Mucosal Metabolomic Profiling and Pathway Analysis Reveal the Metabolic Signature of Ulcerative Colitis

 , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. Subjects Characteristics
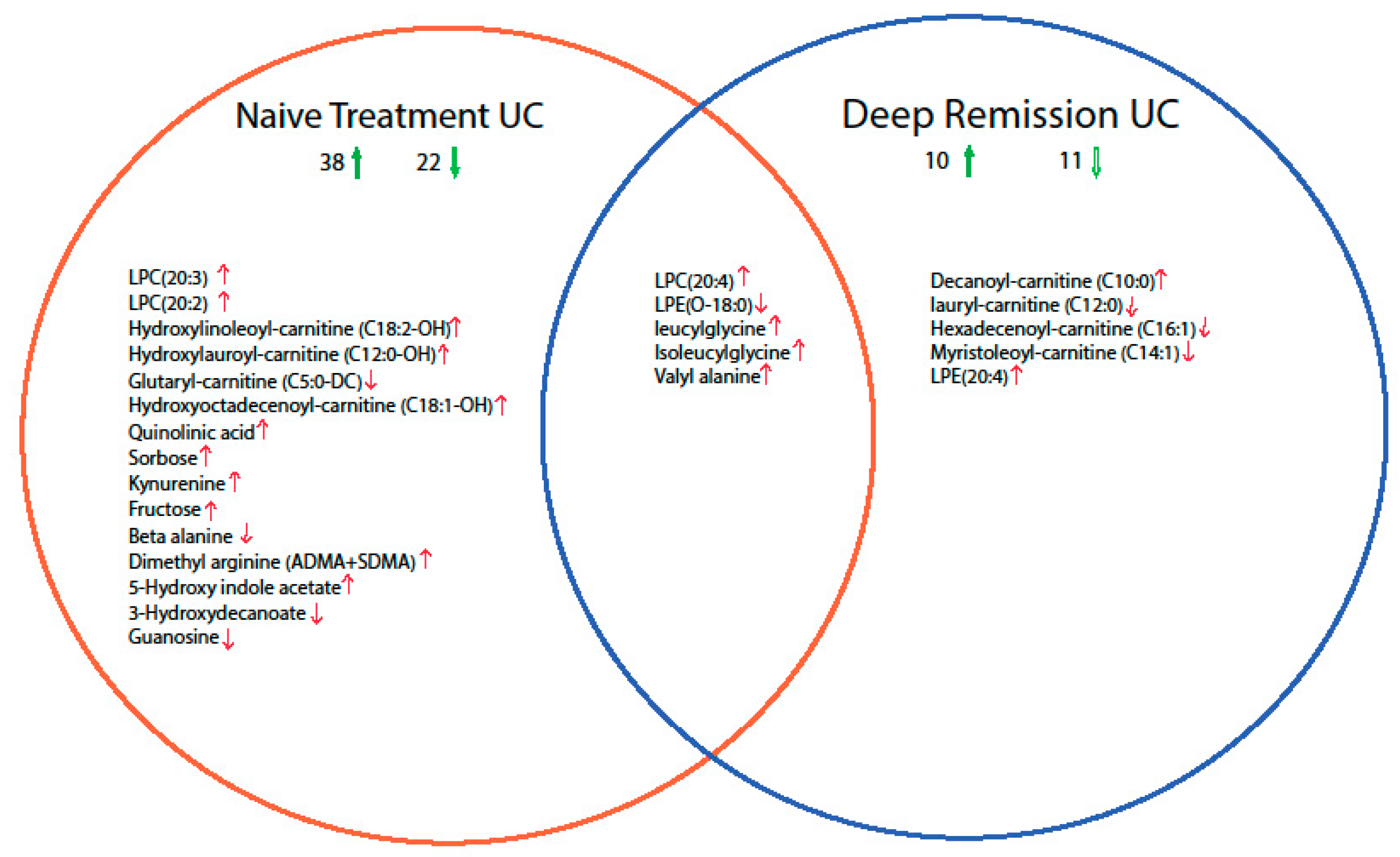
2.2. Mucosal Metabolite Profiles in Treatment-Naïve UC Patients, UC Remission Patients and Controls
2.3. Discriminative Models for UC State
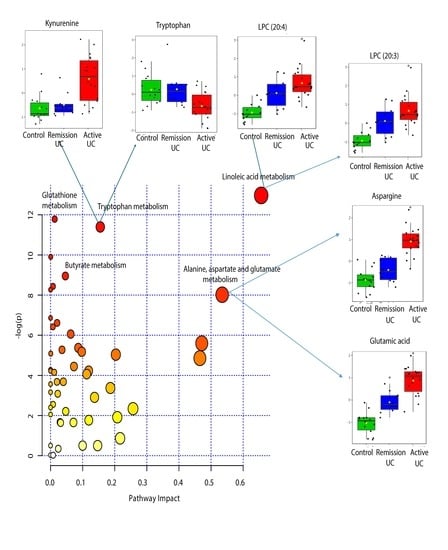
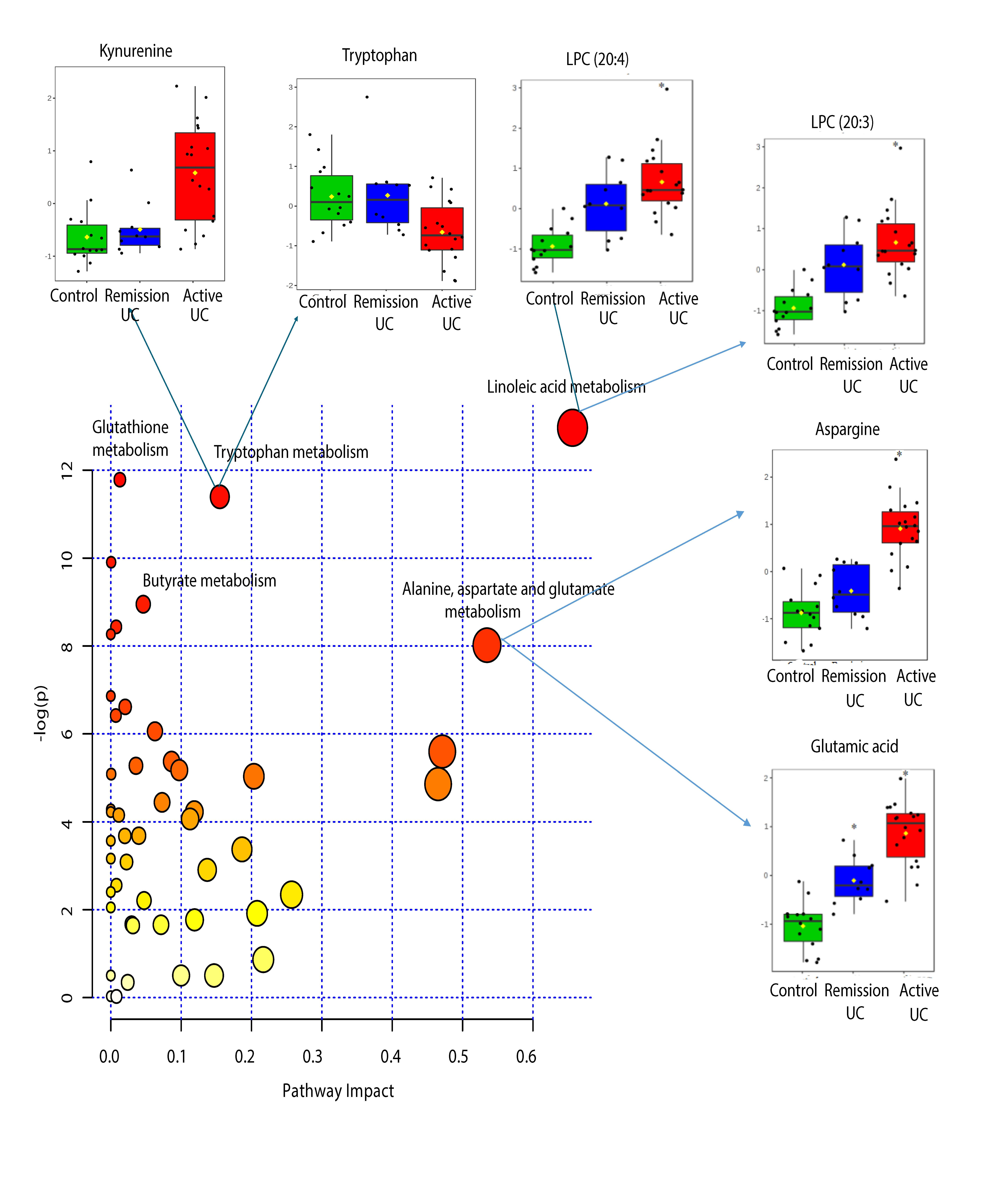
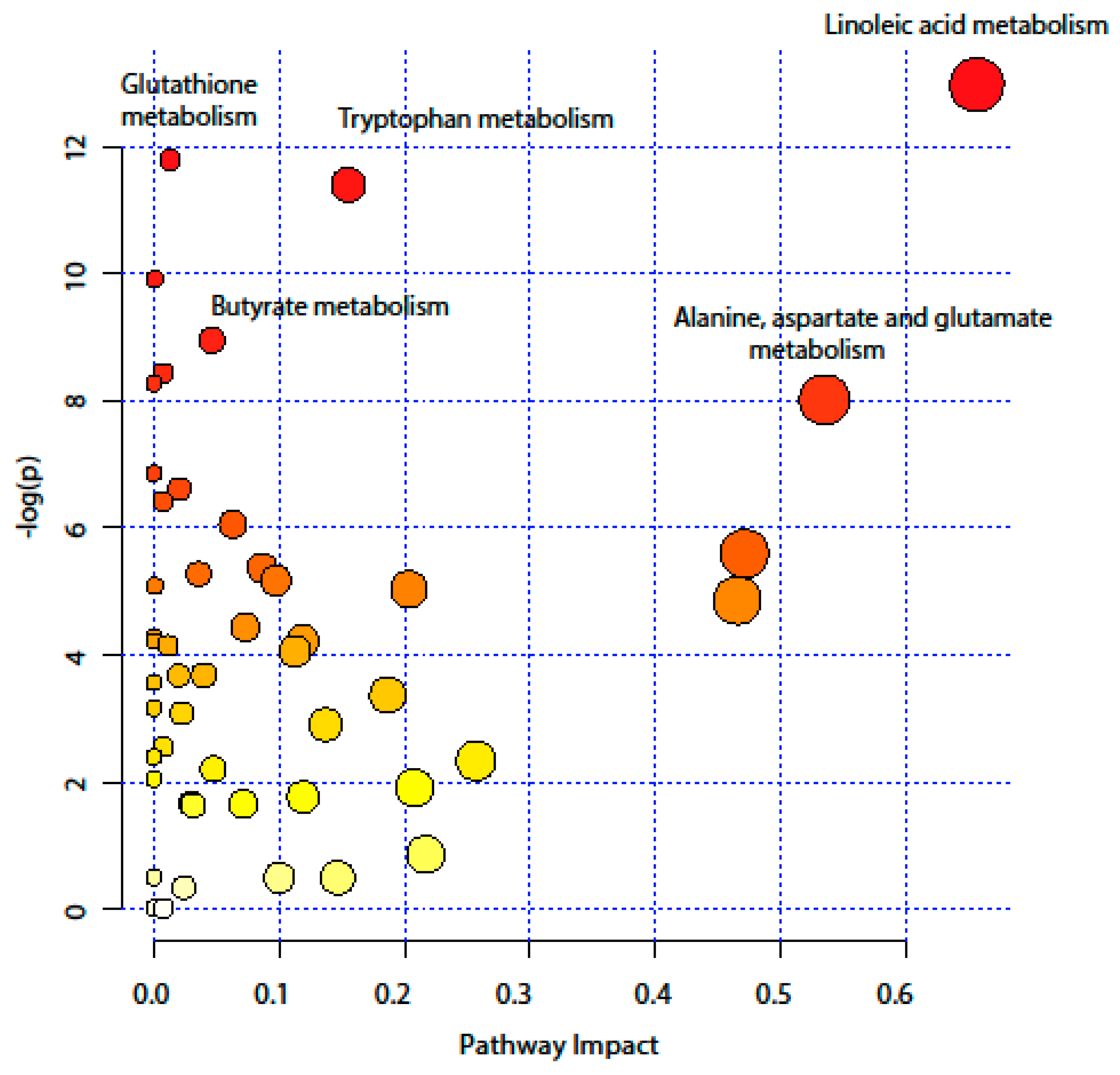
2.4. Pathway Analysis
3. Discussion
4. Materials and Methods
4.1. Patients and Biopsy Collection
4.2. Chemicals and Reagents
4.3. Sample Preparation
4.4. UHPLC-MS Analysis
4.5. GC-MS Analysis
4.6. Metabolites Identification and Data Processing
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Neurath, M.F. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat. Immunol. 2019, 20, 970–979. [Google Scholar] [CrossRef] [PubMed]
- De Souza, H.S.P.; Fiocchi, C.; Iliopoulos, D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Amp Hepatol. 2012, 9, 599. [Google Scholar] [CrossRef] [PubMed]
- Orazio, P.; Tommaso, M.; Stefano, C.; Panza, A.; Latiano, T.; Corritore, G.; Andriulli, A.; Latiano, A. Inflammatory bowel disease meets systems biology: A multi-omics challenge and frontier. OMICS J. Integr. Biol. 2016, 20, 692–698. [Google Scholar]
- Trivedi, D.K.; Hollywood, K.A.; Goodacre, R. Metabolomics for the masses: The future of metabolomics in a personalized world. New Horiz. Transl. Med. 2017, 3, 294–305. [Google Scholar] [CrossRef]
- Beger, R.D.; Dunn, W.; Schmidt, M.A.; Gross, S.S.; Kirwan, J.A.; Cascante, M.; Brennan, L.; Wishart, D.S.; Oresic, M.; Hankemeier, T.; et al. Metabolomics enables precision medicine: “A White Paper, Community Perspective”. Metab. Off. J. Metab. Soc. 2016, 12, 149. [Google Scholar] [CrossRef]
- Scoville, E.A.; Allaman, M.M.; Brown, C.T.; Motley, A.K.; Horst, S.N.; Williams, C.S.; Koyama, T.; Zhao, Z.; Adams, D.W.; Beaulieu, D.B.; et al. Alterations in lipid, amino acid, and energy metabolism distinguish crohn’s disease from ulcerative colitis and control subjects by serum metabolomic profiling. Metabolomics 2018, 14, 17. [Google Scholar] [CrossRef]
- Kohashi, M.; Nishiumi, S.; Ooi, M.; Yoshie, T.; Matsubara, A.; Suzuki, M.; Hoshi, N.; Kamikozuru, K.; Yokoyama, Y.; Fukunaga, K.; et al. A novel gas chromatography mass spectrometry-based serum diagnostic and assessment approach to ulcerative colitis. J. Crohns Colitis 2014, 8, 1010–1021. [Google Scholar] [CrossRef]
- Lai, Y.; Xue, J.; Liu, C.W.; Gao, B.; Chi, L.; Tu, P.; Lu, K.; Ru, H. Serum metabolomics identifies altered bioenergetics, signaling cascades in parallel with exposome markers in Crohn’s disease. Molecules 2019, 24, 449. [Google Scholar] [CrossRef] [PubMed]
- Kolho, K.L.; Pessia, A.; Jaakkola, T.; de Vos, W.M.; Velagapudi, V. Faecal and serum metabolomics in paediatric inflammatory bowel disease. J. Crohns Colitis 2017, 11, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Murgia, A.; Hinz, C.; Liggi, S.; Denes, J.; Hall, Z.; West, J.; Santoru, M.L.; Piras, C.; Manis, C.; Usai, P.; et al. Italian cohort of patients affected by inflammatory bowel disease is characterised by variation in glycerophospholipid, free fatty acids and amino acid levels. Metabolomics 2018, 14, 140. [Google Scholar] [CrossRef] [PubMed]
- Daniluk, U.; Daniluk, J.; Kucharski, R.; Kowalczyk, T.; Pietrowska, K.; Samczuk, P.; Filimoniuk, A.; Kretowski, A.; Lebensztejn, D.; Ciborowski, M. Untargeted metabolomics and inflammatory markers profiling in children with Crohn’s disease and ulcerative colitis—A preliminary study. Inflamm. Bowel Dis. 2019, 25, 1120–1128. [Google Scholar] [CrossRef]
- Stephens, N.S.; Siffledeen, J.; Su, X.; Murdoch, T.B.; Fedorak, R.N.; Slupsky, C.M. Urinary NMR metabolomic profiles discriminate inflammatory bowel disease from healthy. J. Crohns Colitis 2013, 7, e42–e48. [Google Scholar] [CrossRef]
- Viant, M.R.; Ludwig, C.; Günther, U. 1D and 2D NMR spectroscopy: From metabolic fingerprinting to profiling. RSC Biomol. Sci. 2007, 44–70. [Google Scholar] [CrossRef]
- Gowda, G.A.N.; Zhang, S.; Gu, H.; Asiago, V.; Shanaiah, N.; Raftery, D. Metabolomics-based methods for early disease diagnostics. Expert Rev. Mol. Diagn. 2008, 8, 617–633. [Google Scholar] [CrossRef]
- Shulaev, V. Metabolomics technology and bioinformatics. Brief. Bioinform. 2006, 7, 128–139. [Google Scholar] [CrossRef]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef]
- Das, U.N. Inflammatory bowel disease as a disorder of an imbalance between pro- and anti-inflammatory molecules and deficiency of resolution bioactive lipids. Lipids Health Dis. 2016, 15, 11–18. [Google Scholar] [CrossRef]
- Diab, J.; Al-Mahdi, R.; Gouveia-Figueira, S.; Hansen, T.; Jensen, E.; Goll, R.; Moritz, T.; Florholmen, J.; Forsdahl, G. A quantitative analysis of colonic mucosal oxylipins and endocannabinoids in treatment-naïve and deep remission ulcerative colitis patients and the potential link with cytokine gene expression. Inflamm. Bowel Dis. 2018, 25, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Costea, I.; Mack, D.R.; Lemaitre, R.N.; Israel, D.; Marcil, V.; Ahmad, A.; Amre, D.K. Interactions between the dietary polyunsaturated fatty acid ratio and genetic factors determine susceptibility to pediatric Crohn’s disease. Gastroenterology 2014, 146, 929–931.e3. [Google Scholar] [CrossRef] [PubMed]
- Mesbah-Uddin, M.; Elango, R.; Banaganapalli, B.; Shaik, N.A.; Al-Abbasi, F.A. In-silico analysis of inflammatory bowel disease (IBD) GWAS loci to novel connections. PLoS ONE 2015, 10, e0119420. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Kim, C.J.; Kovacs-Nolan, J.A.; Yang, C.; Archbold, T.; Fan, M.Z.; Mine, Y. l-Tryptophan exhibits therapeutic function in a porcine model of dextran sodium sulfate (DSS)-induced colitis. J. Nutr. Biochem. 2010, 21, 468–475. [Google Scholar] [CrossRef]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Häsler, R.; et al. Increased tryptophan metabolism is associated with activity of inflammatory bowel diseases. Gastroenterology 2017, 153, 1504–1516.e2. [Google Scholar] [CrossRef]
- Ni, J.; Shen, T.-C.D.; Chen, E.Z.; Bittinger, K.; Bailey, A.; Roggiani, M.; Sirota-Madi, A.; Friedman, E.S.; Chau, L.; Lin, A.; et al. A role for bacterial urease in gut dysbiosis and Crohn’s disease. Sci. Transl. Med. 2017, 9, eaah6888. [Google Scholar] [CrossRef]
- Martin, F.P.; Su, M.M.; Xie, G.X.; Guiraud, S.P.; Kussmann, M.; Godin, J.P.; Jia, W.; Nydegger, A. Urinary metabolic insights into host-gut microbial interactions in healthy and IBD children. World J. Gastroenterol. 2017, 23, 3643–3654. [Google Scholar] [CrossRef]
- Bjerrum, J.T.; Nielsen, O.H.; Hao, F.; Tang, H.; Nicholson, J.K.; Wang, Y.; Olsen, J. Metabonomics in ulcerative colitis: Diagnostics, biomarker identification, and insight into the pathophysiology. J. Proteome Res. 2010, 9, 954–962. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Huda-Faujan, N.; Abdulamir, A.S.; Fatimah, A.B.; Anas, O.M.; Shuhaimi, M.; Yazid, A.M.; Loong, Y.Y. The impact of the level of the intestinal short chain Fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem. J. 2010, 4, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E.W. The colonic epithellium in ulcerative colitis: An energy-deficiancy disease? Lancet 1980, 316, 712–715. [Google Scholar] [CrossRef]
- Schniers, A.; Goll, R.; Pasing, Y.; Sørbye, S.W.; Florholmen, J.; Hansen, T. Ulcerative colitis: Functional analysis of the in-depth proteome. Clin. Proteom. 2019, 16, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, S.; Bartlett, K.; Pourfarzam, M. Mammalian mitochondrial beta-oxidation. Biochem. J. 1996, 320 Pt 2, 345–357. [Google Scholar] [CrossRef]
- Jackson, D.N.; Theiss, A.L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes 2019, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Florholmen, J. Mucosal healing in the era of biologic agents in treatment of inflammatory bowel disease. Scand. J. Gastroenterol. 2015, 50, 43–52. [Google Scholar] [CrossRef]
- Diab, J.; Hansen, T.; Goll, R.; Stenlund, H.; Ahnlund, M.; Jensen, E.; Moritz, T.; Florholmen, J.; Forsdahl, G. Lipidomics in ulcerative colitis reveal alteration in mucosal lipid composition associated with the disease state. Inflamm. Bowel Dis. 2019, 25, 1780–1787. [Google Scholar] [CrossRef]
- Polytarchou, C.; Koukos, G.; Iliopoulos, D. Systems biology in inflammatory bowel diseases: Ready for prime time. Curr. Opin. Gastroenterol. 2014, 30, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Stange, E.F.; Travis, S.P.; Vermeire, S.; Reinisch, W.; Geboes, K.; Barakauskiene, A.; Feakins, R.; Fléjou, J.F.; Herfarth, H.; Hommes, D.W.; et al. European evidence-based Consensus on the diagnosis and management of ulcerative colitis: Definitions and diagnosis. J. Crohns Colitis 2008, 2, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Marteau, P.; Probert, C.S.; Lindgren, S.; Gassul, M.; Tan, T.G.; Dignass, A.; Befrits, R.; Midhagen, G.; Rademaker, J.; Foldager, M. Combined oral and enema treatment with Pentasa (mesalazine) is superior to oral therapy alone in patients with extensive mild/moderate active ulcerative colitis: A randomised, double blind, placebo controlled study. Gut 2005, 54, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.; Goll, R.; Cui, G.; Husebekk, A.; Vonen, B.; Birketvedt, G.S.; Florholmen, J. Tissue levels of tumor necrosis factor-alpha correlates with grade of inflammation in untreated ulcerative colitis. Scand. J. Gastroenterol. 2007, 42, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.B.; Hart, A.L.; Hindryckx, P.; et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 1: Definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J. Crohns Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.M.; Goll, R.; Hansen, V.; Olsen, T.; Rismo, R.; Heitmann, R.; Gundersen, M.D.; Kvamme, J.M.; Paulssen, E.J.; Kileng, H.; et al. Repeated intensified infliximab induction—Results from an 11-year prospective study of ulcerative colitis using a novel treatment algorithm. Eur. J. Gastroenterol. Hepatol. 2017, 29, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Trygg, J.; Gullberg, J.; Johansson, A.I.; Jonsson, P.; Antti, H.; Marklund, S.L.; Moritz, T. Extraction and GC/MS analysis of the human blood plasma metabolome. Anal. Chem. 2005, 77, 8086–8094. [Google Scholar]
- Lindahl, A.; Forshed, J.; Nordstrom, A. Overlap in serum metabolic profiles between non-related diseases: Implications for LC-MS metabolomics biomarker discovery. Biochem. Biophys. Res. Commun. 2016, 478, 1472–1477. [Google Scholar] [CrossRef] [Green Version]
- Karimpour, M.; Surowiec, I.; Wu, J.; Gouveia-Figueira, S.; Pinto, R.; Trygg, J.; Zivkovic, A.M.; Nording, M.L. Postprandial metabolomics: A pilot mass spectrometry and NMR study of the human plasma metabolome in response to a challenge meal. Anal. Chim. Acta 2016, 908, 121–131. [Google Scholar] [CrossRef]
- Redestig, H.; Fukushima, A.; Stenlund, H.; Moritz, T.; Arita, M.; Saito, K.; Kusano, M. Compensation for systematic cross-contribution improves normalization of mass spectrometry based metabolomics data. Anal. Chem. 2009, 81, 7974–7980. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Dunn, O.J. Multiple comparisons among means. J. Am. Stat. Assoc. 1961, 56, 52–64. [Google Scholar] [CrossRef]
- Bonferroni, C.E. Teoria statistica delle classi e calcolo delle probabilita. Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze 1936, 8, 3–62. [Google Scholar]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiklund, S.; Johansson, E.; Sjöström, L.; Mellerowicz, E.J.; Edlund, U.; Shockcor, J.P.; Gottfries, J.; Moritz, T.; Trygg, J. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal. Chem. 2008, 80, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Su, M.; Lin, J.; Wang, X.; Qiu, Y.; Zhao, A.; Chen, T.; Jia, W. Metabolic profiling reveals disorder of amino acid metabolism in four brain regions from a rat model of chronic unpredictable mild stress. FEBS Lett. 2008, 582, 2627–2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2017, 46, D608–D617. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Goeman, J.J.; van de Geer, S.A.; de Kort, F.; van Houwelingen, H.C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 2004, 20, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Aittokallio, T.; Schwikowski, B. Graph-based methods for analysing networks in cell biology. Brief. Bioinform. 2006, 7, 243–255. [Google Scholar] [CrossRef]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Active UC (Debut) | UC Remission | Healthy Controls |
|---|---|---|---|
| Number of Subjects | 18 | 10 | 14 |
| Age, years (mean, range, P-value *) | 40 (20–68) 0.09 | 48 (31–77) 0.18 | 55 (26–83) |
| Gender (Female/Male) | 6/12 | 4/6 | 4/10 |
| UCDAI Score (Mild, Moderate, Severe) | 12/2/4 | ||
| Biopsy sampling site (Rectum/sigmoid) | 3/15 | 5/5 | 4/10 |
| TNF-α, copies/μg of total RNA (mean, range, P-value *) | 18,122 (4600–31,700) 0.01 | 4675 (800–7300) 0.11 | 5478 (1800–11,300) |
| Fecal calprotectin, μg/g (mean, range, P-value *) | 828 (25–1970) < 0.01 | 53 (25–150) 0.15 | 46 (25–180) |
| C-Reactive protein, mg/L (mean, range, P-value *) | 16.5 (5–92) 0.08 | 5.6 (5–11) 0.31 | 5.2 (5–11) |
| Smoking/non-smoking | 1/17 | 1/9 | 3/11 |
| Omega-3 daily supplementation (Yes/No) | 6/12 | 3/7 | 7/7 |
| Antibiotic in the last 6 months prior to the biopsy (Yes/No) | 3/15 | 0/10 | 2/12 |
| KEEG Pathway | Numb. Metabolites | Matched Metabolites from the Metabolomics Data | Adjusted P-value ** | Impact *** |
|---|---|---|---|---|
| Linoleic Acid Metabolism | 15 | Linoleic acid * | <0.001 | 0.66 |
| Alanine, Aspartate and Glutamate Metabolism | 24 | N-Acetyl-L-aspartic acid *; L-Asparagine *; L-Glutamine *; L-Glutamic acid *; Gamma-Aminobutyric acid; Fumaric acid; Succinic acid | 0.014 | 0.53 |
| Tryptophan Metabolism | 79 | L-Tryptophan *; 5-Hydroxyindoleacetic acid *; L-Kynurenine *; Picolinic acid; Quinolinic acid* | <0.001 | 0.15 |
| Butyrate Metabolism | 40 | Gamma-Aminobutyric acid; L-Glutamic acid *; Fumaric acid | 0.006 | 0.05 |
| Glutathione Metabolism | 38 | L-Glutamic acid *; Cysteinylglycine; Pyroglutamic acid *; Ornithine * | <0.001 | 0.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diab, J.; Hansen, T.; Goll, R.; Stenlund, H.; Jensen, E.; Moritz, T.; Florholmen, J.; Forsdahl, G. Mucosal Metabolomic Profiling and Pathway Analysis Reveal the Metabolic Signature of Ulcerative Colitis. Metabolites 2019, 9, 291. https://doi.org/10.3390/metabo9120291
Diab J, Hansen T, Goll R, Stenlund H, Jensen E, Moritz T, Florholmen J, Forsdahl G. Mucosal Metabolomic Profiling and Pathway Analysis Reveal the Metabolic Signature of Ulcerative Colitis. Metabolites. 2019; 9(12):291. https://doi.org/10.3390/metabo9120291
Chicago/Turabian StyleDiab, Joseph, Terkel Hansen, Rasmus Goll, Hans Stenlund, Einar Jensen, Thomas Moritz, Jon Florholmen, and Guro Forsdahl. 2019. "Mucosal Metabolomic Profiling and Pathway Analysis Reveal the Metabolic Signature of Ulcerative Colitis" Metabolites 9, no. 12: 291. https://doi.org/10.3390/metabo9120291