that bind to all the three viral proteins (NiV-G ∩ NiV-F ∩ NiV-N) were considered multitarget inhibitors. This study identified phytochemical molecules RASE0125 (17-O-Acetyl-nortetraphyllicine) and CARS0358 (NA) as distinct multitarget inhibitors of all three viral proteins, and chemical molecule ND_nw_193 (RSV604) as an inhibitor of NiV-G and NiV-N. We expect the identified compounds to be potential candidates for in vitro and in vivo antiviral studies, followed by clinical treatment of NiV.
1. Introduction
Nipah virus (NiV), a virus of the genus Henipavirus within the Paramyxoviridae family, was initially isolated in 1999 during an outbreak in Malaysia [1]. Since then, there have been sporadic reports of NiV outbreaks in various other Asian countries such as Singapore, the Philippines, Bangladesh, and India, the most recent of which is in the South Indian state of Kerala [2]. NiV is potentially pandemic, highly pathogenic, and capable of infecting most mammalian species. From 1998 to 2015, more than 600 cases of NiV human infections were reported, with a fatality rate of 38% in Malaysia and 43–100% in India and Bangladesh [2].
NiV is classified as a biosafety level 4 (BSL4) pathogen due to its high case fatality rate following human infection and due to the lack of effective therapeutics or vaccines for infection treatment [3]. There have been some efforts to manage NiV by searching for small-molecule therapeutics [4,5]. For example, favipiravir has demonstrated efficacy against a broad spectrum of RNA viruses such as Paramyxoviridae, Filoviridae, Arenaviridae, and Bunyavirales; however, a recent in vitro study also demonstrated favipiravir to be inhibiting NiV and Hendra virus (HeV) virus replication and transcription at micromolar concentrations [6]. R1479 (4′-azidocytidine), a drug previously identified to inhibit flaviviruses, is also found to inhibit henipaviruses, including other paramyxoviruses, with high potency [7]. Small molecules that activate IRF3 and modulate RIG-I-like receptors pathways have also been investigated as potential strategies for targeting NiV infection [8]. However, despite these efforts for developing small-molecule inhibitors, no approved drugs are available yet for efficient use in humans. Though Ribavirin is not a proven treatment for NiV, it is particularly being used in a state of emergency to treat acute NiV encephalitis as a first-line treatment strategy [9]; however, it offers various side effects such as nausea, vomiting, and convulsions during NiV treatment [10]. Additionally, driven by the unwanted pharmacokinetics and numerous side effects of synthetic compounds, there is also a specific interest in using compounds of natural origin (phytochemicals) to overcome the side effects. Therefore, taken altogether, there is a strong need to discover novel small-molecule chemical inhibitors or phytochemicals against NiV with potential therapeutic value and fewer or no side effects in humans.
Contrary to conventional drug discovery approaches that rely on targeting a single protein, the next paradigm in drug discovery is to search for multitarget drugs. Viral entry into the host begins with relatively nonspecific interactions between the virus and attachment factors on the cell surface. The henipaviruses encode two envelope glycoproteins, including G and F glycoproteins. These virus attachment glycoproteins are essential for the recognition of host-cell-surface receptors ephrin-B2 (EFNB2) and ephrin-B3 (EFNB3), which mediate cellular attachment, fusion, and virus entry [11]. NiV glycoprotein G (NiV-G) has a globular head domain formed of a six-bladed beta-sheet propeller, connected via a flexible stalk domain to a transmembrane anchor. The binding of G glycoprotein to its ephrin receptors leads to conformational changes in glycoprotein, followed by subsequent conformational changes in F glycoprotein (NiV-F) that lead to the establishment of a physical link between viral and cellular membranes by stripping of the fusion peptide. Concluding the fusion process, nucleocapsid enters the host cell cytoplasm, resulting in the onset of viral replication [12]. The NiV RNA genome is encapsidated by the nucleoprotein (NiV-N) that protects the viral genome from degradation and also acts as a template for mRNA transcription [13]. Considering the role of each of the three viral proteins (i.e., NiV-G, NiV-F, and NiV-N) in virus replication and spread, they are, therefore, of particular interest in anti-Nipah drug discovery.
Multitarget drug discovery has raised considerable interest in the last decade [14,15,16,17]. Most drugs aim at a single biological molecule, widely known as the “one target, one drug” strategy, whereas a multitarget drug has the potential to modulate the effect of multiple targets. Multitarget drugs have even been approved for clinical use [18], and many complex diseases, including neurodegenerative diseases, cardiovascular diseases, and cancers, are often treated with multidrug therapy or a “cocktail” of drugs [19]. Many computational approaches have been developed to address polypharmacology-guided drug discovery [16,20]; these approaches, including virtual screening, molecular docking, and molecular dynamics, have been widely implemented in modern drug discovery and are capable of screening new compounds with multitarget characteristics. For example, docking simulations have identified a series of novel multitarget compounds (e.g., donepezil–indolyl hybrid, donepezil–pyridyl hybrid, donepezil hybrid, etc.) for the treatment of neurodegenerative diseases [15]. In another study, Zhou et al. (2017) [21] implemented a computational drug design method that aided the synthesis and characterization of a novel multikinase inhibitor molecule. Yousuf et al. (2017) [22] proposed novel multitarget inhibitors for breast cancer treatment by using a virtual screening/molecular docking-based approach. Using multitargeted molecular docking Singh and Bast (2015) [23] also identified various potential natural compounds as inhibitors of tyrosine kinase receptors involved in the development of several types of cancers. Computational approaches for multitarget drug discovery in pathogens have also been reported [24,25,26]. Several studies have been reported in the literature addressing various computational methods (e.g., virtual screening, molecular docking, molecular modeling, machine learning, quantitative structure–activity relationship, in silico ADME, pharmacophore, etc.) targeting multiple targets [27].
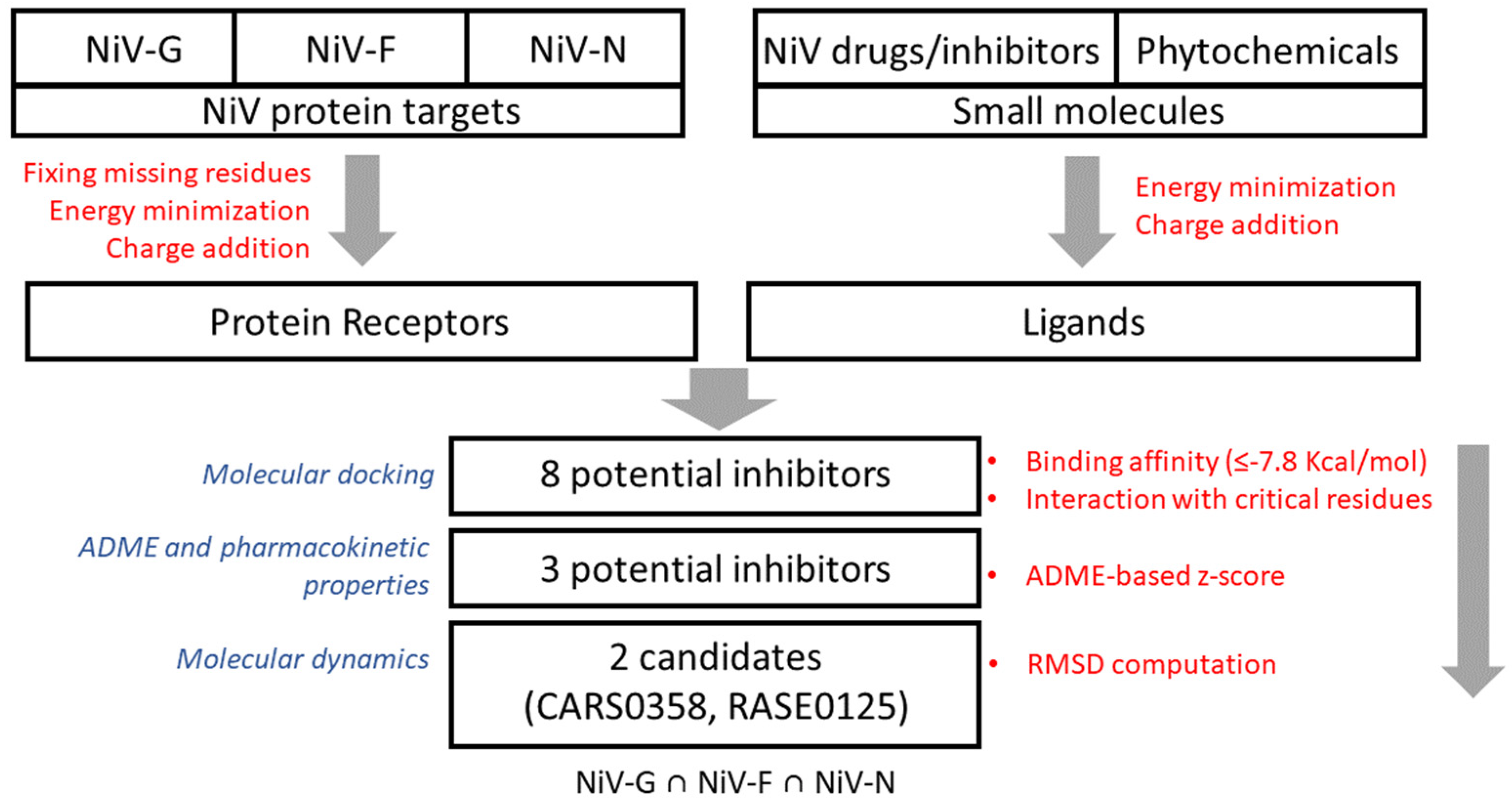
Our group has already developed various therapeutic/antiviral resources [28,29] and methods to predict antiviral peptides [30,31], small molecules [32,33], and siRNAs [34]. Our group has developed a resource of multitargeted putative therapeutics and epitopes for NiV drug discovery as well [35]. Additionally, several small-molecule inhibitors were identified by our group using QSAR [36] and molecular docking-based studies [37]. In our previous study [37], we have identified small-molecule FDA-approved drugs as potential inhibitors of NiV-G using an ensemble of molecular docking and analysis of the chemical–protein interaction network. Computational screening of small-molecule inhibitors/drugs is a useful approach for rapid screening of therapeutic molecules from a large chemical space; this motivated us to shortlist potential leads to meet the urgent demand for repurposing drugs for the treatment of NiV. In this study, to extend the scope of already developed methods and approaches against NiV, an integrative structure-based approach was implemented to identify potential multitarget small-molecule chemical/phytochemical inhibitor(s) against NiV-G, NiV-F, and NiV-N—the three most important NiV proteins targets. The workflow for the proposed integrated approach for multitarget molecule screening is presented in Figure 1.
2. Materials and Methods
2.1. Data Compilation and Small-Molecule Library Preparation
An extensive literature search in PubMed (pubmed.ncbi.nlm.nih.gov/) database was performed to extract the structural information on known NiV inhibitors using the following integrated text query: (((Nipah) AND drug*)) OR ((Nipah) AND inhibit*). Then, 3D chemical structures of small-molecule NiV inhibitors were prepared and edited using MarvinSketch v5.10.0 software (https://chemaxon.com/). To probe for NiV inhibitors that might be associated with adverse side effects, ADME and pharmacokinetic analyses were performed using the SwissADME webserver (http://swissadme.ch/) (accessed on 18 October 2021).
Data on different aspects of phytochemicals, including their 3D structures, were obtained from three well-established databases: (1) SerpentinaDB [38], for a structured compilation of Indian snakeroot (Rauvolfia serpentina) plant-derived molecules (n = 142); (2) Phytochemica [39], for a structured compilation of molecules from the plants Atropa belladonna, Catharanthus roseus, Heliotropium indicum, Picrorhiza kurroa, and Podophyllum hexandrum (n = 868); (3) Phytochemical and Drug Target DataBase (PDTDB, n = 221) [40]; these databases contain a vast amount of information on phytochemicals having therapeutic potential against various diseases. All obtained chemical structures were structurally optimized via energy minimization (500 steps of steepest descent) with Merck Molecular Force Field (MMFF94) while implementing OpenBabel v 2.4.0 software [41].
2.2. Chemical Variability Analysis of Compiled Molecules
The R-ChemmineR package [42] was used to cluster the literature-compiled NiV inhibitors into their discrete similarity groups (on the basis of the Tanimoto similarity measure/index), using fingerprints generated from the descriptor vectors. A maximum common substructure search was also performed to identify the potential representative scaffolds in the inhibitors using the flexible common substructure (FMCS) algorithm [43].
2.3. Retrieval and Assessment of Viral Protein Structures
The 3D structures of Nipah glycoprotein (NiV-G), fusion protein (NiV-F), and nucleoprotein (NiV-N) were obtained from the RCSB Protein Data Bank (PDB) (https://www.rcsb.org/) (accessed on 10 October 2021) having identifiers 2VSM (1.8 Å) [11], 5EVM (3.3 Å) [44], and 4CO6 (2.5 Å) [45], respectively. The 3D structures of proteins were assessed in Chimera v 1.16 (http://cgl.ucsf.edu/chimera/), and any missing residue(s) was/were fixed, and optimized. The structure of NiV-N (4CO6) is a NiV nucleoprotein–phosphoprotein complex, and therefore, the phosphoprotein chains were removed from the complex. Nucleoprotein structure also comprised many modified residues (e.g., MSE/AMSE/BMSE), which were modified to Gly, the simplest amino acid, in Pymol v 2.3.0 (https://pymol.org/2/) software. The modified protein structure was structurally optimized via energy minimization (1000 steps of steepest descent) in Chimera.
2.4. Assessment of Ligand-Binding Pockets
The data on ligand-binding sites in all three viral proteins were obtained from the published literature. The binding-site residues of protein NiV-G (Gln559, Glu579, Tyr581, Ile588) were obtained from Kalbhor et al. (2021) [46], while binding-site residues of NiV-F (His29, Tyr30, Val39, Lys40, Asn380, Tyr432, Leu433) and NiV-N (Lys34, Arg36, Phe38, Val58, Ala65, Ser67, Glu124, Leu128, Ile131) were obtained from Sen et al. (2019) [47]. To assess the conformational/structural differences among the ligand-binding pockets in the viral protein structures, their respective pocket volume sizes were assessed using Pocket Volume Measurer (POVME) [48] and Pymol software.
2.5. Molecular Docking
For molecular docking, the cleaned and energy-minimized structures (called receptors henceforth) were considered (see Section 2.3). The three-step docking comprises the following preparations: (1) receptor preparation—using Autodock Tools 4.2.6 software [49], polar hydrogen atoms were added, and the protonation states were assigned by including appropriate Gasteiger charges to the respective protein models (NiV-G, 8.9803; NiV-F, 2.986; NiV-N,0.9779). All other receptor preparation options were kept at default; (2) ligand preparation—charges were added to small molecules (chemical inhibitors and phytochemicals), and all bonds of ligands were set to be rotatable except N–C bonds; (3) molecular docking—all ligands were docked into the respective ligand-binding sites using the QuickVina v 2.0 [50] software, a docking tool that accelerates AutoDock Vina software [51] by implementing an already benchmarked (on CCDC/ASTEX dataset) molecular docking approach [37]. The small molecules that bind to all the three viral proteins (NiV-G ∩ NiV-F ∩ NiV-N) were considered multitarget inhibitors. Intermolecular interactions among receptors and ligands were analyzed with LigPlot+ v 2.2 software (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/), and 3D images were rendered with Pymol.
As a positive control, molecular docking studies were also performed among viral proteins and the drug remdesivir (GS-5734) [52]. The 2D structure of remdesivir (DB14761) was obtained from the DrugBank database (https://www.drugbank.com/) (accessed on 18 April 2022), which was 3D optimized (using the obminimize module), with 500 steps of conjugate gradient and steepest descent methods. Remdesivir was then docked into the respective binding sites of all three viral proteins using the implemented docking pipeline.
2.6. Computing ADME and Pharmacokinetics Properties of Small Molecules
ADME and pharmacokinetics properties of docking-based prioritized molecules were computed using ADMETlab 2.0 webserver [53]. ADMETlab computes a total of 88 molecular property descriptors based on molecule’s physicochemical properties (n = 17), medicinal chemistry (n = 13), and ADMET properties [A (n = 7), D (n = 4), M (n = 10), E (n = 2), T (n = 35)]. To prioritize these molecules for next step, the respective z-scores for each molecule were computed based on ADME descriptors. Briefly, the most relevant property value values were first transformed into a data matrix of binary variables (0 or 1). During data transformation, the values within the standard limits were transformed into binary variable 1, else 0. All values were then transformed into a normalized score for each molecule, which was computed as , where X is the value being analyzed, μ is the mean, and σ is the standard deviation. In addition, z-scores for molecules were computed in R v 3.6.3 (https://www.r-project.org/) statistical environment.
2.7. Assessing Gene Expression Induction by Molecules
Based on the positive z-scores, the following molecules were selected: ND_nw_193 (RSV604), CARS0358 (NA), and RASE0125 (17-O-Acetyl-nortetraphyllicine). The potential up- and downregulated human protein targets of these molecules were predicted based on their predicted pharmacological activity (Pa (activity probability) > 0.6) using the DIGEP-Pred server [54]. DIGEP-Pred uses the prediction of activity spectra for substances (PASS) algorithm to estimate various kinds of biological activities (including the interaction of small molecules with gene/proteins) by querying the 2D structure with those of well-known biologically active substances (i.e., activity = f (structure)). PASS prediction results are represented by the list of activities with probabilities ‘‘to be active’’ Pa and ‘‘to be inactive’’ Pi. The background data (i.e., drug-induced changes in gene expression) for DIGEP-Pred was taken from the Comparative Toxicogenomics Database (CTD) [55], which provides per se general drug-induced changes in gene expression [56], and such changes in gene expression represent the intrinsic properties of a drug molecule.
2.8. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations were performed to ascertain the structural binding stability of 3 potential multitarget NiV inhibitors—RASE0125, ND_nw_193, and CARS0358—with respective viral protein structures (i.e., NiV-G-ND_nw_193 complex, NiV-G-CARS0358 complex, and NiV-G-RASE0125 complex; NiV-F-ND_nw_193 complex, NiV-F-CARS0358 complex, and NiV-F-RASE0125 complex; NiV-N-ND_nw_193 complex, NiV-N-CARS0358 complex, and NiV-N-RASE0125 complex). As respective controls, MD simulations were also performed for free protein structures (apo-NiV-G; apo-NiV-N; apo-NiV-N). In total, 12 MD simulations were performed. Groningen Machine for Chemical Simulation (GROMACS) v 2020.1 software [57] was used for simulations utilizing CHARMM36 force field v 2019 [58] at 300 K. The topology parameters for inhibitor were produced in the CGenFF server (http://cgenff.umaryland.edu/) (accessed on 11 April 2022) and complexed into the protein topologies to make protein–inhibitor complexes. Systems were solvated using the simple point charge water model (spc216) in cubic boxes and counter ions (Na+ or Cl−) were added to neutralize the systems. Solvated systems were then minimized with 1000 steps using the steepest descent method, followed by the equilibration run (NVT + NPT), for 100 picoseconds. After equilibration, a 5 ns production simulation (MD run) was performed for all the 12 established systems (i.e., 9 complexes and 3 free proteins). The trajectories were analyzed by assessing C-α root-mean-square deviations (RMSDs) and root-mean-square fluctuation (RMSF) using “gmx rmsd” and “gmx rmsf” modules, respectively. The trajectories were plotted gnuplot-x11 program (http://gnuplot.info/).
2.9. Principal Component Analysis and Gibbs Free Energy Calculation
To explore the conformational changes docked molecules—RASE0125, ND_nw_193, CARS0358—brought into the viral protein structures, principal component analysis (PCA) was carried out utilizing the essential dynamics approach. Using the GROMACS “gmx anaeig” and “gmx covar” modules, the covariance matrix was calculated (considering the protein backbone atoms), followed by the diagonalization to calculate the eigenvectors and eigenvalues. Principal component analysis (PCA) was calculated from the trajectory, and the first two components (PC1 and PC2) were used for the plotting. The Gibbs free energy landscapes (FELs) were developed using the “g_sham” module to capture the lowest energy stable state. The first two principal components were used to calculate the FEL based on the equation . PC1 and PC2 are reaction coordinates, KB symbolizes the Boltzmann constant, and P (PC1, PC2) illustrates the probability distribution of the system over the first two principal components.
3. Results and Discussion
3.1. Compiling Small-Molecule Inhibitors of NiV
An extensive literature search was performed to obtain information on small-molecule NiV inhibitors or drug molecules. The PubMed database was queried by combining suitable keywords and Boolean operators to first obtain relevant articles. Next, these articles were manually curated to archive molecules’ data and their additional details, including chemical name, IUPAC name, 2D structure, references, etc. Finally, information on a total of 206 NiV inhibitors was compiled (Table S1).
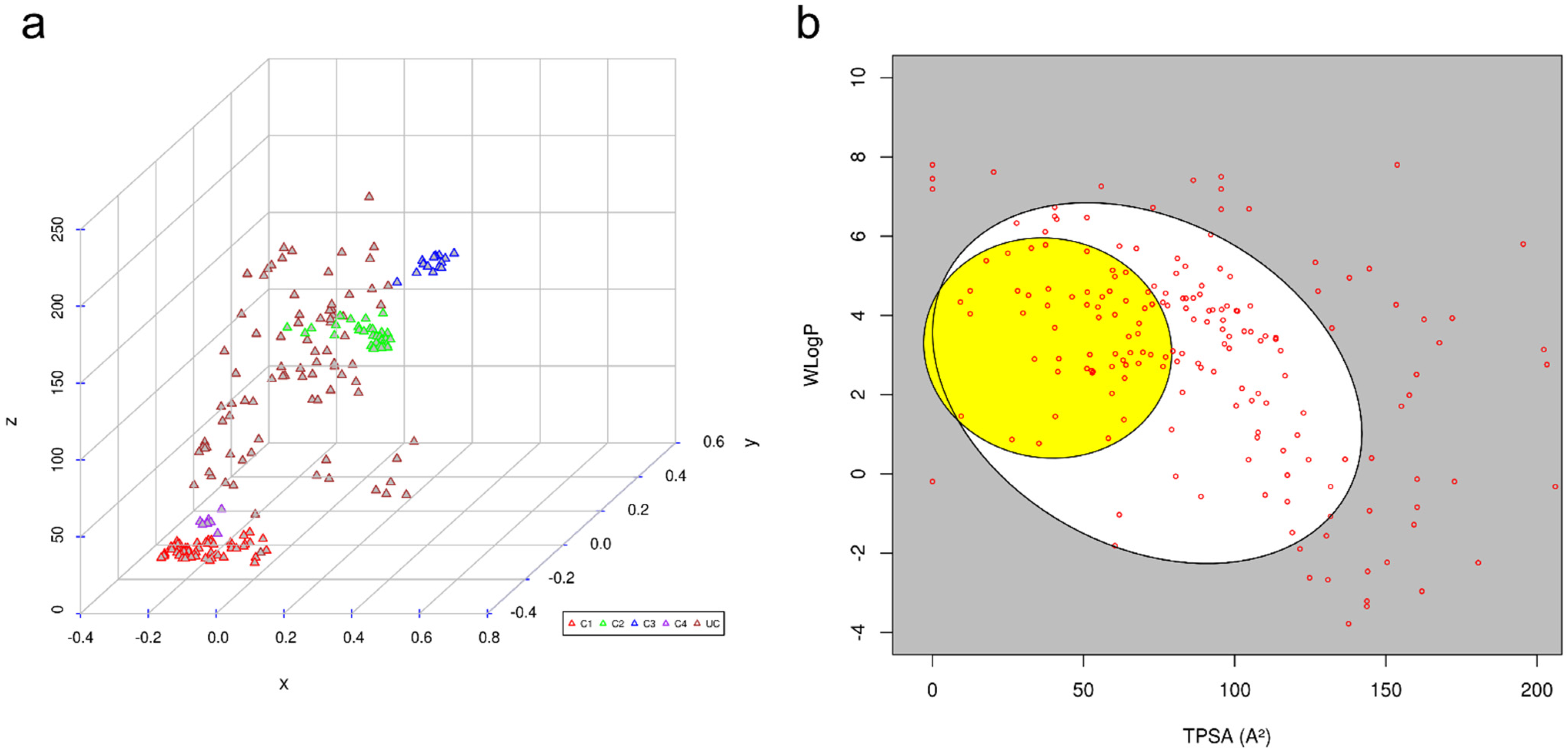
To assess the diversity and unique molecular scaffolds that were highly prevalent in compiled NiV inhibitors, these were clustered into their discrete groups on the basis of the Tanimoto similarity measure [59], one of the most widely used similarity measures for comparing chemical structures in cheminformatics. Clustering results were visualized by inspecting the multidimensional scaling (MDS) plot that indicated 206 inhibitors to be broadly grouped into four distinct clusters: Cluster 1 (n = 57), Cluster 2 (n = 35), Cluster 3 (n = 16), and Cluster 4 (n = 7). The molecules in each of the clusters comprised compounds with similar structural patterns (Figure 2), while the rest of the 91 molecules were present as independent entries.
ADME and Pharmacokinetic Properties of Small Molecules
To probe for NiV inhibitors that might be associated with adverse side effects, ADME and pharmacokinetic properties of literature-compiled small-molecule inhibitors were computed using the SwissADME webserver (Table S2). SwissADME uses comparative information derived from poorly and highly absorbed drugs to predict passive intestinal absorption and brain penetration, as a function of lipophilicity and apparent polarity (described by WLOGP and TPSA, respectively). For all compiled inhibitors, the predictions were displayed as a bi-plot, also called the BOILED-Egg plot [60] (Figure 2). A total of 160 molecules (~78%) fall inside the whole bi-plot, where white and yellow ellipses indicate molecules having a high probability of good intestinal absorption and blood–brain barrier (BBB) crossing, respectively. A total of 46 molecules (~22%) were predicted as not absorbed by the GI and BBB non-permeant, which are located in the grey area of the plot; these molecules were explicitly excluded from our analyses, as they are likely associated with side effects. The ellipsoidal regions defining the intestine and brain permeation were drawn as initially reported. The selected molecules were also analyzed for drug-relevant properties on the basis of “Lipinski’s rule of five”, which indicated a total of 149 (72%) to have drug-like properties.
3.2. Compiling Information on Small-Molecule Phytochemicals
Phytochemicals are plant-derived small-molecule inhibitors, which are rich sources of diverse scaffolds that could serve as a basis for rational drug design. Many plants have shown effective antiviral and immune-boosting potentials against viruses including SARS-CoV, Zika, Ebola, Nipah, and other highly pathogenic viruses [61]. Therefore, we also prospected for potential phytochemical molecules against NiV proteins. Information on different features of phytochemicals, including their 3D structures, was obtained from three well-established small-molecule databases—namely, SerpentinaDB (n = 142), Phytochemica (n = 868), and Phytochemical and Drug Target DataBase (PDTDB, n = 221); these databases contain extensive information on phytochemicals having therapeutic potential against various diseases. The molecules in these databases already have information on pharmacokinetic properties and, therefore, were not computed in our analyses.
3.3. Volumetric Analyses of Ligand-Binding Protein Pockets
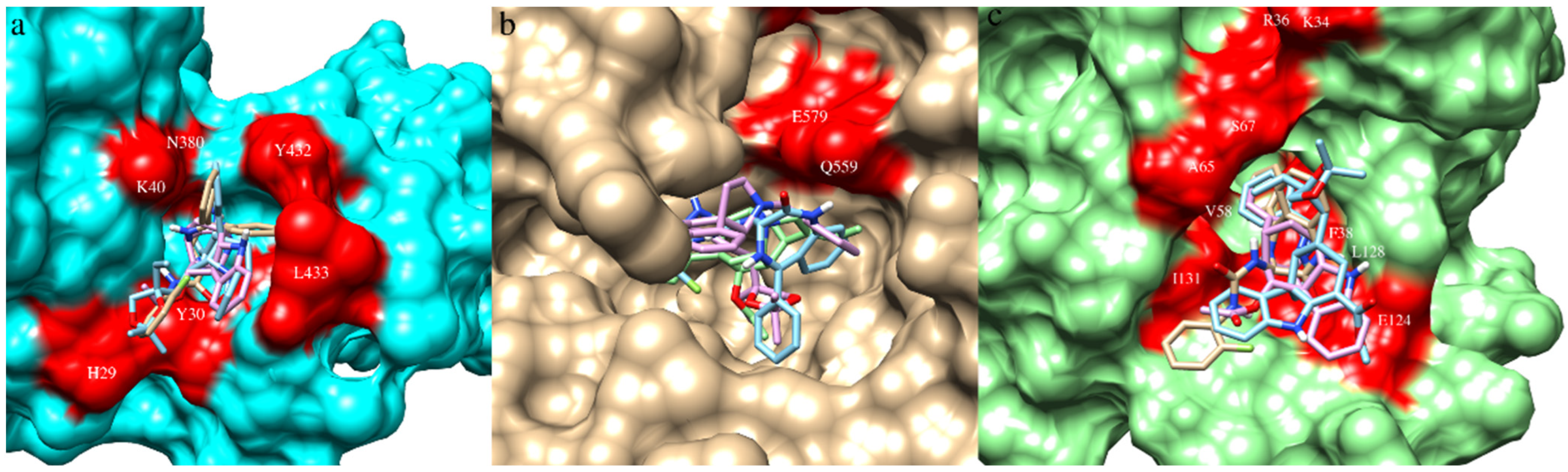
The data on ligand-binding sites in all three viral proteins were obtained from the published literature. The binding-site residues of protein NiV-G (Gln559, Glu579, Tyr581, Ile588) were obtained from Kalbhor et al. (2021) [46]. The residues Ile588 and Tyr581 participate in hydrophobic interactions and contribute to the binding pocket that accommodates Phe120 of human EFNB2; hence, they are considered critical for inhibiting interactions by which NiV attach to EFN receptors [62]. The binding-site residues of NiV-F (His29, Tyr30, Val39, Lys40, Asn380, Tyr432, and Leu433) and NiV-N (Lys34, Arg36, Phe38, Val58, Ala65, Ser67, Glu124, Leu128, and Ile131) were obtained from Sen et al. (2019) [47]. Pocket volumes of NiV-F, NiV-G, and Niv-N were 69 ų, 23 ų, and 60 ų, respectively; this indicated availability of similar conformational space for ligands in NiV-F and Niv-N but a smaller space for NiV-G.
3.4. Molecular Docking
Docking studies were carried out in order to find optimal conformations of all ligands into the respective binding pockets of NiV-G, NiV-F, and NiV-N. As a post-docking filter, molecules with the best binding affinity (≤−7.0 Kcal/mol) were considered, which predicted 300, 827, 685 molecules to be docking inside NiV-N, NiV-G, and NiV-F, respectively. The molecules were then shortlisted based on their interaction with the binding-site residues. Of 300 molecules in NiV-N, 227 molecules bind to ligand-binding sites (Lys34, Arg36, Phe38, Val58, Ala65, Ser67, Glu124, Leu128, and Ile131), whereas of 827 molecules in NiV-G, 620 molecules bind to the ligand-binding sites (Gln559, Glu579, Tyr581, and Ile588). Similarly, of 685 molecules in NiV-F, 681 molecules bind to ligand-binding sites (His29, Tyr30, Val39, Lys40, Asn380, Tyr432, Leu433).
To gain an understanding of differences in the number of docked small-molecule inhibitors, volumetric differences among ligand-binding pockets of proteins were analyzed. The difference in binding of a comparatively larger number of small molecules in NiV-F and NiV-N could be attributed to a comparatively wide open and large pocket, compared with NiV-G. The results are also contrary to the pocket volumes of viral proteins (Niv-G, 23 ų; NiV-F, 69 ų; Niv-N, 60 ų). The inhibitor molecules that bind to all three viral proteins (NiV-G ∩ NiV-F ∩ NiV-N) were considered. Overall, based on binding energy threshold (≤−7.0 Kcal/mol), interactions with ligand-binding-site residues, a total of 156 molecules are selected as common inhibitors of NiV-G, NiV-F, and NiV-N. Since 156 was a large number for further analyses, the binding-energy threshold was raised to −7.8 Kcal/mol, which predicted a comparatively smaller number of lists as common inhibitors of three viral proteins. Based on the readjusted threshold, a total of eight molecules were selected, of which two were chemical inhibitors, and six were phytochemicals. The binding affinities of eight molecules within the ligand-binding pocket are provided in Table 1, and the information on intermolecular interactions, including hydrogen bonding and hydrophobic interactions, is provided in Table S3. Chemical details of molecules, including IUPAC and SMILES, are provided in Table S4.
As a positive control, molecular docking studies were also performed with the drug remdesivir (GS-5734). Remdesivir is a nucleotide prodrug that has broad antiviral activity against viruses from different families and therapeutic efficacy in nonhuman primate models of lethal Nipah virus infection [63] and is, therefore, likely effective against Nipah virus infection in humans [53]. Remdesivir was also found to interact with all three viral proteins but with comparatively low binding affinity. Remdesivir binds to NiV-G with a binding affinity of −7.5 Kcal/mol, and it binds to NiV-F (−6.6 Kcal/mol) and Niv-N (−5.6 Kcal/mol) with comparatively poor affinity, which is attributed to weak intermolecular interactions among remdesivir and proteins.
3.5. Computing ADME Properties for Molecule Prioritization
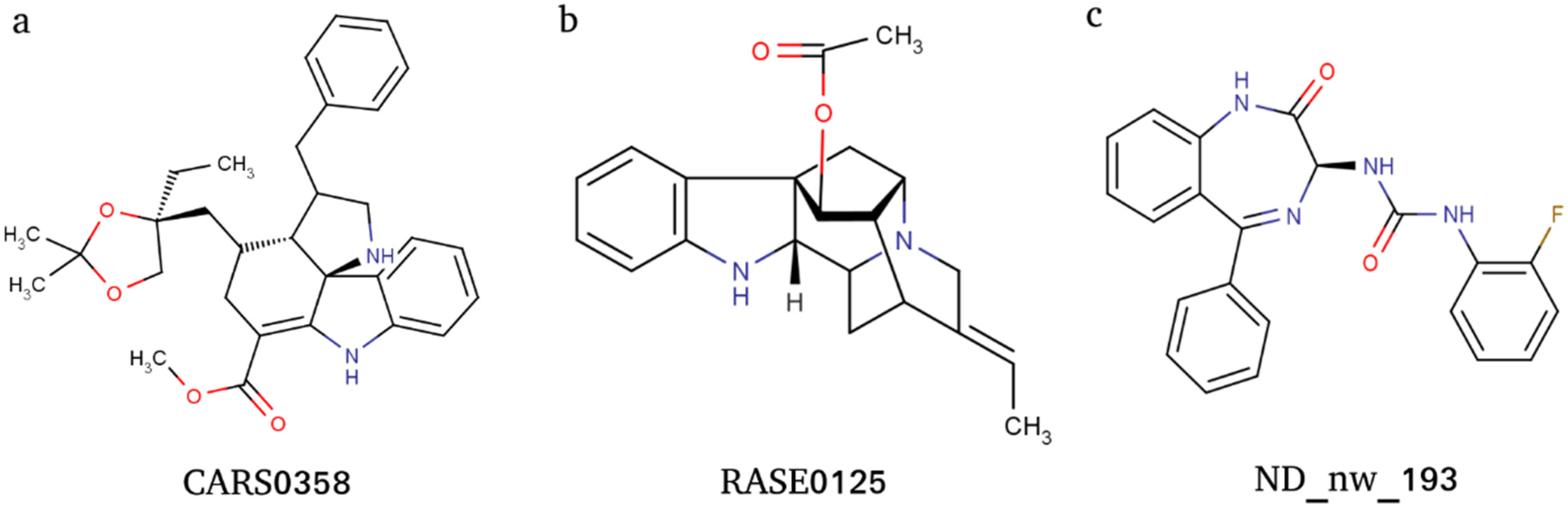
ADME and pharmacokinetic properties of docking-based prioritized molecules were computed using the ADMETlab webserver. The pharmacokinetic properties were transformed into z-scores and considered for further molecule prioritization (Table S5). A positive z-score indicates molecules that have more drug-like properties, and therefore, molecules were ranked and prioritized based on positive z-scores; this threshold shortlisted a total of three molecules: ND_nw_193 (RSV604), CARS0358 (NA), and RASE0125 (17-O-acetyl-nortetraphyllicine) (Figure 3). Information on ADME-based z-score is provided in Table 2. The docked conformations of the three molecules into their respective ligand-binding sites are provided in Figure 4.
3.6. Assessment of Gene Expression Induction by Small Molecules
Drug molecules interact with many off-targets that could lead to various side effects [64]. In fact, interactions between drug molecules and their targets/off-targets can often induce changes in expression profiles of many genes/proteins [65] that can either result in activation, repression, or dysregulation of downstream signaling pathways [66]. Therefore, to prioritize the molecules further, only those molecules were considered that could lead to dysregulation of expression profiles of a comparatively smaller number of human proteins. The potential up- and downregulated human protein targets of molecules—RASE0125, ND_nw_193, CARS0358—were predicted based on their predicted pharmacological activity (Pa (activity probability) > 0.6) using the DIGEP-Pred. It was observed that molecule RASE0125 could upregulate gene expression of proteins KRT18 (Pa = 0.670) and RAC1 (Pa = 0.621), while it downregulated CHEK1 (Pa = 0.655). Similarly, ND_nw_193 could only upregulate gene expression of ATG5 (Pa = 0.670). CARS0358 was not found to be regulating the expression profiles of any of the proteins. Overall, these three small-molecule inhibitors could induce the expression profiles of only a fewer number of proteins that could ultimately avoid dysregulation of critically important signaling pathways, thus reducing the risk of drug-induced severe side effects.
3.7. Molecular Dynamics Simulations
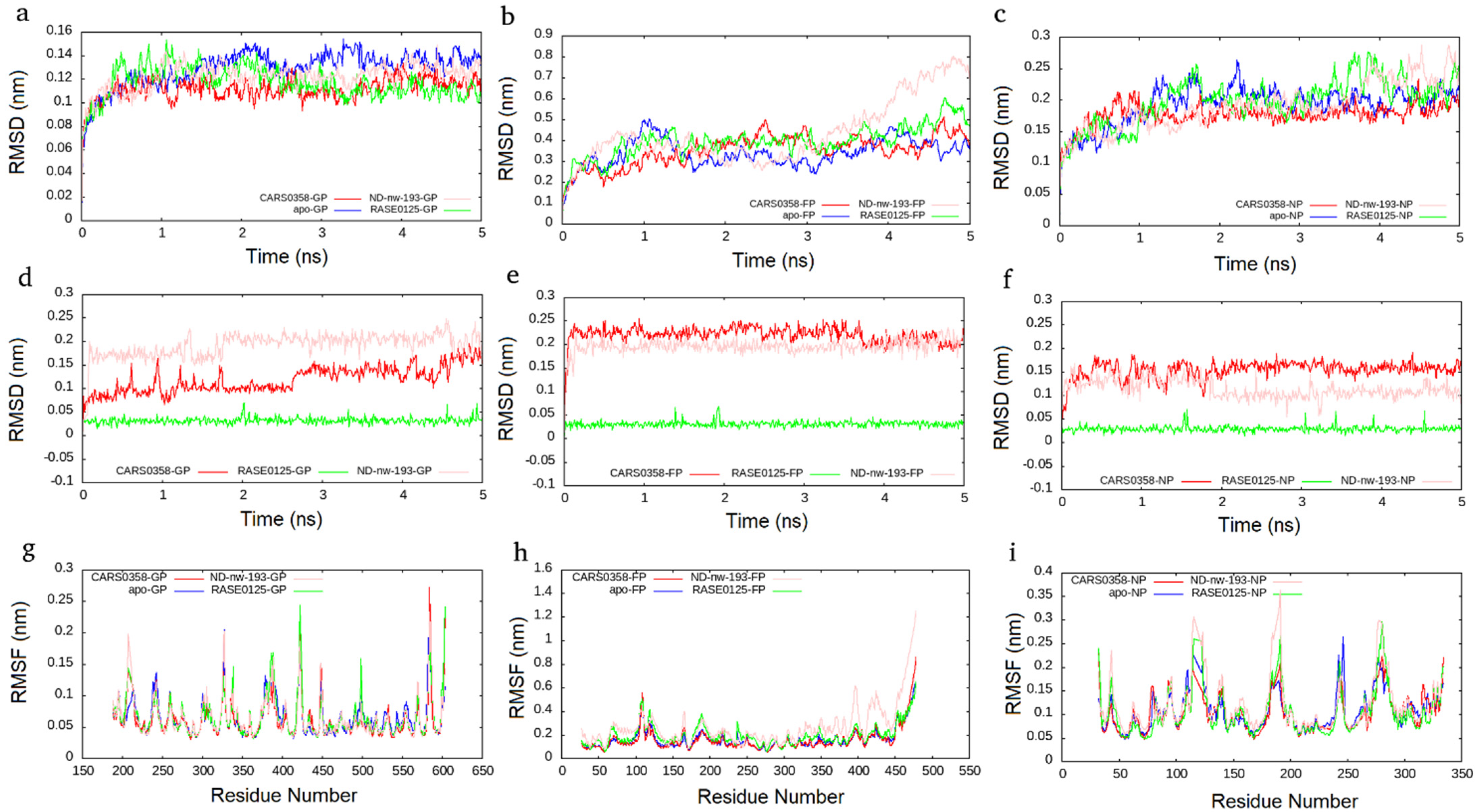
MD simulation presents an approach for the structural refinement of docked complexes [67]. In order to refine and examine the stability of the three selected molecules (RASE0125, ND_nw_193, and CARS0358), MD simulations of corresponding protein–ligand complexes, as well as free proteins, were performed for 5 ns. Since remdesivir, the positive control, was not able to well bind to all of the three viral protein targets, we did not perform its MD simulations. The stability of complexes was assessed by computing two main structural parameters: Cα-RMSD and -RMSF. RMSD is a standard measure of computing structural distance between coordinates and measures the average distance between a group of atoms. As a control, Cα RMSD values of all free proteins were also computed. Complexes of molecules RASE0125 and CARS0358 were found to be stable in all the three viral protein structures and plateaued to average RMSDs of ~0.12 nm, ~0.35 nm, and ~0.12 nm in NiV-G, NiV-F, and NiV-N protein structures (Figure 5). It was interesting to observe that the docked complexes were more stable than the free proteins. Apart from protein complexes, the RMSDs of bound molecules were also computed during the simulations, and both the molecules were found to be quite stable. The complex derived from molecule ND_nw_193 was stable in NiV-G and NiV-N and plateaued to an average RMSD similar to that of RASE0125 and CARS0358. Apart from whole-molecule RMSDs, RMSDs of bound molecules alone were also computed, which also presented stable conformations (Figure 5). RMSF, a measure of the displacement of atom(s) relative to the reference structure, is another measure of structure assessment that captures local changes in protein structures. As a control, Cα RMSF values of all free proteins were also computed. Cα RMSF values indicated that the molecules RASE0125 and CARS0358 did not cause much fluctuations in the amino acids in NiV-G and NiV-N, indicating these to be not disturbed during ligand binding (Figure 5). Molecule ND_nw_193 also did not cause fluctuations in NiV-G and NiV-N, but the overall flexibility of the NiV-F protein structure increased upon ligand binding; these results are also in concordance with the results of RMSDs. Importantly, none of the critical binding-site residues showed large flexibility. The high peaks in the RMSF plots indicate fluctuations in loop regions that are comparatively higher than those in the structured regions due to their inherent structural flexibility.
3.8. Details of Selected Multitarget Molecules
Overall, the molecules RASE0125 and CARS0358 were well capable of binding to NiV-G, NiV-F, and NiV-N proteins, whereas ND_nw_193 could only bind well to NiV-G and NiV-N. Both RASE0125 (17-O-Acetyl-nortetraphyllicine) and CARS0358 are indole alkaloids (phytochemicals), derived from plant Rauvolfia serpentina and Catharanthus roseus, respectively. Indole alkaloid derivatives are known to inhibit dengue and Zika virus infection by modulating the virus replication complex [68]. Therefore, we also speculate that indole alkaloids RASE0125 and CARS0358 might possess the same mode of action on NiV. Literature search confirmed that these molecules have never been considered even for antiviral drug discovery. Contrarily, ND_nw_193 (CHEMBL223402) is a chemical drug RSV604, which is a known inhibitor of human respiratory syncytial virus (RSV) [69].
RASE0125 and CARS0358 established many hydrophobic contacts and H-bonds with binding-site residues with all three viral protein targets. In NiV-G, RASE0125 established three hydrophobic contacts with Gln559, whereas CARS0358 established six hydrophobic contacts with Gln559, and ND_nw_193 established five hydrophobic contacts with Gln559. It is interesting to note that Gln559 is the common amino acid residue in NiV-G that binds to all three molecules. In NiV-N, RASE0125, CARS0358, and ND_nw_193 established 27, 40, and 34 hydrophobic contacts with binding-site residues (Table S3), having residues Ile31, Val58, Leu128, Phe38, and Arg36 in common. In NiV-F, RASE0125 and CARS0358 established 28 and 22 hydrophobic contacts with binding-site residues (Table S3), having residues Tyr30, His29, Lys40, Val39, Tyr432, and Leu433 in common. The common binding-site residues could also be of prime interest to virologists for anti-NiV drug discovery.
3.9. Principal Component Analysis and Gibbs Free Energy Calculation
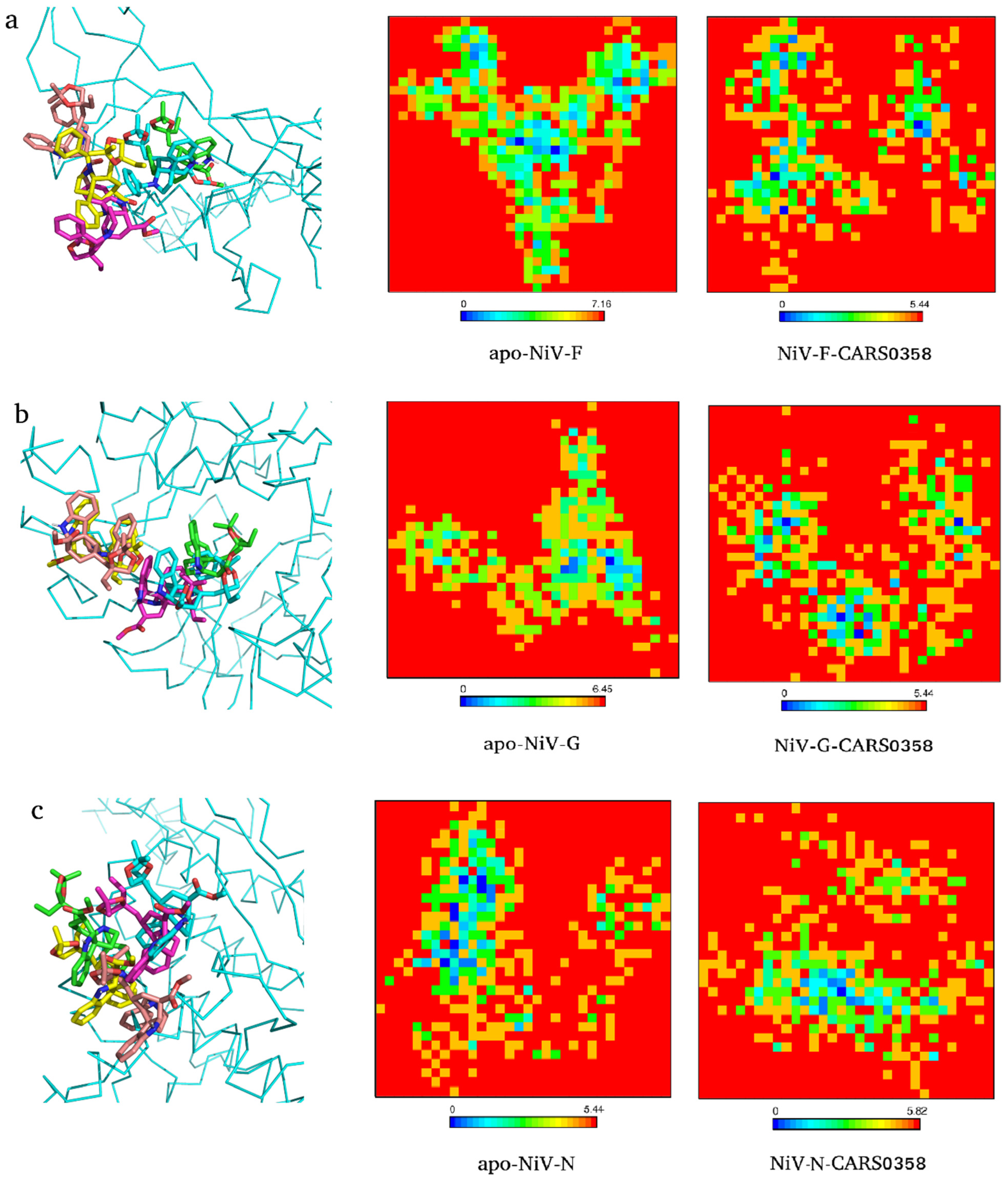
To explore the conformational changes docked molecules (RASE0125, ND_nw_193, and CARS0358) brought into the viral protein structures, PCA was carried out utilizing the essential dynamics approach. The high fluctuations in residues of proteins are captured through PCA [70], while variation in GFE values is computed to assess protein stability [71,72]. Proteins regulate their functions via entering into different conformations. The overall conformational change is governed by the collective movements of the atoms in a protein and this internal motion can be measured using PCA analysis. Therefore, to study the collective motion of three selected molecules before and after binding with viral protein targets occupied in the conformational subspace during the simulation, PC1 and PC2 were computed. FELs deliver a precise portrayal of a protein’s most stable conformational ensembles, which are certainly important for the study of conformational changes underlying protein−ligand interactions. The FEL plots were constructed and analyzed using the first two PCs (eigenvectors). The corresponding free energy contour map with a deeper blue color indicates lower energy (global minima) and energetically favored protein conformations, and yellow spots reflect unfavorable conformations [73]. As an example, we extracted the conformations of docked complexes CARS0358-NiV-G/NiV-F/NiV-N at different time points and superimposed the respective ligand conformations (Figure 6). In all of the complexes, minor differences in ligand conformations were observed at each time point corresponding to changes in energy basins during the course of the simulation. The FELs of free proteins and CARS0358-bound proteins (final conformations) are provided in Figure 6. The FELs of RASE0125 and ND_nw_193 complexes with respective viral proteins are provided in Figure S1. In all of the FEL plots, free and molecule-bound complexes had different patterns for the free energies, and protein structures were still able to attain comparable energetically and structurally stable conformations. FEL analyses suggested that the presence of small molecules affected the size and the position of the sampled energy basin to achieve stable equilibriums.
4. Conclusions
In this study, a sequential molecular docking and molecular dynamics-based approach was implemented to prospect for potential multitarget chemical/phytochemical inhibitor(s) against NiV by simultaneously targeting NiV-G, NiV-F, and NiV-N. The study identified phytochemical molecules RASE0125 (17-O-acetyl-nortetraphyllicine) and CARS0358 (NA), which are indole alkaloids, as distinct multitarget inhibitors of all three viral proteins, while chemical molecule ND_nw_193 (RSV604) was revealed to be an inhibitor of NiV-G and NiV-N.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms10061181/s1, Table S1: Information on NiV inhibitors compiled from the literature, Table S2: ADME and pharmacokinetic properties of NiV inhibitors, Table S3: Intermolecular interactions among docking-based prioritized molecules and viral proteins, Table S4: Information on docking-based prioritized molecules, Table S5: The pharmacokinetic properties and transformed z-scores of docking-based prioritized molecules, Figure S1: Two-dimensional (2D) graphs of the Gibbs free energy landscape for NiV-F (a), NiV-G (b), and NiV-N (c) (apo- and docked complexes).
Author Contributions
Conceptualization, M.K.; methodology, V.R. and S.P.; formal analysis, V.R. and S.P.; investigation, V.R., S.P. and M.K.; data curation, V.R., S.P. and M.K.; writing—original draft preparation, V.R. and S.P.; writing—review and editing, V.R., S.P. and M.K.; supervision, M.K.; funding acquisition, M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the DST-Science and Engineering Research Board (SERB), the Government of India (VR, N-PDF file number PDF/2016/002009; SP, N-PDF file number PDF/2016/002422), and the grants from the CSIR-Institute of Microbial Technology, Council of Scientific and Industrial Research (CSIR) (GAP0001 and STS0038).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We acknowledge the support of the CSIR-Institute of Microbial Technology and the Department of Biotechnology, Government of India for providing computational infrastructure.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analysis, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
CCDC, Cambridge Crystallographic Data Centre; MD, Molecular dynamics; NiV, Nipah virus; NiV-F, NiV-fusion glycoprotein; NiV-G, NiV-attachment glycoprotein; NiV-N, NiV-nucleoprotein; RMSD, root-mean-square deviation; RMSF, root-mean-square fluctuation.
References
- Chua, K.B.; Bellini, W.J.; Rota, P.A.; Harcourt, B.H.; Tamin, A.; Lam, S.K.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W.; et al. Nipah Virus: A Recently Emergent Deadly Paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Chattu, V.K.; Kumar, R.; Kumary, S.; Kajal, F.; David, J.K. Nipah Virus Epidemic in Southern India and Emphasizing “One Health” Approach to Ensure Global Health Security. J. Fam. Med. Prim. Care 2018, 7, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, A.N.; Worthy, M.N.; Lee, B.; Holbrook, M.R. Combined Chloroquine and Ribavirin Treatment Does Not Prevent Death in a Hamster Model of Nipah and Hendra Virus Infection. J. Gen. Virol. 2010, 91, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Tigabu, B.; Rasmussen, L.; White, E.L.; Tower, N.; Saeed, M.; Bukreyev, A.; Rockx, B.; LeDuc, J.W.; Noah, J.W. A BSL-4 High-Throughput Screen Identifies Sulfonamide Inhibitors of Nipah Virus. Assay Drug Dev. Technol. 2014, 12, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Niedermeier, S.; Singethan, K.; Rohrer, S.G.; Matz, M.; Kossner, M.; Diederich, S.; Maisner, A.; Schmitz, J.; Hiltensperger, G.; Baumann, K.; et al. A Small-Molecule Inhibitor of Nipah Virus Envelope Protein-Mediated Membrane Fusion. J. Med. Chem. 2009, 52, 4257–4265. [Google Scholar] [CrossRef]
- Dawes, B.E.; Kalveram, B.; Ikegami, T.; Juelich, T.; Smith, J.K.; Zhang, L.; Park, A.; Lee, B.; Komeno, T.; Furuta, Y.; et al. Favipiravir (T-705) Protects against Nipah Virus Infection in the Hamster Model. Sci. Rep. 2018, 8, 7604. [Google Scholar] [CrossRef] [Green Version]
- Hotard, A.L.; He, B.; Nichol, S.T.; Spiropoulou, C.F.; Lo, M.K. 4’-Azidocytidine (R1479) Inhibits Henipaviruses and Other Paramyxoviruses with High Potency. Antivir. Res. 2017, 144, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, V.; Contamin, H.; Loth, P.; Grosjean, I.; Courbot, M.C.G.; Deubel, V.; Buckland, R.; Wild, T.F. Antibody Prophylaxis and Therapy against Nipah Virus Infection in Hamsters. J. Virol. 2006, 80, 1972–1978. [Google Scholar] [CrossRef] [Green Version]
- Snell, N.J.C. Ribavirin Therapy for Nipah Virus Infection. J. Virol. 2004, 78, 10211. [Google Scholar] [CrossRef] [Green Version]
- Chong, H.T.; Kamarulzaman, A.; Tan, C.T.; Goh, K.J.; Thayaparan, T.; Kunjapan, S.R.; Chew, N.K.; Chua, K.B.; Lam, S.K. Treatment of Acute Nipah Encephalitis with Ribavirin. Ann. Neurol. 2001, 49, 810–813. [Google Scholar] [CrossRef]
- Bowden, T.A.; Aricescu, A.R.; Gilbert, R.J.C.; Grimes, J.M.; Jones, E.Y.; Stuart, D.I. Structural Basis of Nipah and Hendra Virus Attachment to Their Cell-Surface Receptor Ephrin-B2. Nat. Struct. Mol. Biol. 2008, 15, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Biering, S.B.; Huang, A.; Vu, A.T.; Robinson, L.R.; Bradel-Tretheway, B.; Choi, E.; Lee, B.; Aguilar, H.C. N-Glycans on the Nipah Virus Attachment Glycoprotein Modulate Fusion and Viral Entry as They Protect against Antibody Neutralization. J. Virol. 2012, 86, 11991–12002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ker, D.S.; Jenkins, H.T.; Greive, S.J.; Antson, A.A. CryoEM Structure of the Nipah Virus Nucleocapsid Assembly. PLoS Pathog. 2021, 17, e1009740. [Google Scholar] [CrossRef]
- Csermely, P.; Ágoston, V.; Pongor, S. The Efficiency of Multi-Target Drugs: The Network Approach Might Help Drug Design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A Perspective on Multi-Target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Viana, J.D.O.; Félix, M.B.; Maia, M.D.S.; Serafim, V.d.L.; Scotti, L.; Scotti, M.T. Drug Discovery and Computational Strategies in the Multitarget Drugs Era. Braz. J. Pharm. Sci. 2018, 54. [Google Scholar] [CrossRef]
- Makhoba, X.H.; Viegas, C.; Mosa, R.A.; Viegas, F.P.D.; Pooe, O.J. Potential Impact of the Multi-Target Drug Approach in the Treatment of Some Complex Diseases. Drug Des. Devel. Ther. 2020, 14, 3235. [Google Scholar] [CrossRef]
- Qiu, C.; Kivipelto, M.; Von Strauss, E. Epidemiology of Alzheimer’s Disease: Occurrence, Determinants, and Strategies toward Intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology. ChemMedChem 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Pei, J.; Lai, L. Computational Multitarget Drug Design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef]
- Zhou, Y.; Shan, S.; Li, Z.B.; Xin, L.J.; Pan, D.S.; Yang, Q.J.; Liu, Y.P.; Yue, X.P.; Liu, X.R.; Gao, J.Z.; et al. CS2164, a Novel Multi-Target Inhibitor against Tumor Angiogenesis, Mitosis and Chronic Inflammation with Anti-Tumor Potency. Cancer Sci. 2017, 108, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousuf, Z.; Iman, K.; Iftikhar, N.; Mirza, M.U. Structure-Based Virtual Screening and Molecular Docking for the Identification of Potential Multi-Targeted Inhibitors against Breast Cancer. Breast Cancer 2017, 9, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Bast, F. Screening of Multi-Targeted Natural Compounds for Receptor Tyrosine Kinases Inhibitors and Biological Evaluation on Cancer Cell Lines, in Silico and in Vitro. Med. Oncol. 2015, 32, 233. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.X.; Shang, W.J.; Yin, W.C.; Ge, H.; Wang, L.; Zhang, X.L.; Li, B.Q.; Li, H.L.; Xu, Y.C.; Xu, E.H.; et al. A Multi-Targeting Drug Design Strategy for Identifying Potent Anti-SARS-CoV-2 Inhibitors. Acta Pharmacol. Sin. 2021, 43, 483–493. [Google Scholar] [CrossRef]
- Joshi, R.S.; Jagdale, S.S.; Bansode, S.B.; Shankar, S.S.; Tellis, M.B.; Pandya, V.K.; Chugh, A.; Giri, A.P.; Kulkarni, M.J. Discovery of Potential Multi-Target-Directed Ligands by Targeting Host-Specific SARS-CoV-2 Structurally Conserved Main Protease. J. Biomol. Struct. Dyn. 2021, 39, 3099–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Jiang, W.; Jia, H.; Zheng, L.; Xing, J.; Liu, A.; Du, G. Discovery of Multitarget-Directed Ligands Against Influenza A Virus From Compound Yizhihao Through a Predictive System for Compound-Protein Interactions. Front. Cell. Infect. Microbiol. 2020, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.H.; Shi, Z.; Tan, C.; Jiang, Y.; Go, M.L.; Low, B.C.; Chen, Y.Z. In-Silico Approaches to Multi-Target Drug Discovery: Computer Aided Multi-Target Drug Design, Multi-Target Virtual Screening. Pharm. Res. 2010, 27, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A Database of Experimentally Validated Antiviral Peptides Targeting Medically Important Viruses. Nucleic Acids Res. 2014, 42, D1147–D1153. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Kaur, K.; Rajput, A.; Dhanda, S.K.; Sehgal, M.; Khan, M.S.; Monga, I.; Dar, S.A.; Singh, S.; Nagpal, G.; et al. ZikaVR: An Integrated Zika Virus Resource for Genomics, Proteomics, Phylogenetic and Therapeutic Analysis. Sci. Rep. 2016, 6, 32713. [Google Scholar] [CrossRef]
- Thakur, N.; Qureshi, A.; Kumar, M. AVPpred: Collection and Prediction of Highly Effective Antiviral Peptides. Nucleic Acids Res. 2012, 40, W199–W204. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, A.; Tandon, H.; Kumar, M. AVP-IC50 Pred: Multiple Machine Learning Techniques-Based Prediction of Peptide Antiviral Activity in Terms of Half Maximal Inhibitory Concentration (IC50). Biopolymers 2015, 104, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.; Kaur, G.; Kumar, M. AVCpred: An Integrated Web Server for Prediction and Design of Antiviral Compounds. Chem. Biol. Drug Des. 2017, 89, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.; Rajput, A.; Kaur, G.; Kumar, M. HIVprotI: An Integrated Web Based Platform for Prediction and Design of HIV Proteins Inhibitors. J. Cheminform. 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, A.; Thakur, N.; Kumar, M. VIRsiRNApred: A Web Server for Predicting Inhibition Efficacy of SiRNAs Targeting Human Viruses. J. Transl. Med. 2013, 11, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; Kumar, A.; Rajput, A.; Kaur, K.; Dar, S.A.; Thakur, A.; Megha, K.; Kumar, M. NipahVR: A Resource of Multi-Targeted Putative Therapeutics and Epitopes for the Nipah Virus. Database 2020, 2020, baz159. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Kumar, A.; Kumar, M. Computational Identification of Inhibitors Using QSAR Approach against Nipah Virus. Front. Pharmacol. 2019, 10, 71. [Google Scholar] [CrossRef] [Green Version]
- Pathania, S.; Randhawa, V.; Kumar, M. Identifying Potential Entry Inhibitors for Emerging Nipah Virus by Molecular Docking and Chemical-Protein Interaction Network. J. Biomol. Struct. Dyn. 2020, 38, 5108–5125. [Google Scholar] [CrossRef]
- Pathania, S.; Ramakrishnan, S.M.; Randhawa, V.; Bagler, G. SerpentinaDB: A Database of Plant-Derived Molecules of Rauvolfia Serpentina. BMC Complement. Altern. Med. 2015, 15, 262. [Google Scholar] [CrossRef] [Green Version]
- Pathania, S.; Ramakrishnan, S.M.; Bagler, G. Phytochemica: A Platform to Explore Phytochemicals of Medicinal Plants. Database J. Biol. Databases Curation 2015, 2015, bav075. [Google Scholar] [CrossRef] [Green Version]
- Ashok Kumar, T. PDTDB—An Integrative Structural Database and Prediction Server for Plant Metabolites and Therapeutic Drug Targets. Int. J. Curr. Res. 2017, 9, 46537–46541. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Charisi, A.; Cheng, L.-C.; Jiang, T.; Girke, T. ChemmineR: A Compound Mining Framework for R. Bioinformatics 2008, 24, 1733–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Backman, T.W.H.; Horan, K.; Girke, T. FmcsR: Mismatch Tolerant Maximum Common Substructure Searching in R. Bioinformatics 2013, 29, 2792–2794. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Chan, Y.-P.; Bradel-Tretheway, B.; Akyol-Ataman, Z.; Zhu, Y.; Dutta, S.; Yan, L.; Feng, Y.; Wang, L.-F.; Skiniotis, G.; et al. Crystal Structure of the Pre-Fusion Nipah Virus Fusion Glycoprotein Reveals a Novel Hexamer-of-Trimers Assembly. PLoS Pathog. 2015, 11, e1005322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabukarski, F.; Lawrence, P.; Tarbouriech, N.; Bourhis, J.M.; Delaforge, E.; Jensen, M.R.; Ruigrok, R.W.H.; Blackledge, M.; Volchkov, V.; Jamin, M. Structure of Nipah Virus Unassembled Nucleoprotein in Complex with Its Viral Chaperone. Nat. Struct. Mol. Biol. 2014, 21, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Kalbhor, M.S.; Bhowmick, S.; Alanazi, A.M.; Patil, P.C.; Islam, M.A. Multi-Step Molecular Docking and Dynamics Simulation-Based Screening of Large Antiviral Specific Chemical Libraries for Identification of Nipah Virus Glycoprotein Inhibitors. Biophys. Chem. 2021, 270, 106537. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Kanitkar, T.R.; Roy, A.A.; Soni, N.; Amritkar, K.; Supekar, S.; Nair, S.; Singh, G.; Madhusudhan, M.S. Predicting and Designing Therapeutics against the Nipah Virus. PLoS Negl. Trop. Dis. 2019, 13, e0007419. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; de Oliveira, C.A.F.; McCammon, J.A. POVME: An Algorithm for Measuring Binding-Pocket Volumes. J. Mol. Graph. Model. 2011, 29, 773–776. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Alhossary, A.; Handoko, S.D.; Mu, Y.; Kwoh, C.-K. Fast, Accurate, and Reliable Molecular Docking with QuickVina 2. Bioinformatics 2015, 31, 2214–2216. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; Feldmann, F.; Cronin, J.; Jordan, R.; Okumura, A.; Thomas, T.; Scott, D.; Cihlar, T.; Feldmann, H. Prophylactic and Therapeutic Remdesivir (GS-5734) Treatment in the Rhesus Macaque Model of MERS-CoV Infection. Proc. Natl. Acad. Sci. USA 2020, 117, 6771–6776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Ivanov, S.; Rudik, A.; Filimonov, D.; Poroikov, V. DIGEP-Pred: Web Service for in Silico Prediction of Drug-Induced Gene Expression Profiles Based on Structural Formula. Bioinformatics 2013, 29, 2062–2063. [Google Scholar] [CrossRef] [Green Version]
- Mattingly, C.J.; Rosenstein, M.C.; Colby, G.T.; Forrest, J.N.; Boyer, J.L. The Comparative Toxicogenomics Database (CTD): A Resource for Comparative Toxicological Studies. J. Exp. Zool. Part A Comp. Exp. Biol. 2006, 305, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.A.; Rajpal, D.K. Applications of Connectivity Map in Drug Discovery and Development. Drug Discov. Today 2012, 17, 1289–1298. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular Similarity in Medicinal Chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Anand, A.V.; Balamuralikrishnan, B.; Kaviya, M.; Bharathi, K.; Parithathvi, A.; Arun, M.; Senthilkumar, N.; Velayuthaprabhu, S.; Saradhadevi, M.; Al-Dhabi, N.A.; et al. Medicinal Plants, Phytochemicals, and Herbs to Combat Viral Pathogens Including SARS-CoV-2. Molecules 2021, 26, 1775. [Google Scholar] [CrossRef] [PubMed]
- Bowden, T.A.; Crispin, M.; Harvey, D.J.; Aricescu, A.R.; Grimes, J.M.; Jones, E.Y.; Stuart, D.I. Crystal Structure and Carbohydrate Analysis of Nipah Virus Attachment Glycoprotein: A Template for Antiviral and Vaccine Design. J. Virol. 2008, 82, 11628–11636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.K.; Feldmann, F.; Gary, J.M.; Jordan, R.; Bannister, R.; Cronin, J.; Patel, N.R.; Klena, J.D.; Nichol, S.T.; Cihlar, T.; et al. Remdesivir (GS-5734) Protects African Green Monkeys from Nipah Virus Challenge. Sci. Transl. Med. 2019, 11, eaau9242. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug Discovery for the Future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Noh, H.; Shoemaker, J.E.; Gunawan, R. Network Perturbation Analysis of Gene Transcriptional Profiles Reveals Protein Targets and Mechanism of Action of Drugs and Influenza A Viral Infection. Nucleic Acids Res. 2018, 46, e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melas, I.N.; Sakellaropoulos, T.; Iorio, F.; Alexopoulos, L.G.; Loh, W.-Y.; Lauffenburger, D.A.; Saez-Rodriguez, J.; Bai, J.P.F. Identification of Drug-Specific Pathways Based on Gene Expression Data: Application to Drug Induced Lung Injury. Integr. Biol. 2015, 7, 904–920. [Google Scholar] [CrossRef] [Green Version]
- Pathania, S.; Randhawa, V.; Bagler, G. Prospecting for Novel Plant-Derived Molecules of Rauvolfia Serpentina as Inhibitors of Aldose Reductase, a Potent Drug Target for Diabetes and Its Complications. PLoS ONE 2013, 8, e61327. [Google Scholar] [CrossRef] [Green Version]
- Fikatas, A.; Vervaeke, P.; Meyen, E.; Llor, N.; Ordeix, S.; Boonen, I.; Bletsa, M.; Kafetzopoulou, L.E.; Lemey, P.; Amat, M.; et al. A Novel Series of Indole Alkaloid Derivatives Inhibit Dengue and Zika Virus Infection by Interference with the Viral Replication Complex. Antimicrob. Agents Chemother. 2021, 65, e0234920. [Google Scholar] [CrossRef]
- Chapman, J.; Abbott, E.; Alber, D.G.; Baxter, R.C.; Bithell, S.K.; Henderson, E.A.; Carter, M.C.; Chambers, P.; Chubb, A.; Cockerill, G.S.; et al. RSV604, a Novel Inhibitor of Respiratory Syncytial Virus Replication. Antimicrob. Agents Chemother. 2007, 51, 3346–3353. [Google Scholar] [CrossRef] [Green Version]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential Dynamics of Proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Khan, M.T.; Ali, S.; Zeb, M.T.; Kaushik, A.C.; Malik, S.I.; Wei, D.Q. Gibbs Free Energy Calculation of Mutation in PncA and RpsA Associated With Pyrazinamide Resistance. Front. Mol. Biosci. 2020, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, A.; Shahwan, M.; Khan, M.S.; Husain, F.M.; Alhumaydhi, F.A.; Aljohani, A.S.M.; Rehman, M.T.; Hassan, M.I.; Islam, A. Elucidating the Interaction of Human Ferritin with Quercetin and Naringenin: Implication of Natural Products in Neurodegenerative Diseases: Molecular Docking and Dynamics Simulation Insight. ACS Omega 2021, 6, 7922–7930. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.X.; Trovato, A.; Seno, F.; Banavar, J.R.; Maritan, A. Geometry and Symmetry Presculpt the Free-Energy Landscape of Proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 7960–7964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Workflow for the integrated approach for multitarget molecule screening in NiV.

Figure 2.
(a) Clustering results of molecules compiled from literature, where red-, green-, blue-, and purple-colored triangles represent Clusters 1–4. Unclustered molecules are represented as brown triangles; (b) graphical distribution of molecules compiled from literature as BOILED-Egg plot.
Figure 2.
(a) Clustering results of molecules compiled from literature, where red-, green-, blue-, and purple-colored triangles represent Clusters 1–4. Unclustered molecules are represented as brown triangles; (b) graphical distribution of molecules compiled from literature as BOILED-Egg plot.

Figure 3.
Two-dimensional (2D) images of molecules CARS0358 (a), RASE0125 (b), and ND_nw_193 (c).

Figure 4.
Molecule CARS0358 (blue), RASE0125 (pink), and ND_nw_193 (bisque) docked inside the respective ligand-binding sites (red colored) of NiV-F (a), NiV-G (b), and NiV-N (c).
Figure 4.
Molecule CARS0358 (blue), RASE0125 (pink), and ND_nw_193 (bisque) docked inside the respective ligand-binding sites (red colored) of NiV-F (a), NiV-G (b), and NiV-N (c).

Figure 5.
Cα−RMSD plots for selected molecules (CARS0358 (red); RASE0125 (green), ND_nw_193 (pink)), complexed with NiV−G (a), NiV-F (b) and NiV-N (c). Cα-RMSD plots for free proteins (blue) are also presented. RMSD plots for individual molecules in NiV−G (d), NiV−F (e), and NiV−N (f). Cα−RMSF plot for NiV−G (g), NiV−F (h), and NiV−N (i) as a function of time.
Figure 5.
Cα−RMSD plots for selected molecules (CARS0358 (red); RASE0125 (green), ND_nw_193 (pink)), complexed with NiV−G (a), NiV-F (b) and NiV-N (c). Cα-RMSD plots for free proteins (blue) are also presented. RMSD plots for individual molecules in NiV−G (d), NiV−F (e), and NiV−N (f). Cα−RMSF plot for NiV−G (g), NiV−F (h), and NiV−N (i) as a function of time.

Figure 6.
Conformations (1 ns (green), 2 ns (cyan), 3 ns (pink), 4 ns (yellow), and 5 ns (brick)) of molecule CARS0358 inside binding pockets of NiV-F (a), NiV-G (b), and NiV-N (c) during whole MD simulations (left panel). The respective 2D graphs of the Gibbs free energy landscape for apo- (middle panel) and final ligand-bound conformation (right panel) are also provided.
Figure 6.
Conformations (1 ns (green), 2 ns (cyan), 3 ns (pink), 4 ns (yellow), and 5 ns (brick)) of molecule CARS0358 inside binding pockets of NiV-F (a), NiV-G (b), and NiV-N (c) during whole MD simulations (left panel). The respective 2D graphs of the Gibbs free energy landscape for apo- (middle panel) and final ligand-bound conformation (right panel) are also provided.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Binding affinity (BA, Kcal/mol) values of 8 selected molecules against viral protein targets.
Table 1.
Binding affinity (BA, Kcal/mol) values of 8 selected molecules against viral protein targets.
| Mol. ID | Mol./Drug Name | Mol. Type | BA (NiV-F) | BA (NiV-G) | BA (NiV-N) |
|---|---|---|---|---|---|
| ND_nw_193 | RSV604 | Chemical | −8.6 | −8.8 | −9.0 |
| ND_nw_93 | AC1MH6FW | Chemical | −8.2 | −8.3 | −8.8 |
| CARS0358 | NA | Phytochemical | −7.9 | −8.0 | −8.4 |
| CARS0394 | NA | Phytochemical | −7.8 | −7.8 | −8.9 |
| CARS0456 | 24-Olefinic sterol | Phytochemical | −7.8 | −8.2 | −8.0 |
| pdtdblig00047 | Naringin | Phytochemical | −8 | −7.9 | −8.4 |
| RASE0125 | 17-O-Acetyl-nortetraphyllicine | Phytochemical | −7.8 | −7.8 | −8.6 |
Table 2.
ADME-based z-scores for each molecule. Bolds are the top-ranked molecules selected for molecular dynamics simulations.
Table 2.
ADME-based z-scores for each molecule. Bolds are the top-ranked molecules selected for molecular dynamics simulations.
| Mol. ID | Mol./Drug Name | Mol. Type |
|---|---|---|
| RASE0125 | 17-O-Acetyl-nortetraphyllicine | 1.55 |
| ND_nw_193 | RSV604 | 1.05 |
| CARS0358 | NA | 0.79 |
| CARS0456 | 24-Olefinic sterol | −1.27 |
| pdtdblig00040 | Procyanidin B2 | −0.73 |
| CARS0394 | NA | −0.73 |
| ND_nw_93 | AC1MH6FW | −0.48 |
| pdtdblig00047 | Naringin | −0.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Randhawa, V.; Pathania, S.; Kumar, M. Computational Identification of Potential Multitarget Inhibitors of Nipah Virus by Molecular Docking and Molecular Dynamics. Microorganisms 2022, 10, 1181. https://doi.org/10.3390/microorganisms10061181
AMA Style
Randhawa V, Pathania S, Kumar M. Computational Identification of Potential Multitarget Inhibitors of Nipah Virus by Molecular Docking and Molecular Dynamics. Microorganisms. 2022; 10(6):1181. https://doi.org/10.3390/microorganisms10061181
Chicago/Turabian StyleRandhawa, Vinay, Shivalika Pathania, and Manoj Kumar. 2022. "Computational Identification of Potential Multitarget Inhibitors of Nipah Virus by Molecular Docking and Molecular Dynamics" Microorganisms 10, no. 6: 1181. https://doi.org/10.3390/microorganisms10061181
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.