
Prostanoid Metabolites as Biomarkers in Human Disease


1
Division of Rheumatology, Department of Medicine, Solna, Karolinska Institute, Karolinska University Hospital, SE-171 76 Stockholm, Sweden
2
Cardiovascular Medicine Unit, Department of Medicine, Solna, Karolinska Institute, SE-171 76 Stockholm, Sweden
3
Division of Vascular and Coronary Disease, Theme Heart and Vessels, Karolinska University Hospital, SE-171 76 Stockholm, Sweden
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Metabolites 2022, 12(8), 721; https://doi.org/10.3390/metabo12080721
Submission received: 14 July 2022
/
Revised: 27 July 2022
/
Accepted: 27 July 2022
/
Published: 4 August 2022
(This article belongs to the Special Issue Prostaglandin Metabolites)
Abstract
:Prostaglandins (PGD2, PGE2, PGF2α), prostacyclin (PGI2), and thromboxane A2 (TXA2) together form the prostanoid family of lipid mediators. As autacoids, these five primary prostanoids propagate intercellular signals and are involved in many physiological processes. Furthermore, alterations in their biosynthesis accompany a wide range of pathological conditions, which leads to substantially increased local levels during disease. Primary prostanoids are chemically instable and rapidly metabolized. Their metabolites are more stable, integrate the local production on a systemic level, and their analysis in various biological matrices yields valuable information under different pathological settings. Therefore, prostanoid metabolites may be used as diagnostic, predictive, or prognostic biomarkers in human disease. Although their potential as biomarkers is great and extensive research has identified major prostanoid metabolites that serve as target analytes in different biofluids, the number of studies that correlate prostanoid metabolite levels to disease outcome is still limited. We review the metabolism of primary prostanoids in humans, summarize the levels of prostanoid metabolites in healthy subjects, and highlight existing biomarker studies. Since analysis of prostanoid metabolites is challenging because of ongoing metabolism and limited half-lives, an emphasis of this review lies on the reliable measurement and interpretation of obtained levels.
Keywords:
prostanoid; prostaglandin; prostacyclin; thromboxane; eicosanoid; biomarker; metabolism; creatinine; LC–MS/MS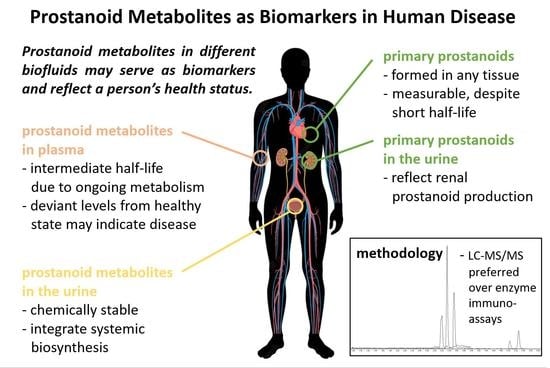
1. Introduction
The prostanoid family of bioactive lipid mediators comprises five members, prostaglandin (PG)E2, PGD2, PGF2α, prostacyclin (PGI2), and thromboxane A2 (TXA2). These five prostanoids mediate biological function and are therefore denoted as primary prostanoids. They are effective as bioactive lipid mediators and involved in many critical physiological and pathophysiological processes, among others, blood pressure homeostasis [1,2,3], sleep regulation [4,5], labor [6], induction of fever [7,8,9,10], and pain [11,12] as well as inflammatory reactions [13,14,15,16] and malignancies [10,17,18]. Although their functions have been studied for decades, a complete understanding of their importance in health and disease is still elusive due to the complexity of their signaling. Their locally and temporally restricted biosynthesis, which is limited by the expression of their synthesizing enzymes to usually one or two prostanoids per cell type, causes complex profiles that are specific for each tissue. Furthermore, target cells can express multiple types of prostanoid receptors with sometimes opposing downstream functions, which contributes to sophisticated signaling. The importance of primary prostanoids as mediators was previously empathized and recently reviewed with a focus on enzyme [19,20] and receptor [21] expression, respectively. Another layer that contributes to the complexity of prostanoid signaling is the fact that primary prostanoids are short lived under physiological conditions. Rapid metabolic conversion of prostanoids leads to their functional inactivation and thus restricts their biological action to auto- or paracrine effects in the immediate vicinity of their origin.
Metabolites of primary prostanoid are usually biologically inactive, but may integrate changes in the local prostanoid levels and may therefore be useful biomarkers to indicate human disease. In this review, we survey the metabolism of each of the primary prostanoids, identify the major metabolites and their concentrations in different biofluids of healthy human subjects, and evaluate their stability in vivo and in vitro with respect to their suitability as biomarkers. Although our emphasis lies on frequently sampled biofluids such as plasma and urine, we also review other specimens in which prostanoid metabolites were measured. We give examples of prostanoid metabolites that were studied in the context of human disease and suggest interpretations of what their levels in different body compartments may reflect. We furthermore comment on analytical approaches for the detection of prostanoid metabolites and give practical guidance that will help to expand the use of prostanoid metabolites as biomarkers for human disease.
2. Metabolism of Prostanoids
Prostanoids are derived from the ω-6 polyunsaturated fatty acid arachidonic acid (AA; 20:4Δ5Z,8Z,11Z,14Z). Cyclooxygenase (COX), the key enzyme in the biogenesis of prostanoids, converts free AA in a two-step enzymatic reaction to form the labile intermediates PGG2 and PGH2 (Figure 1). The intermediate PGH2 is the common substrate to various terminal synthases, which give rise to the five primary prostanoids. The enzymatic mechanism of this catalysis was studied in detail [22,23,24,25], and the anabolism of prostanoids was previously reviewed [19,20,26]. As a consequence of their shared molecular origin, all primary prostanoids are characterized by a fatty acid backbone of 20 carbon atoms and thus belong to the class of eicosanoid signaling molecules in which they constitute their own subfamily. All primary prostanoids, with the exception of TXA2, also maintain a cyclopentane ring, which is formed by COX and is a structural characteristic of prostaglandins.
Figure 1 provides, in a nutshell, a schematic overview of the catabolic pathways involved in the metabolism of each primary prostanoid and identifies the major metabolites in plasma and urine, respectively. Besides these major metabolites, several other metabolites can be detected at different levels in the various specimens. Their concentrations and basic characteristics are summarized in Table 1, and Supplementary Table S1 lists their molecular identity, including molecular structure, as well as possible synonym names that were used throughout the literature.
The metabolic fate of primary prostanoids in humans was investigated by infusion studies of radioactively labeled molecules [28,37,50,51]. PGE2, PGD2, and PGF2α have half-lives in the circulation that are reported to be in the range of minutes [24,28,52] (Table 1). The ubiquitously expressed enzyme 15-hydroxyprostaglandin dehydrogenase (15-PGDH) [27] oxidizes these three prostanoids at the hydroxyl group at C-15 and thereby renders them biologically inactive. PGI2 and TXA2 follow a somewhat different path with even shorter half-lives than that of the other prostanoids, which were reported to be in the range of seconds [39] (Table 1). Due to their chemical instability, these two prostanoids decompose non-enzymatically to form the biologically inactive metabolites 6-keto PGF1α and TXB2, respectively. PGE2, PGD2, and PGF2α are, therefore, under certain conditions, amenable to analytical methods, while 6-keto PGF1α and TXB2 are almost always measured as direct substitutes for PGI2 and TXA2, respectively. For analytical purposes and throughout this review, primary prostanoids are thus referred to as PGE2, PGD2, PGF2α, 6-keto PGF1α and TXB2.
2.1. Metabolites of PGE2
PGE2 has a biological half-life of about 1.5 min [28,52] and is metabolically inactivated by 15-PGDH to form 15-keto PGE2, which is subsequently reduced at the double bond between C-13 and C-14 by 15-oxo-prostaglandin Δ13-reductase [53]. Its primary plasma metabolite is 13,14-dihydro-15-keto PGE2 (Figure 2), which accumulates to detectable levels in the range of about 20 pg/mL 32. Although 13,14-dihydro-15-keto PGE2 has a longer plasma half-life than PGE2 of about 9 min, it undergoes further metabolism and is therefore rather unreliable as a measure to estimate the production of PGE2 in humans [29,32]. One of the catabolic steps in its further metabolism is the non-enzymatic dehydration of 13,14-dihydro-15-keto PGE2 to form 13,14-dihydro-15-keto PGA2 [30,54]. This reaction is facilitated by albumin, which is abundant in plasma and thus yet another factor for the rapid metabolism of prostanoids. Both 13,14-dihydro-15-keto PGE2 and 13,14-dihydro-15-keto PGA2 can be converted to the stable, base-catalyzed breakdown product, bicyclo PGE2, to integrate the intermediates that are subjected to ongoing metabolism 29. Bicyclo PGE2 should be quantified to reflect plasma levels of PGE2 [54,55].
Further metabolic pathways of PGE2, but also PGE1, involve the removal of four carbons at the α-terminus via β-oxidation and additional oxidation of the terminal ω-carbon to yield tetranor-PGEM, which is excreted by the kidneys as the major urinary metabolite of PGE2. Tetranor-PGEM is commonly used as a surrogate marker to estimate the systemic biosynthesis of PGE2 [56,57]. The levels of tetranor-PGEM in healthy humans range between 2.6 and 7.4 ng/mg creatinine but can be doubled under pathological conditions [31] (Table 1).
2.2. Metabolites of PGD2
PGD2 has an approximate plasma half-life of 0.9 min [33] (Table 1). It gives rise to different metabolites that either retain the D-series cyclopentane ring with a hydroxy group at position C-9 and a keto group at position C-11, adopt the F-series ring with a hydroxy group at both positions, or adopt the cyclopentanone ring of the J-series (Figure 3). In vivo studies on the metabolic fate of radiolabeled, exogenous PGD2 revealed 25 different urinary metabolites; 23 of which belonged to the F-series [50]. The major human metabolite, however, belonged to the D-series [50]. This major metabolite, tetranor-PGDM, is metabolized analog to PGE2 via the 15-PGDH pathway to initially form 13,14-dihydro-15-keto PGD2 in plasma [58] before undergoing β- and ω-oxidation and being excreted in the urine [59]. Tetranor-PGDM reflects the systemic biosynthesis of PGD2 in vivo. Its concentration in the urine of healthy human subjects is low; between 0.3 and 2.5 ng/mg of creatinine [31] (Table 1).
Reduction of the keto group of the cyclopentane ring of PGD2 yields 11β-PGF2α and is catalyzed by the NADPH-dependent enzyme 11-ketoreductase [60], which is expressed in the lungs [61] and the liver [60].
In an aqueous solution or in plasma, PGD2 degrades non-enzymatically to yield Δ12-PGD2 and PGJ2, respectively [33]. This transformation is reinforced by plasma albumin. Further non-enzymatic degradation gives rise to their respective downstream metabolites, 15-deoxy-Δ12,14-PGD2, Δ12-PGJ2, and 15-deoxy-Δ12,14-PGD2 (Figure 3). The formation of Δ12-PGJ2 from PGJ2 strictly depends on albumin [33].
Unlike the metabolites of all other primary prostanoids, many of the PGD2 metabolites retain biological activity, although not via the PGD2 receptors DP1 and DP2 [62]. 11β-PGF2α was shown to contract bronchial smooth muscle cells [63] as well as coronary arteries [64] and induce phosphorylation of the serine/threonine-specific protein kinase ERK [65]. 15-deoxy-Δ12,14-PGD2 was shown to activate the nuclear receptor PPARγ in macrophages and has anti-inflammatory properties [66]. Δ12-PGJ2 was assigned anti-tumor and anti-viral properties [67]. 15-deoxy-Δ12,14-PGJ2 also ligates PPARγ and promotes the differentiation of fibroblasts to adipocytes [68,69]. The mechanism of action of the cyclopentenone prostanoids of the A and J series was previously reviewed [70].
Although the demonstrated biological activity of PGD2 metabolites required much higher concentrations than those measured in physiological samples, the local tissue concentrations may differ markedly from the circulating levels, among others due to adhesion to albumin or conjugation with glutathione. For instance, while plasma and urinary levels of 15-deoxy-Δ12,14-PGJ2 in healthy human individuals were reported in the range of 2 to 350 pg/mL [34] and 6.3 ± 2.7 pg/mg of creatinine [35], its tissue levels of 15 µM (4.7 µg/mL) are magnitudes higher and similar to those of arachidonic acid in tissues [53].
2.3. Metabolites of PGF2α
The same pathway that metabolizes PGE2 and PGD2 converts also PGF2α to form, through modification by 15-PGDH and 15-oxo-prostaglandin Δ13-reductase, its initial, biologically inactive plasma metabolite, 13,14-dihydro-15 keto PGF2α (Figure 4) [71,72]. 13,14-dihydro-15 keto PGF2α accumulates in human plasma to concentrations of 0.08–20 pmol/mL [36] but prevails only briefly. Further metabolism involves β- and ω-oxidation and leads to tetranor-PGFM [73], which is detectable in urine, but also in plasma [74]. In fact, about 20 min after injection of radiolabeled PGF2α, tetranor-PGFM was the dominating plasma metabolite and stayed longer in the circulation than its precursor 13,14-dihydro-15 keto PGF2α [37]. It was therefore suggested to preferably measure tetranor-PGFM as a more reliable plasma biomarker. PGF2α is vastly produced by females during labor. Consequently, the plasma levels of 13,14-dihydro-15 keto PGF2α rise from low basal levels of about 40–60 pg/mL during late pregnancy to peak concentrations of 1200–4100 pg/mL and decrease rapidly to almost baseline levels 2 h after parturition. Interestingly, plasma levels of tetranor-PGFM follow a slightly different pattern and increase from basal levels of about 60–100 pg/mL during late pregnancy to reach a peak concentrations between 1000 and 2000 pg/mL; however, unlike 13,14-dihydro-15 keto PGF2α, tetranor-PGFM peaks later, about 2 h after parturition, and its levels stay at these high concentrations for several hours and remain elevated (about 100–300 pg/mL) 24 h after parturition [37].
Tetranor-PGFM is excreted by the kidneys and constitutes the major urinary metabolite of PGF2α. The mean urinary concentration of tetranor-PGFM in healthy subjects is 1.2 ± 7.1 µg/24 h in urine for women and 1.6 ± 6.0 µg/24 h in urine for men [38].
2.4. Metabolites of TXA2
TXA2 is an unusual bicyclic prostanoid, displaying an oxane ring instead of the cyclopentane ring that is characteristic of the other four prostanoids, coupled to a cyclic ether. TXA2 is released in substantial quantities from aggregating platelets and is highly unstable under physiological conditions. The cyclic ether function on its oxane ring is non-enzymatically hydrated in aqueous solution within about 30 s [39,75] to form a hemiacetal and thus the biologically inactive TXB2 (Figure 5). The levels of TXB2 are very low in plasma from uninduced blood, around 1–2 pg/mL (3–6 fmol/mL), in line with TXA2’s pro-aggregatory, vaso-, and bronchoconstrictive function. However, these low levels can increase tremendously during hemostasis to 300–400 ng/mL (0.8–1.0 nmol/mL) in serum from fully coagulated blood [40]. While TXB2 is relatively stable with a half-life of about 7 min [41] and readily detectable in plasma and urine, it is not necessarily well suited as a biomarker to assess in vivo TXA2 formation due to its further metabolism.
There are a total of twenty metabolites of TXB2 that can be detected in urine [51]; however, two pathways were identified to account for the majority of metabolites. The first pathway is the direct oxidation of TXB2 by 11-hydroxy Thromboxane Dehydrogenase and leads to 11-dehydro TXB2 [76,77]. The second major pathway is the β-oxidation of the carboxy terminus of TXB2, leading to 2,3-dinor-TXB2 [78].
Both metabolites are detectable in plasma and urine; however, 11-dehydro TXB2 was identified as the main plasma metabolite while 2,3-dinor TXB2 was the main urinary metabolite [79]. In plasma, 11-dehydro-TXB2 has a half-life of 45–60 min and accumulates to levels of 0.9–4.3 pg/mL. Its levels in urine are about 70 ng/mmol of creatinine [80]. The median level of excreted 2,3-dinor TXB2 in the urine ranges from 138 pg/mg of creatine (10.3 ng/h) in healthy male volunteers [78] to about 175 pg/mg creatinine in healthy subjects of both sexes [49]. 2,3-dinor TXB2 displays neither a significant sex difference nor any significant diurnal variation [49].
2.5. Metabolites of PGI2
PGI2 is also a chemically unstable bicyclic molecule and has a similar short half-life in vivo as compared to TXA2. The vinyl ether moiety between C-6 and C-9 of PGI2 is hydrolyzed to form 6-keto PGF1α within about 30 s in vivo. Ex vivo, PGI2 is substantially more stable with a half-life of about 6–10 min in whole blood or plasma [45], and in pharmacological preparations at basic pH, PGI2 is stable for at least 48 h [81]. 6-keto PGF1α is biologically inactive and with a plasma half-life of about 30 min [81] relatively stable and can be used as a biomarker for the PGI2 biosynthesis in vivo [82]. Plasma concentrations of 6-keto PGF1α in healthy individuals were determined to be 1.9 ± 0.8 pg/mL [48].
6-keto PGF1α can not only be detected in human plasma, but also in human urine; however, the levels of urinary 6-keto PGF1α, which were determined to be 92 ± 51 pg/mL, or, as reported normalized to creatinine, as 168 ± 91 pg/mg creatinine [48], account for only about 6% of all urinary metabolites of PGI2 in healthy volunteers [82] and were suggested to reflect renal production of PGI2 rather than its systemic biosynthesis [83]. Further metabolism of 6-keto PGF1α yields at least 16 compounds, which are primarily excreted in the urine [82]. The major urinary metabolite in humans is 2,3-dinor-6-keto PGF1α (Figure 6) [84], which was found to be 3-fold more abundant than 6-keto PGF1α [48]. The average excreted level of 2,3-dinor-6-keto PGF1α in healthy individuals is around 100 pg/mg creatinine. There is neither a significant sex difference in the excreted levels of 2,3-dinor-6-keto PGF1α nor any significant diurnal variation [49]. Urinary 2,3-dinor-6-keto PGF1α was thought of as a marker for systemic production of PGI2 [85]; however, this concept has been recently challenged [86].
3. Analysis of Prostanoid Metabolites
It is critical to select appropriate biological samples and sufficiently stable target analytes when designing a study or clinical trial to obtain meaningful and robust results. A good understanding of the pathway and detailed knowledge about the continuous metabolism of primary prostanoids, especially regarding the expression and location of metabolizing enzymes as well as excretion of the respective metabolites, is crucial to making the right choices. Furthermore, proper short- and long-term storage of the samples and handling during the analytical process is critical to ensure that all samples of a given cohort that are collected over a longer period are treated equally and analyte losses are avoided.
3.1. Sample Selection and Choice of Analyte(s)
In many non-physiological matrices devoid of 15-PGDH and 15-oxo-prostaglandin Δ13-reductase, e.g., in the supernatant of cultured cells, metabolism of primary prostanoids can be excluded. Under these conditions, PGE2, PGD2, and PGF2α are relatively stable and may be readily measured to estimate the biosynthetic capacity of the respective cells in vitro [16,87,88]. 6-keto PGF1α and TXB2, the immediate non-enzymatical breakdown products of PGI2 and TXA2, respectively, serve under these circumstances as surrogate markers [87]. These prostanoids were also measured in samples of exhaled breath condensate [89] and in tissue samples [90] instead of their downstream metabolites.
Plasma and serum on the other hand are generally problematic biological samples for the analysis of prostanoids. Although some metabolites can be detected in these matrices [29,32], their levels will change over time due to ongoing metabolism, resulting in a variable temporal flux that will influence the study results. Therefore, although blood samples are frequently analyzed for other biomarkers, one should avoid measuring prostanoids in plasma or serum, respectively if other options are available. One noticeable exception from this general rule is the ex vivo analysis of serum TXB2, which has proven useful to assess COX-1 dependent formation of TXA2 in fully coagulated blood and its inhibition by Aspirin, respectively [40].
A more reliable source to measure and interpret in vivo formation of prostanoids is urine. Unlike the primary prostanoids with auto- or paracrine biological activity but a very limited operating range, the urinary prostanoid metabolites have undergone several steps of metabolism during which they have lost their biological activity, but as final metabolic products, they are relatively stable and integrate systemic biosynthesis. Therefore, it is very informative to quantify their levels in disease as compared to the healthy state or changes in their levels that were caused by pharmaceutical intervention.
3.2. Sample Collection, Handling, and Storage
Accurate quantification of prostanoids relies on many factors. Correct sampling, handling, and short- as well as long-term storage, will contribute to the accuracy of the analysis results, reflecting in part the unstable nature of prostanoids.
Avoiding artifacts during sample collection is of particular importance for the analysis of 6-keto PGF1α and TXB2, respectively, in blood samples. Venipuncture may activate endothelial cells to produce PGI2, leading to artifactually increased levels of 6-keto PGF1α in plasma samples, which reflects incorrect sample handling rather than real biosynthesis [91]. Similarly, platelets are easily activated ex vivo during the blood draw, leading to erroneously increased levels of TXB2 in the samples [42]. For more reliable measurements of the systemic TXA2 formation, analysis of downstream metabolites was suggested [41,92]. In this context, and as discussed below in Section 4, the consumption of Aspirin and other Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) should be closely monitored in the subjects because these over-the-counter drugs hamper prostanoid biosynthesis, and are widely used but oftentimes not considered genuine drugs by the study participants and therefore not always correctly declared. Due to its irreversible inhibition and preferred binding to platelet COX-1, even low doses of Aspirin have a profound cumulative and long-lasting effect on the formation of TXA2 [93].
While sampling artifacts can lead to increased levels of TXB2, inappropriate storage after sample collection may result in substantial losses of the analytes of interest due to their chemical instability and subsequent decomposition. For example, storage of frozen urine samples at −20 °C resulted already after only one week in significantly decreased levels of tetranor-PGEM and losses of up to 80% of the initial amounts after 1.5 to 2 years [8]. On the contrary, the tetranor-PGEM levels in urine samples that were stored at −80 °C instead for the same time were indistinguishable from samples that were quantified directly after sampling [8]. This observation is particularly important for large cohort studies with samples collected at different time points. To avoid inconsistencies and false results, a few principles should be considered: the time from sampling to freezing should be minimized; samples should be aliquoted to avoid repetitive freeze–thaw cycles; ideally, the volume of the aliquots corresponds to the volume that will be later analyzed, and authentic internal standards are added directly at the time of freezing; and the samples should be stored at low temperatures. If possible, a pilot study should be run as part of a proper method validation before the study or clinical trial begins to characterize the stability of the analyte(s) of interest.
3.3. Methodology for the Analysis of Prostanoid Lipid Mediators
Enzyme immunoassays (EIA) were extensively used to analyze primary prostanoids as well as their downstream metabolites. Immunoassays combine the advantage of high sensitivity and thus the ability to detect minute analyte levels with a fairly easy implementation that does not require advanced detection technology or specialized analytical skills. They are commercially available for many prostanoid metabolites, but not for all, and they are usually very reliable and reproducible. Furthermore, immunoassays have the advantage that they are amenable in the analysis of large sample numbers. Therefore, this method is still frequently employed. However, immunoassays do not provide a direct measurement of the analyte but rather an indirect measurement of a tracer molecule. Moreover, they are not amenable for the analysis of several analytes at the same time, and the quality of each immunoassay is highly dependent on the specificity of the implemented antibody. This may result in a loss of accuracy due to cross-reactivity with other compounds than the target analyte, which is a particular concern in the analysis of structurally similar prostanoids.
Nowadays, liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS) is regarded as the gold standard method for the detection of prostanoids and downstream metabolites. Mass spectrometry was the key methodology in the discovery of prostanoids, their structural elucidation [94], and early quantitation studies [95,96]. Mass spectrometers are broadly applicable to many analytes in biological matrices, they are very sensitive and highly accurate, especially when several characteristic ion transitions per analyte are scrutinized in a typical multiple reaction monitoring (MRM) method. Furthermore, it is possible to simultaneously detect and quantify several target analytes in the same sample, and with advanced LC–MS/MS methods with optimized chromatography, it is even possible to screen larger cohorts [97]. Hence, LC–MS/MS may be approaching the medium to high throughput capacity of immunoassays while gaining accuracy, specificity, and the number of analytes.
In LC–MS/MS, the analyte molecules are separated in an initial chromatographic step based on their interaction with a stationary phase according to their chemical properties. This separation step is crucial, especially when constitutional isomeric prostanoids with identical mass-to-charge ratios such as, e.g., PGE2 and PGD2 or tetranor-PGEM and tetranor-PGDM are among the target analytes [98,99]. Analyte identification is based on a characteristic combination of the retention time during the chromatography step and the specific mass transition(s) in the MRM tandem mass spectrometry step, which takes into account the molecular masses both of the precursor ion and one or several fragment ions. This ensures the high specificity and accuracy of the method. In real life, however, only one transition pair might be monitored or only the dominant fragment ion may be detectable at the low concentrations in the samples. Longer LC gradients generally decrease the risk of wrongly assigned analyte peaks but decrease sample throughput at the same time. In addition, chiral chromatography might be needed to separate stereoisomers. This includes the separation of enzymatically and non-enzymatically formed species. Biological samples may undergo oxidation during prolonged storage, which may create prostanoid-like, non-enzymatically formed isoprostanes [100]. Difficulties that may be encountered in LC–MS/MS methods, including unique MRM transitions, in-source fragmentation, and matrix effects were previously discussed [98].
Some prostanoids, such as TXB2 or 2,3-dinor-6-keto PGF1α, produce several tautomeric forms, which appear as a conglomerate of individual signals and cause a broad peak shape during chromatographic separation. While quantification of these tautomers is usually unproblematic at sufficiently high concentrations, splitting a small signal at lower levels into several tautomeric peaks may result in a broad but shallow signal that falls below the required signal-to-noise ratio (S/N) of 10 for reliable quantification. For these compounds, derivatization may help to avoid tautomerization and enhance sensitivity [101].
The levels at which prostanoids are present in human biological samples are in the pmol to fmol range, making the sensitivity of the LC–MS/MS instrument an important feature to obtain a low limit of quantification. Sensitivity may be further enhanced by an initial sample preparation that purifies and concentrates the analytes of interest. Proper sample preparation can be a challenging task for a method that aims to quantify several metabolites in a single run and reach a decent overall sensitivity for all metabolites. Depending on chemical properties and interaction with different solvents and sorbents used in liquid–liquid extraction (LLE) or solid phase extraction (SPE), respectively, as well as depending on method optimization, losses may be more than 50% for individual analytes. Therefore, deuterated internal standards for every monitored analyte should be included in all methods that involve extraction to account for losses during sample preparation and compensate for matrix effects such as ion suppression [102]. The internal standard should be added as early as possible in the sample preparation procedure, and incubation time in the biological matrix should be long enough to be able to compensate for possible protein binding.
4. Frequently Encountered Obstacles When Analyzing Prostanoid Metabolites
4.1. Accuracy of Analytical Results Obtained by Immunoassay versus LC–MS/MS
Enzyme immunoassays and mass spectrometry-based assays are the two prime methodologies that are employed for the measurement of primary prostanoids and their metabolites as well as to quantify other lipid mediators. When comparing the literature values, it is eye-catching that results obtained by immunoassays are usually several fold higher than those obtained by LC–MS/MS [31]. A study that measured urinary 2,3-dinor-5,6-dihydro-15-F2t-isoprostane and directly compared the obtained results from two different methods found on average about 30-fold greater values when using an immunoassay as compared to gas chromatography (GC)–MS/MS analysis and an overall poor correlation (Pearson’s r = 0.51) between the two methods [103]. The tendency to overestimate analyte levels with immunoassays is most likely characteristic of the inherent weaknesses of this method, i.e., lack of analyte clean-up due to missing sample separation combined with a varying degree of antibody specificity that may lead to cross-reactivity with related compounds. Therefore, the measured levels may rather represent the sum of several metabolites than only a single target analyte and thus appear higher than their real levels because they are quantified based on the comparison to clean, synthetic, external standards. This may be a subsidiary analytical problem as long as relative changes in samples within one study are regarded that are all handled and measured by the same assay; however, it prevents the comparison of absolute values between studies. On the other hand, immunoassays of good quality, employing highly specific antibodies may well be able to take on results obtained by mass spectrometry [16]. Furthermore, biological samples may undergo oxidation during storage and sample handling, leading to isoprostanes, which are non-enzymatically formed, prostanoid-like molecules that can interfere with enzyme immunoassays and LC–MS/MS analysis, and result in measuring artifacts [100].
4.2. Undisclosed Use of NSAIDs May Compromise Study Results
When analyzing prostanoid metabolites as biomarkers in disease the biosynthetic pathway leading to their generation should be considered. COX is the molecular target of the widely used class of NSAIDs [13,104], which exert their analgesic, antipyretic, and anti-inflammatory effects through suppression of prostanoid biosynthesis, in line with the prominent role of prostanoids in the signaling of pain, inflammation, and the induction of fever. In 2010, about 12.8% of adults in the US consumed NSAIDs on a regular basis. An additional 19.0% of US adults chronically consumed low-dose aspirin for secondary prevention of cardiovascular disease [105]. Therefore, the levels of prostanoid metabolites measured in a given cohort may be underestimated due to inhibition of their biosynthesis in a substantial part of the general population, and the use of NSAIDs should be carefully controlled in any study that aims to quantify prostanoid metabolites as biomarkers for disease. However, non-reported NSAID usage may be unavoidable.
4.3. Normalization of Urinary Metabolites
Most biological fluids that may serve as samples for the analysis of prostanoid metabolites, including plasma, serum, and cerebrospinal fluid (CSF), are tightly controlled in their physiological composition. Therefore, mere concentrations of analyte per volume may suffice to compare different individuals, cohorts, or even studies from different labs. Urine, on the other hand, the biological fluid that contains the most stable prostanoid metabolites, which hence constitutes the best-suited biomarkers for disease, may differ substantially from sample to sample. This depends largely on the intake of water, which affects the total volume of excreted urine and thus the concentration of solutes. As a result, the investigation of specific target analytes requires normalization to compensate for these effects.
Among the most widely accepted normalization approaches are the use of urinary creatinine concentrations and urinary osmolality. These two methods yield comparable results; however, both methods have also certain drawbacks [106], and other normalization approaches might be better suited for specific study populations [8]. In the best analytical scenario, several normalization approaches can be used to yield similar results and thus validate the results [107].
Measurement of urinary analytes as a ratio to the creatinine concentration is considered the gold standard in normalization. Ideally, creatinine is released at a constant rate from muscle and protein metabolism and cleared from the blood at a constant rate in healthy patients with regular kidney function. Hence, the urinary creatinine concentration depends solely on the total volume of excreted urine and is therefore suitable for volume corrections of any other solute in the same sample. This implies, however, a healthy renal function and a constant glomerular filtration of creatinine. Moreover, since creatine release into the blood depends, among other factors, on the muscle mass (which generally differs between sexes), exercise status, and age of the subjects, the groups that are to be compared in a given study need to be carefully matched with regard to these parameters, and kidney function, as well as possible differences in creatinine excretion, need to be considered during the conception of a study.
Osmolality measures the urine concentration by considering all solutes that contribute to the osmotic pressure of a given urine sample and thus lower its freezing point compared to water. The above-mentioned factors that may compromise urinary creatinine concentrations do not affect osmolality [107]. However, a freezing point depression osmometer is necessary to determine osmolality, which is not available in all laboratories. Furthermore, insoluble particles or inhomogeneous samples may hamper accurate measurements of osmolality.
Alternative normalization strategies include a consistent and uniform dilution of all urine samples based on their optical absorption at 300 nm [101] and the use of the total useful MS signal [107], which requires elaborate equipment but is easily available when the analysis is performed using LC–MS/MS. In infants, whose kidney function is not yet fully developed and hence creatinine concentrations are not meaningful, the body surface area (BSA) was used before to normalize for metabolic status and excretion attainment [8].
4.4. Collection of 24 h Urine versus Spot Urine Samples
Initially, urinary prostanoid metabolites were studied in 24 h urine [108]. However, this method is intricate, relies on good subject compliance, and limited stability of some metabolites [8] at storage conditions available during the sampling period may impair the results. To circumvent these difficulties, normalization methods allow the use of spot urine samples instead because analytes that are normalized to, e.g., creatinine accurately reflects the 24 h excretion of the respective analyte. For instance, the creatinine-normalized levels of 2,3-dinor-6-keto PGF1α and 2,3-dinor-TXA2 in spot urine samples from 3 h long time intervals during a 24 h sampling period correlated highly with the total amounts determined for the respective interval. Simple concentrations in these spot urine samples, on the other hand, were poor indices of the 24 h excretion of both analytes [49]. General directions on how to sample, handle, store, and prepare urine samples prior to analysis were thoroughly reviewed [102].
4.5. Renal versus Extrarenal Origin of Urinary Prostanoid Metabolites and Data Interpretation
For reasons discussed in this article, i.e., rapid and continuous metabolism of prostanoids in vivo, the measurement of excreted stable end products of prostanoid metabolism has proven to be a useful approach. Thus, urine has lent itself as a valuable and informative source of biological samples. However, to judge what the measured levels really reflect in a given biological context, a careful interpretation of the results is demanded. Biosynthesis, metabolism, and excretion happen continuously and in parallel; therefore, many of the prostanoid metabolites are detectable both in plasma and in urine (Table 1). Here, we have reviewed the metabolites that are commonly regarded and measured as the major ones in the respective biofluids; however, most of the minor metabolites can also be detected in these samples. Analysis of several metabolites at the same time, which is easily achieved when using LC–MS/MS methodology, may serve as an internal verification of the results or even yield additional information and address thought-provoking aspects of the research question.
When analyzing and interpreting data from urine samples, one needs to carefully consider that this is an indirect way to estimate the systemic generation of prostanoids in vivo. Biosynthesis of prostanoids is locally restricted and occurs in a cell- and tissue-specific way. Prostanoid metabolites detected in the urine integrate biosynthesis of prostanoids in the entire organism but do not indicate their origin. Kidney function is particularly important when analyzing prostanoid metabolites in the urine; not only for its excretion activity, which affects both the analyte and the normalized creatinine but also because the kidneys have a high capacity to form prostanoids [109,110]. Therefore, it is generally assumed that the stable end metabolites reflect systemic biosynthesis, while more intermediate metabolites rather originate directly from the kidney. Although urinary prostanoid metabolites are synonymously addressed as systemic prostanoids, one should actually distinguish between circulatory and systemic prostanoids, which include kidney-derived prostanoids. For instance, both 6-keto PGF1α and 2,3-dinor-6-keto PGF1α are detectable in human urine, although at different levels. There is a statistically significant but only moderate correlation (r2 = 0.55) between these two metabolites of PGI2 [48], which indicates that the fraction that is fully metabolized to yield 2,3-dinor-6-keto PGF1α largely represents the circulatory PGI2 levels, while the fraction that is only partly metabolized to 6-keto PGF1α likely is a mix of systemic prostanoids that originates from both the circulation and the kidneys. This concept has been recently challenged by a case report [111] of one patient with inherited human group IV A cPLA2α deficiency due to a rare homozygous deletion in the PLA2G4A gene, causing a premature stop codon that results in an almost complete loss of systemic formation of eicosanoids. After suffering end-stage renal failure due to tubulointerstitial nephritis, this patient received an unrelated donor kidney from their spouse, which created a unique human whole-body cPLA2α knock-out, kidney-specific knock-in model. Interestingly, the urinary levels of 2,3-dinor-6-keto PGF1α and 11-dehydro TXB2 were restored from very low levels before transplantation to similar and even higher levels, respectively, than those in healthy volunteers [111]. The authors of the case report concluded that the urinary metabolites had purely renal origin [111] and that they poorly reflect production within the circulation in general [112]. Although this conclusion was criticized [113], among other reasons for possible adaptation processes within the kidney after transplantation and for comparing this patient to healthy volunteers instead of other renal failure patients after kidney transplantation, it raises awareness to critically consider established concepts, e.g., to what extent kidney-derived prostanoids are metabolized. Unfortunately, the case report provided only limited data; additional information on urinary levels of 6-keto PGF1α or TXB2 and 2,3-dinor TXB2 might have been revealing. This report raises furthermore awareness to regard eicosanoid lipid mediators as an interconnected network and evaluate it with appropriate controls on different levels. Ideally, observed changes in one or a few metabolites should be backed-up in a broader lipidomic approach and can be reflected in the expression and activity levels of the involved enzymatic machinery.
5. Prostanoid Metabolites as Relevant Biomarkers in Human Disease
The role of primary prostanoids as lipid mediators in disease is well described, e.g., in inflammatory pain [114], autoimmunity [115] and cancer [116], and their potential as a therapeutic target has recently been reviewed [117]. Here, we summarize what is known about the use of prostanoid metabolites as potential biomarkers in human disease. Biomarkers are defined as endogenous molecules that reflect the health status of a person. They are present in biological fluids or soft tissues and change from their baseline concentrations in response to physiological or pathological processes, respectively. Hence, they can be measured as an index of disease. Furthermore, they also respond to pharmacological intervention. Thus, biomarkers can be used as diagnostic, predictive, or prognostic tools to evaluate a patient’s condition or response to treatment.
Prostanoids emerge locally in a cell- and tissue-specific way, their concentration may differ substantially between different tissues, but also change immensely with physiological responses. Therefore, prostanoids can be viewed as biomarkers; however, interpretation of measured concentrations is not trivial.
A few studies investigated primary prostanoids in local tissues for their potential to serve as biomarkers of disease. It should be noted that this approach has some caveats that limit a wide application. Invasive procedures are usually necessary to obtain tissue samples from human subjects. Furthermore, the measured prostanoid levels may differ from the true levels in vivo due to ongoing metabolism. Therefore, studying the biosynthetic capacity of a certain tissue and its changes during disease by investigating induced prostanoid formation after in vitro stimulation might be a better alternative to directly measuring primary prostanoids as biomarkers. In patients with familial adenomatous polyposis (FAP), exogenously added arachidonic acid to mucosa samples induced the formation of all five primary prostanoids. Chemoprevention for colorectal neoplasia with NSAIDs leads to a reduction in the levels of all prostanoids, which correlated with the number and size of adenomas in these patients and could thus be used to monitor the clinical progression of polyposis [118]. In CSF from patients with amyotrophic lateral sclerosis (ALS) [119], infants with hypoxic damage [120], and patients with multiple sclerosis (MS) [121], the levels of PGE2 were found to be increased and could serve as a biomarker for disease severity. However, sampling of CSF is invasive, which restricts the use of this biomarker to a relatively small group of patients.
Because sampling of plasma and urine, respectively, is less invasive, these types of samples are more broadly applicable to large patient cohorts and therefore generally preferred to analyze prostanoids as biomarkers for human disease. Prostanoid metabolites detected in plasma/urine and associated with human diseases are summarized in Table 2. Studies on systemic production of prostanoids in humans are mainly performed in urine.
5.1. PGE2 Metabolites as Biomarkers
As a mainly pro-inflammatory mediator, biosynthesis of PGE2 is upregulated by inflammatory stimuli [141,142], which makes PGE2 and its metabolites a relevant marker for human diseases that involve chronic inflammation. Plasma levels of PGE2 were, however, also investigated as biomarkers in the context of other pathologic conditions, e.g., autism [143], acetylsalicylic acid (ASA)/Aspirin exposure [144], and obesity [31]. PGE2 was furthermore measured in saliva and associated with arterial stiffness [126] and markedly correlated with age [145]. Several of these studies used immunoassays that report both PGE2 and several of its metabolites combined as a measure of inflammation. Therefore, it is not always clear which specific analyte was actually reported. However, many of these studies claim that it is not important to distinguish between different metabolites. More defined biomarker studies focused on bicyclo-PGE2 in plasma and reported it as a marker for malaria pathogenesis [127]. Prostanoids also play a prominent role during pregnancy and labor, which has been recently reviewed [6]. In this context, plasma levels of PGE2, 13,14-dihydro-15-keto-PGE2, and bicyclo-PGE2 yielded, however, inconsistent results [6].
While the measurement of unmetabolized PGE2 is uncommon in urine, it was suggested as a biomarker for recurrent urinary tract infections [146]. Urinary metabolites of PGE2 on the other hand, foremost tetranor-PGEM, were suggested as potential biomarkers for inflammation in many human diseases, e.g., for ulcerative colitis [122], viral infections in infants [8], and as a prognostic marker for the development of breast cancer [123]. A pre-validation study that investigated tetranor-PGEM as a potential biomarker for the detection of advanced colorectal neoplasia found increased levels of this metabolite in patients with colorectal cancers and large adenomas as compared to healthy patients with small or without adenomas [124]. Importantly, this study also described a sex difference in the levels of tetranor-PGEM with higher levels in men. A subsequent clinical trial with the aim to establish tetranor-PGEM as a biomarker for Crohn’s disease activity (ClinicalTrials.gov Identifier: NCT00496548) was completed in 2015, but results are still pending. Tetranor-PGEM was furthermore shown to be moderately increased in obese subjects and predominantly associated with abdominal obesity and parameters of pre-diabetes [31]. In this study, the combination of tetranor-PGEM with tetranor-PGDM was particularly informative. Both tetranor-PGEM with tetranor-PGDM were also increased in patients with cystic fibrosis [125].
5.2. PGD2 Metabolites as Biomarkers
Several plasma metabolites of PGD2 were investigated in the context of human disease. 13,14-dihydro-15-keto-prostaglandin D2 was suggested as a potential biomarker for the diagnosis of nonalcoholic fatty liver (NASH) [133] and serum levels of 11β-PGF2α as a marker of anaphylaxis [134]. There are a few studies with plasma levels of 15-deoxy-Δ12,14-PGJ2 in their focus, but only one of them suggested it as a biomarker [34]. In this study, 15-deoxy-Δ12,14-PGJ2 was increased in the plasma of diabetic patients and inversely correlated to CRP [34].
Biomarker studies on urinary PGD2 metabolites are more numerous. Prostanoids, especially PGD2, are important mediators released by activated mast cells and thus involved in severe allergic and anaphylactic reactions. Tetranor-PGDM was shown to be increased in patients with food allergies [128] as well as in the urine of patients with Aspirin-intolerant asthma and may be considered a marker for mast cell activation [129]. Recently, the U-BIOPRED (Unbiased Biomarkers for the Prediction of Respiratory Diseases Outcomes) study revealed that the levels of urinary PGD2 metabolites (both tetranor-PGDM and 2,3-dinor-11β-PGF2α) were, along with urinary levels of leukotriene (LT)E4, increased in patients with mild and severe asthma and associated with a lower lung function. Hence, these urinary eicosanoids were suggested as new, non-invasive biomarkers to detect type 2 inflammation and to phenotype asthma patients on a molecular level [130]. NSAIDs inhibit prostanoid biosynthesis and are a leading cause of drug hypersensitivity reactions [147]. Urinary tetranor-PGDM was suggested as a biomarker for NSAID-induced hypersensitivity [132]. In obese subjects, tetranor-PGDM was markedly increased and associated with serum triglycerides, which led to the hypothesis that elevated levels of systemic PGD2 might aggravate obesity [31]. It was also suggested as a urinary marker for the progression of Duchenne muscular dystrophy [131]. Furthermore, urinary tetranor-PGDM was investigated as a marker for atopic dermatitis but was not found to be altered compared to healthy controls [148]. In the field of cancer, tetranor-PGDM was identified as a biomarker in animal studies, e.g., tetranor-PGDM is increased in mice with colitis-associated colorectal cancer [149], but there are no studies in humans.
5.3. PGF2α Metabolites as Biomarkers
The in vivo synthesis of PGF2α may be measured in human plasma as 13,14-dihydro-15-keto PGF2α (so-called “PGFM”) [150]. The levels of both PGF2α and PGFM were found to be increased during labor in amniotic fluid, while the same increase was apparent in the blood only for PGFM [6].
Urinary tetranor-PGFM was investigated in humans to study the effect of diet on lipid peroxidation; however, no changes in the levels of this metabolite could be found [151]. In another study, patients with psoriasis were investigated, and tetranor-PGFM was found to be associated with increased psoriasis area and severity index [135].
5.4. TXA2 Metabolites as Biomarkers
Increased serum levels of TXB2 might predict myocardial infarction [152], but metabolites of TXB2 in plasma have not been shown to be potential biomarkers for this condition.
While it was previously proposed that 2,3-dinor-TXB2 is the main urinary metabolite in mice and 11-dhydro-TXB2 is the main urinary metabolite in humans [153], it is now suggested that both metabolites are formed in similar amounts in humans [40]. Their alterations in human disease were recently reviewed [40].
In urine, 2,3-dinor-TXB2 is considered a marker of in vivo platelet activation [40]. Urinary 11-dehydro-TXB2 was shown to be increased in patients with different stages of chronic kidney disease [136]. This metabolite was also associated with an increased risk of cardiovascular events as measured at baseline in patients with nonvalvular atrial fibrillation [154], and vascular inflammation [138]. Other studies on this biomarker in cardiovascular disease were reviewed [137].
5.5. PGI2 Metabolites as Biomarkers
Plasma levels of 6-keto PGF1α were investigated during labor; the results were, however, inconsistent [6].
While 6-keto PGF1α can also be detected in urine, the main urinary metabolite that reflects the systemic biosynthesis of PGI2 is 2,3-dinor-6-keto PGF1α, which is also called PGIM. The ratio between 2,3-dinor-6-keto PGF1α and 11-dehydro-TXB2 was suggested to reflect the pathological state in diabetic patients [139] and to evaluate pregnancy after in vitro fertilization [155]. 2,3-dinor-6-keto PGF1α was also suggested as a prognostic marker for acute kidney injury [140].
6. Conclusions
The five primary prostanoids, PGE2, PGD2, PGF2α, PGI2, and TXA2, are important mediators and play a central role in many physiological and pathophysiological processes. Inherent to their role as evanescent autacoids in vivo, they have a limited action range and rapidly lose their biological activity through metabolic rendering. Pioneering work has elucidated a plethora of metabolites, of which major plasma and urinary metabolites were identified for each of the primary prostanoids. Using sensitive methodology, these metabolites can be measured as substitutes to reflect integrated prostanoid biosynthesis in vivo and to serve as diagnostic, predictive, or prognostic biomarkers for human diseases. However, only a limited number of biomarker studies are available to date that explore the potential of measuring prostanoid metabolites and their fluctuations during pathological changes in humans in relation to disease outcomes.
Metabolism of prostanoids is continuously ongoing; hence, measuring the more stable end products in the urine, i.e., the tetranor-metabolites of PGE2, PGD2, and PGF2α, and the dinor-metabolites of PGI2, and TXA2, yields the most consistent and reliable results. However, depending on the specific research question, analysis of other biofluids with their respective major prostanoid metabolites can be informative, too. Urine and plasma are the most commonly studied biofluids for prostanoid metabolite analysis, and they lend themselves to biomarker studies in large cohorts because they are relatively easily sampled. Analyzing prostanoid metabolites in these samples is, however, an indirect approach to estimating systemic in vivo generation of primary prostanoids. Their levels are integrated pools of production throughout the organism and do not reveal any information about specific levels in their tissue(s) of origin. Initial proof-of-principal studies that elucidate the pathogenic mechanism of primary prostanoids under a given disease condition, identify their biological source, trace their metabolism, and verify their induced concentration range as compared to the healthy state, are therefore a prerequisite for the identification of reliable biomarkers and form, combined with larger clinical studies that demonstrate the correlation of systemic levels of their metabolites with specific symptoms, a powerful translational approach.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/metabo12080721/s1, Table S1: Molecular identity and frequently used synonym names in the literature.
Author Contributions
H.I. and S.-C.P. contributed equally to this work by conceptualizing and drafting the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Karolinska Institute’s Research Foundation Grants (H.I.) and the Stiftelsen Professor Nanna Svartz fund (S.-C.P.).
Data Availability Statement
The data presented in this study are available in the main article and the Supplementary Materials.
Acknowledgments
H.I. and S.-C.P. would like to thank Marina Korotkova and Magnus Bäck from Karolinska Institute for critically reading the manuscript and providing valuable comments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mitchell, J.A.; Kirkby, N.S.; Ahmetaj-Shala, B.; Armstrong, P.C.; Crescente, M.; Ferreira, P.; Lopes Pires, M.E.; Vaja, R.; Warner, T.D. Cyclooxygenases and the cardiovascular system. Pharmacol. Ther. 2021, 217, 107624. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, B.; Gowda, S.; Welty, S.E.; Barrington, K.J.; Pammi, M. Prostanoids and their analogues for the treatment of pulmonary hypertension in neonates. Cochrane Database Syst. Rev. 2019, 10, CD012963. [Google Scholar] [CrossRef]
- Yang, T.; Liu, M. Regulation and function of renal medullary cyclooxygenase-2 during high salt loading. Front. Biosci. 2017, 22, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, A.S.; Ottallah, H.; Maciel, C.B.; Strickland, M.; Doré, S. Role of the L-PGDS-PGD2-DP1 receptor axis in sleep regulation and neurologic outcomes. Sleep 2019, 42, zsz073. [Google Scholar] [CrossRef] [PubMed]
- Urade, Y.; Hayaishi, O. Prostaglandin D2 and sleep/wake regulation. Sleep Med. Rev. 2011, 15, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.M.; Hornaday, K.K.; Slater, D.M. Prostaglandins in biofluids in pregnancy and labour: A systematic review. PLoS ONE 2021, 16, e0260115. [Google Scholar] [CrossRef] [PubMed]
- Garami, A.; Steiner, A.A.; Romanovsky, A.A. Fever and hypothermia in systemic inflammation. Handb. Clin. Neurol. 2018, 157, 565–597. [Google Scholar] [CrossRef] [PubMed]
- Idborg, H.; Pawelzik, S.C.; Perez-Manso, M.; Bjork, L.; Hamrin, J.; Herlenius, E.; Jakobsson, P.J. Evaluation of urinary prostaglandin E2 metabolite as a biomarker in infants with fever due to viral infection. Prostaglandins Leukot. Essent. Fat. Acids 2014, 91, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Engström, L.; Mackerlova, L.; Jakobsson, P.J.; Blomqvist, A. Impaired febrile responses to immune challenge in mice deficient in microsomal prostaglandin E synthase-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1100–R1107. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Omori, K.; Murata, T. Role of prostaglandins in tumor microenvironment. Cancer Metastasis Rev. 2018, 37, 347–354. [Google Scholar] [CrossRef]
- Jang, Y.; Kim, M.; Hwang, S.W. Molecular mechanisms underlying the actions of arachidonic acid-derived prostaglandins on peripheral nociception. J. Neuroinflamm. 2020, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Grösch, S.; Niederberger, E.; Geisslinger, G. Investigational drugs targeting the prostaglandin E2 signaling pathway for the treatment of inflammatory pain. Expert Opin. Investig. Drugs 2017, 26, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Funk, C.D. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, P.; Pawelzik, S.C.; Idborg, H.; Spahiu, L.; Larsson, C.; Stenberg, P.; Korotkova, M.; Jakobsson, P.J. Characterization of a new mPGES-1 inhibitor in rat models of inflammation. Prostaglandins Other Lipid Mediat. 2013, 102–103, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Morris, R.J.; Bode, A.M.; Zhang, T. Prostaglandin Pathways: Opportunities for Cancer Prevention and Therapy. Cancer Res. 2022, 82, 949–965. [Google Scholar] [CrossRef]
- Hanaka, H.; Pawelzik, S.C.; Johnsen, J.I.; Rakonjac, M.; Terawaki, K.; Rasmuson, A.; Sveinbjörnsson, B.; Schumacher, M.C.; Hamberg, M.; Samuelsson, B.; et al. Microsomal prostaglandin E synthase 1 determines tumor growth in vivo of prostate and lung cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 18757–18762. [Google Scholar] [CrossRef] [Green Version]
- Biringer, R.G. The enzymology of the human prostanoid pathway. Mol. Biol. Rep. 2020, 47, 4569–4586. [Google Scholar] [CrossRef]
- Smith, W.L.; Urade, Y.; Jakobsson, P.J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [Green Version]
- Biringer, R.G. A Review of Prostanoid Receptors: Expression, Characterization, Regulation, and Mechanism of Action. J. Cell Commun. Signal. 2020, 15, 155–184. [Google Scholar] [CrossRef]
- Smith, W.L.; Malkowski, M.G. Interactions of fatty acids, nonsteroidal anti-inflammatory drugs, and coxibs with the catalytic and allosteric subunits of cyclooxygenases-1 and -2. J. Biol. Chem. 2019, 294, 1697–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Donk, W.A.; Tsai, A.L.; Kulmacz, R.J. The cyclooxygenase reaction mechanism. Biochemistry 2002, 41, 15451–15458. [Google Scholar] [CrossRef]
- Hamberg, M.; Samuelsson, B. On the mechanism of the biosynthesis of prostaglandins E-1 and F-1-alpha. J. Biol. Chem. 1967, 242, 5336–5343. [Google Scholar] [CrossRef]
- Hamberg, M.; Samuelsson, B. Oxygenation of unsaturated fatty acids by the vesicular gland of sheep. J. Biol. Chem. 1967, 242, 5344–5354. [Google Scholar] [CrossRef]
- Seo, M.J.; Oh, D.K. Prostaglandin synthases: Molecular characterization and involvement in prostaglandin biosynthesis. Prog. Lipid Res. 2017, 66, 50–68. [Google Scholar] [CrossRef]
- Sun, C.-C.; Zhou, Z.-Q.; Yang, D.; Chen, Z.-L.; Zhou, Y.-Y.; Wen, W.; Feng, C.; Zheng, L.; Peng, X.-Y.; Tang, C.-F. Recent advances in studies of 15-PGDH as a key enzyme for the degradation of prostaglandins. Int. Immunopharmacol. 2021, 101, 108176. [Google Scholar] [CrossRef] [PubMed]
- Hamberg, M.; Samuelsson, B. On the Metabolism of Prostaglandins E1 and E2 in Man. J. Biol. Chem. 1971, 246, 6713–6721. [Google Scholar] [CrossRef]
- Bothwell, W.; Verburg, M.; Wynalda, M.; Daniels, E.G.; Fitzpatrick, F.A. A radioimmunoassay for the unstable pulmonary metabolites of prostaglandin E1 and E2: An indirect index of their in vivo disposition and pharmacokinetics. J. Pharmacol. Exp. Ther. 1982, 220, 229–235. [Google Scholar]
- Fitzpatrick, F.A.; Aguirre, R.; Pike, J.E.; Lincoln, F.H. The stability of 13,14-dihydro-15 keto-PGE2. Prostaglandins 1980, 19, 917–931. [Google Scholar] [CrossRef]
- Pawelzik, S.C.; Avignon, A.; Idborg, H.; Boegner, C.; Stanke-Labesque, F.; Jakobsson, P.J.; Sultan, A.; Back, M. Urinary prostaglandin D2 and E2 metabolites associate with abdominal obesity, glucose metabolism, and triglycerides in obese subjects. Prostaglandins Other Lipid Mediat. 2019, 145, 106361. [Google Scholar] [CrossRef]
- Leonhardt, A.; Krauss, M.; Gieler, U.; Schweer, H.; Happle, R.; Seyberth, H.W. In vivo formation of prostaglandin E1 and prostaglandin E2 in atopic dermatitis. Br. J. Dermatol. 1997, 136, 337–340. [Google Scholar]
- Schuligoi, R.; Schmidt, R.; Geisslinger, G.; Kollroser, M.; Peskar, B.A.; Heinemann, A. PGD2 metabolism in plasma: Kinetics and relationship with bioactivity on DP1 and CRTH2 receptors. Biochem. Pharmacol. 2007, 74, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, J.; Fleming, T.; Kadiyska, I.; Brings, S.; Groener, J.B.; Nawroth, P.; Hecker, M.; Brune, M. Sensitive mass spectrometric assay for determination of 15-deoxy-Δ12,14-prostaglandin J2 and its application in human plasma samples of patients with diabetes. Anal. Bioanal. Chem. 2017, 410, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Bell-Parikh, L.C.; Ide, T.; Lawson, J.A.; McNamara, P.; Reilly, M.; FitzGerald, G.A. Biosynthesis of 15-deoxy-Δ12,14-PGJ2 and the ligation of PPARγ. J. Clin. Investig. 2003, 112, 945–955. [Google Scholar] [CrossRef] [Green Version]
- Meyer, H.H.; Eisele, K.; Osaso, J. A biotin-streptavidin amplified enzymeimmunoassay for 13,14-dihydro-15-keto-PGF2 alpha. Prostaglandins 1989, 38, 375–383. [Google Scholar] [CrossRef]
- Granström, E.; Kindahl, H.; Swahn, M.L. Profiles of prostaglandin metabolites in the human circulation. Identification of late-appearing, long-lived products. Biochim. Biophys. Acta 1982, 713, 46–60. [Google Scholar] [CrossRef]
- Medina, S.; Domínguez-Perles, R.; Gil, J.I.; Ferreres, F.; García-Viguera, C.; Martínez-Sanz, J.M.; Gil-Izquierdo, A. A ultra-pressure liquid chromatography/triple quadrupole tandem mass spectrometry method for the analysis of 13 eicosanoids in human urine and quantitative 24 hour values in healthy volunteers in a controlled constant diet. Rapid Commun. Mass Spectrom. 2012, 26, 1249–1257. [Google Scholar] [CrossRef]
- Needleman, P.; Moncada, S.; Bunting, S.; Vane, J.R.; Hamberg, M.; Samuelsson, B. Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature 1976, 261, 558–560. [Google Scholar] [CrossRef]
- Patrono, C.; Rocca, B. Measurement of Thromboxane Biosynthesis in Health and Disease. Front. Pharmacol. 2019, 10, 1244. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C.; Ciabattoni, G.; Pugliese, F.; Pierucci, A.; Blair, I.A.; FitzGerald, G.A. Estimated rate of thromboxane secretion into the circulation of normal humans. J. Clin. Investig. 1986, 77, 590–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catella, F.; Healy, D.; Lawson, J.A.; FitzGerald, G.A. 11-Dehydrothromboxane B2: A quantitative index of thromboxane A2 formation in the human circulation. Proc. Natl. Acad. Sci. USA 1986, 83, 5861–5865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santilli, F.; Davì, G.; Basili, S.; Lattanzio, S.; Cavoni, A.; Guizzardi, G.; De Feudis, L.; Traisci, G.; Pettinella, C.; Paloscia, L.; et al. Thromboxane and prostacyclin biosynthesis in heart failure of ischemic origin: Effects of disease severity and aspirin treatment. J. Thromb. Haemost. 2010, 8, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Machleidt, C.; Förstermann, U.; Anhut, H.; Hertting, G. Formation and elimination of prostacyclin metabolites in the cat in vivo as determined by radioimmunoassay of unextracted plasma. Eur. J. Pharmacol. 1981, 74, 19–26. [Google Scholar] [CrossRef]
- Orchard, M.A.; Robinson, C. Stability of prostacyclin in human plasma and whole blood: Studies on the protective effect of albumin. Thromb. Haemost. 1981, 46, 645–647. [Google Scholar] [CrossRef]
- Polterauer, P.; Sinzinger, H.; Peskar, B.A. Biological half-life of prostacyclin and 6-oxo-PGF1 alpha levels in plasma of patients with colonic cancer. Prostaglandins Leukot. Med. 1986, 22, 249–258. [Google Scholar] [CrossRef]
- Lucas, F.V.; Skrinska, V.A.; Chisolm, G.M.; Hesse, B.L. Stability of prostacyclin in human and rabbit whole blood and plasma. Thromb. Res. 1986, 43, 379–387. [Google Scholar] [CrossRef]
- Chappell, D.L.; Xiao, X.; Radziszewski, W.; Laterza, O.F. Development and validation of a LC/MS/MS method for 6-keto PGF1α, a metabolite of prostacyclin (PGI2). J. Pharm. Biomed. Anal. 2011, 56, 600–603. [Google Scholar] [CrossRef]
- Wennmalm, A.; Benthin, G.; Granström, E.F.; Persson, L.; Winell, S. 2,3-Dinor metabolites of thromboxane A2 and prostacyclin in urine from healthy human subjects: Diurnal variation and relation to 24h excretion. Clin. Sci. 1992, 83, 461–465. [Google Scholar] [CrossRef]
- Liston, T.E.; Roberts, L.J., 2nd. Metabolic fate of radiolabeled prostaglandin D2 in a normal human male volunteer. J. Biol. Chem. 1985, 260, 13172–13180. [Google Scholar] [CrossRef]
- Roberts, L.J., 2nd; Sweetman, B.J.; Oates, J.A. Metabolism of thromboxane B2 in man. Identification of twenty urinary metabolites. J. Biol. Chem. 1981, 256, 8384–8393. [Google Scholar] [CrossRef]
- Bygdeman, M. Pharmacokinetics of prostaglandins. Best Pract. Res. Clin. Obstet. Gynaecol. 2003, 17, 707–716. [Google Scholar] [CrossRef]
- Chou, W.L.; Chuang, L.M.; Chou, C.C.; Wang, A.H.; Lawson, J.A.; FitzGerald, G.A.; Chang, Z.F. Identification of a novel prostaglandin reductase reveals the involvement of prostaglandin E2 catabolism in regulation of peroxisome proliferator-activated receptor gamma activation. J. Biol. Chem. 2007, 282, 18162–18172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granström, E.; Hamberg, M.; Hansson, G.; Kindahl, H. Chemical instability of 15-keto-13,14-dihydro-PGE 2: The reason for low assay reliability. Prostaglandins 1980, 19, 933–957. [Google Scholar] [CrossRef]
- Murphy, R.C.; FitzGerald, G.A. Current approaches to estimation of eicosanoid formation in vivo. Adv. Prostaglandin Thromboxane Leukot. Res. 1994, 22, 341–348. [Google Scholar] [PubMed]
- Hamberg, M. Inhibition of prostaglandin synthesis in man. Biochem. Biophys. Res. Commun. 1972, 49, 720–726. [Google Scholar] [CrossRef]
- Honda, H.; Fukawa, K.; Sawabe, T. Influence of adjuvant arthritis on main urinary metabolites of prostaglandin F and E in rats. Prostaglandins 1980, 19, 259–269. [Google Scholar] [CrossRef]
- Song, W.-L.; Wang, M.; Ricciotti, E.; Fries, S.; Yu, Y.; Grosser, T.; Reilly, M.; Lawson, J.A.; FitzGerald, G.A. Tetranor PGDM, an Abundant Urinary Metabolite Reflects Biosynthesis of Prostaglandin D2 in Mice and Humans. J. Biol. Chem. 2008, 283, 1179–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangachari, P.K.; Betti, P.A. Biological activity of metabolites of PGD2 on canine proximal colon. Am. J. Physiol. 1993, 264, G886–G894. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Spokas, E.G.; Marcinkiewicz, E.; Wong, P.Y. Hepatic transformation of prostaglandin D2 to a new prostanoid, 9 alpha, 11 beta-prostaglandin F2, that inhibits platelet aggregation and constricts blood vessels. J. Biol. Chem. 1985, 260, 14621–14625. [Google Scholar] [CrossRef]
- Seibert, K.; Sheller, J.R.; Roberts, L.J., 2nd. (5Z,13E)-(15S)-9 alpha, 11 beta,15-trihydroxyprosta-5,13-dien-1-oic acid (9 alpha, 11 beta-prostaglandin F2): Formation and metabolism by human lung and contractile effects on human bronchial smooth muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liston, T.E.; Roberts, L.J., 2nd. Transformation of prostaglandin D2 to 9 alpha, 11 beta-(15S)-trihydroxyprosta-(5Z,13E)-dien-1-oic acid (9 alpha, 11 beta-prostaglandin F2): A unique biologically active prostaglandin produced enzymatically in vivo in humans. Proc. Natl. Acad. Sci. USA 1985, 82, 6030–6034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.A.; Sheldrick, R.L.G. Prostanoid-induced contraction of human bronchial smooth muscle is mediated by TP-receptors. Br. J. Pharmacol. 1989, 96, 688–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, L.J., 2nd; Seibert, K.; Liston, T.E.; Tantengco, M.V.; Robertson, R.M. PGD2 is transformed by human coronary arteries to 9 alpha, 11 beta-PGF2, which contracts human coronary artery rings. Adv. Prostaglandin Thromboxane Leukot. Res. 1987, 17A, 427–429. [Google Scholar] [PubMed]
- Yoda, T.; Kikuchi, K.; Miki, Y.; Onodera, Y.; Hata, S.; Takagi, K.; Nakamura, Y.; Hirakawa, H.; Ishida, T.; Suzuki, T.; et al. 11β-Prostaglandin F2α, a bioactive metabolite catalyzed by AKR1C3, stimulates prostaglandin F receptor and induces slug expression in breast cancer. Mol. Cell. Endocrinol. 2015, 413, 236–247. [Google Scholar] [CrossRef]
- Söderström, M.; Wigren, J.; Surapureddi, S.; Glass, C.K.; Hammarström, S. Novel prostaglandin D 2-derived activators of peroxisome proliferator-activated receptor-γ are formed in macrophage cell cultures. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2003, 1631, 35–41. [Google Scholar] [CrossRef]
- Fukushima, M.; Sasaki, H.; Fukushima, S. Prostaglandin J2 and Related Compounds. Ann. N. Y. Acad. Sci. 1994, 744, 161–165. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Lenhard, J.M.; Willson, T.M.; Patel, I.; Morris, D.C.; Lehmann, J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 1995, 83, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Straus, D.S.; Glass, C.K. Cyclopentenone prostaglandins: New insights on biological activities and cellular targets. Med. Res. Rev. 2001, 21, 185–210. [Google Scholar] [CrossRef]
- Levine, L.; Gutierrez-Cernosek, R.M. Levels of 13,14-dihydro-15-keto-PGF2α in biological fluids as measured by radioimmunoassay. Prostaglandins 1973, 3, 785–804. [Google Scholar] [CrossRef]
- Samuelsson, B.; Goldyne, M.; Granström, E.; Hamberg, M.; Hammarström, S.; Malmsten, C. Prostaglandins and thromboxanes. Annu. Rev. Biochem. 1978, 47, 997–1029. [Google Scholar] [CrossRef]
- Granstrom, E.; Samuelsson, B. Structure of a urinary metabolite of prostaglandin F2.alpha. in man. J. Am. Chem. Soc. 1969, 91, 3398–3400. [Google Scholar] [CrossRef]
- Neves, A.C.O.; Morais, C.L.M.; Mendes, T.P.P.; Vaz, B.G.; Lima, K.M.G. Mass spectrometry and multivariate analysis to classify cervical intraepithelial neoplasia from blood plasma: An untargeted lipidomic study. Sci. Rep. 2018, 8, 3910–3954. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, B. Role of basic science in the development of new medicines: Examples from the eicosanoid field. J. Biol. Chem. 2012, 287, 10070–10080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westlund, P.; Fylling, A.C.; Cederlund, E.; Jörnvall, H. 11-Hydroxythromboxane B2 dehydrogenase is identical to cytosolic aldehyde dehydrogenase. FEBS Lett. 1994, 345, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Westlund, P.; Granström, E.; Kumlin, M.; Nordenström, A. Identification of 11-dehydro-TXB2 as a suitable parameter for monitoring thromboxane production in the human. Prostaglandins 1986, 31, 929–960. [Google Scholar] [CrossRef]
- Lawson, J.A.; Brash, A.R.; Doran, J.; FitzGerald, G.A. Measurement of urinary 2,3-dinor-thromboxane B2 and thromboxane B2 using bonded-phase phenylboronic acid columns and capillary gas chromatography--negative-ion chemical ionization mass spectrometry. Anal. Biochem. 1985, 150, 463–470. [Google Scholar] [CrossRef]
- Lellouche, F.; Fradin, A.; Fitzgerald, G.; Maclouf, J. Enzyme immunoassay measurement of the urinary metabolites of thromboxane A2 and prostacyclin. Prostaglandins 1990, 40, 297–310. [Google Scholar] [CrossRef]
- Perneby, C.; Granström, E.; Beck, O.; Fitzgerald, D.; Harhen, B.; Hjemdahl, P. Optimization of an enzyme immunoassay for 11-dehydro-thromboxane B(2) in urine: Comparison with GC-MS. Thromb. Res. 1999, 96, 427–436. [Google Scholar] [CrossRef]
- Lewis, P.J.; Dollery, C.T. Clinical pharmacology and potential of prostacyclin. Br. Med. Bull. 1983, 39, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R.; Jackson, E.K.; Saggese, C.A.; Lawson, J.A.; Oates, J.A.; FitzGerald, G.A. Metabolic disposition of prostacyclin in humans. J. Pharmacol. Exp. Ther. 1983, 226, 78–87. [Google Scholar] [PubMed]
- Flavahan, N.A. Balancing prostanoid activity in the human vascular system. Trends Pharmacol. Sci. 2006, 28, 106–110. [Google Scholar] [CrossRef]
- Rosenkranz, B.; Fischer, C.; Reimann, I.; Weimer, K.E.; Beck, G.; Frölich, J.C. Identification of the major metabolite of prostacyclin and 6-ketoprostaglandin F1α in man. Biochim. Biophys. Acta Lipids Lipid Metab. 1980, 619, 207–213. [Google Scholar] [CrossRef]
- Patrono, C.; Dunn, M.J. The clinical significance of inhibition of renal prostaglandin synthesis. Kidney Int. 1987, 32, 1–12. [Google Scholar] [CrossRef] [Green Version]
- FitzGerald, G.A.; Brash, A.R.; Falardeau, P.; Oates, J.A. Estimated rate of prostacyclin secretion into the circulation of normal man. J. Clin. Investig. 1981, 68, 1272–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, S.R.; Kleiveland, C.R.; Kassem, M.; Lea, T.; Lundanes, E.; Greibrokk, T. Detecting pM concentrations of prostaglandins in cell culture supernatants by capillary SCX-LC-MS/MS. J. Sep. Sci. 2008, 31, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Matsunuma, S.; Handa, S.; Kamei, D.; Yamamoto, H.; Okuyama, K.; Kato, Y. Oxaliplatin induces prostaglandin E2 release in vascular endothelial cells. Cancer Chemother. Pharmacol. 2019, 84, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Popov, T.A.M.D.P. Human exhaled breath analysis. Ann. Allergy Asthma Immunol. 2011, 106, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Degousee, N.; Fazel, S.; Angoulvant, D.; Stefanski, E.; Pawelzik, S.C.; Korotkova, M.; Arab, S.; Liu, P.; Lindsay, T.F.; Zhuo, S.; et al. Microsomal prostaglandin E2 synthase-1 deletion leads to adverse left ventricular remodeling after myocardial infarction. Circulation 2008, 117, 1701–1710. [Google Scholar] [CrossRef] [Green Version]
- Catella, F.; Nowak, J.; Fitzgerald, G.A. Measurement of renal and non-renal eicosanoid synthesis. Am. J. Med. 1986, 81, 23–29. [Google Scholar] [CrossRef]
- Granström, E.; Kumlin, M. Assay of thromboxane production in biological systems: Reliability of TXB2 versus 11-dehydro-TXB2 as targets for measurement. Adv. Prostaglandin Thromboxane Leukot. Res. 1987, 17, 587–594. [Google Scholar]
- Patrignani, P.; Tacconelli, S.; Piazuelo, E.; Di Francesco, L.; Dovizio, M.; Sostres, C.; Marcantoni, E.; Guillem-Llobat, P.; Del Boccio, P.; Zucchelli, M.; et al. Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action. J. Thromb. Haemost. 2014, 12, 1320–1330. [Google Scholar] [CrossRef] [Green Version]
- Bergstroem, S.; Ryhage, R.; Samuelsson, B.; Sjoevall, J. Prostaglandins and related factors. 15. the structures of prostaglandin e1, f1-alpha, and f1-beta. J. Biol. Chem. 1963, 238, 3555–3564. [Google Scholar] [PubMed]
- Blair, I.A. Measurement of eicosanoids by gas chromatography and mass spectrometry. Br. Med. Bull. 1983, 39, 223–226. [Google Scholar] [CrossRef]
- Blair, I.A. Electron-capture negative-ion chemical ionization mass spectrometry of lipid mediators. Methods Enzymol. 1990, 187, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.; Gonzalez-Riano, C.; Barbas, C.; Kolmert, J.; Hyung Ryu, M.; Carlsten, C.; Dahlén, S.-E.; Wheelock, C.E. Quantitative metabolic profiling of urinary eicosanoids for clinical phenotyping. J. Lipid Res. 2019, 60, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Chhonker, Y.S.; Kanvinde, S.; Ahmad, R.; Singh, A.B.; Oupický, D.; Murry, D.J. Simultaneous Quantitation of Lipid Biomarkers for Inflammatory Bowel Disease Using LC–MS/MS. Metabolites 2021, 11, 106. [Google Scholar] [CrossRef]
- Zhang, T.-Q.; Kuroda, H.; Nagano, K.; Terada, S.; Gao, J.-Q.; Harada, K.; Hirata, K.; Tsujino, H.; Higashisaka, K.; Matsumoto, H.; et al. Development and evaluation of a simultaneous and efficient quantification strategy for final prostanoid metabolites in urine. Prostaglandins Leukot. Essent. Fat. Acids 2020, 157, 102032. [Google Scholar] [CrossRef] [PubMed]
- Brose, S.A.; Thuen, B.T.; Golovko, M.Y. LC/MS/MS method for analysis of E2 series prostaglandins and isoprostanes. J. Lipid Res. 2011, 52, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Balgoma, D.; Larsson, J.; Rokach, J.; Lawson, J.A.; Daham, K.; Dahlén, B.; Dahlén, S.-E.; Wheelock, C.E. Quantification of Lipid Mediator Metabolites in Human Urine from Asthma Patients by Electrospray Ionization Mass Spectrometry: Controlling Matrix Effects. Anal. Chem. 2013, 85, 7866–7874. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Peralbo, M.A.; de Castro, M.D.L. Preparation of urine samples prior to targeted or untargeted metabolomics mass-spectrometry analysis. Trac-Trends Anal. Chem. 2012, 41, 75–85. [Google Scholar] [CrossRef]
- Il’yasova, D.; Morrow, J.D.; Ivanova, A.; Wagenknecht, L.E. Epidemiological marker for oxidant status: Comparison of the ELISA and the gas chromatography/mass spectrometry assay for urine 2,3-dinor-5,6-dihydro-15-F2t-isoprostane. Ann. Epidemiol. 2004, 14, 793–797. [Google Scholar] [CrossRef]
- Mazaleuskaya, L.L.; Ricciotti, E. Druggable Prostanoid Pathway. Adv. Exp. Med. Biol. 2020, 1274, 29–54. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fries, S.; Li, R.; Lawson, J.A.; Propert, K.J.; Diamond, S.L.; Blair, I.A.; FitzGerald, G.A.; Grosser, T. Differential impairment of aspirin-dependent platelet cyclooxygenase acetylation by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 16830–16835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khamis, M.M.; Holt, T.; Awad, H.; El-Aneed, A.; Adamko, D.J. Comparative analysis of creatinine and osmolality as urine normalization strategies in targeted metabolomics for the differential diagnosis of asthma and COPD. Metabolomics 2018, 14, 115. [Google Scholar] [CrossRef]
- Warrack, B.M.; Hnatyshyn, S.; Ott, K.H.; Reily, M.D.; Sanders, M.; Zhang, H.; Drexler, D.M. Normalization strategies for metabonomic analysis of urine samples. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 547–552. [Google Scholar] [CrossRef]
- Schurch, B.; Capaul, M.; Vallotton, M.B.; Rossier, A.B. Prostaglandin E2 measurements: Their value in the early diagnosis of heterotopic ossification in spinal cord injury patients. Arch. Phys. Med. Rehabil. 1997, 78, 687–691. [Google Scholar] [CrossRef]
- Remuzzi, G.; FitzGerald, G.A.; Patrono, C. Thromboxane synthesis and action within the kidney. Kidney Int. 1992, 41, 1483–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonvalet, J.P.; Pradelles, P.; Farman, N. Segmental synthesis and actions of prostaglandins along the nephron. Am. J. Physiol. 1987, 253, F377–F387. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Knowles, R.B.; Kirkby, N.S.; Reed, D.M.; Edin, M.L.; White, W.E.; Chan, M.V. Kidney Transplantation in a Patient Lacking Cytosolic Phospholipase A2 Proves Renal Origins of Urinary PGI-M and TX-M. Circ. Res. 2018, 122, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Knowles, R.B.; Kirkby, N.S.; Reed, D.M.; Edin, M.L.; White, W.E.; Chan, M.V.; Longhurst, H.; Yaqoob, M.M.; Milne, G.L.; et al. Letter by Mitchell et al. Regarding Article, “Urinary Prostaglandin Metabolites: An Incomplete Reckoning and a Flush to Judgment”. Circ. Res. 2018, 122, e84–e85. [Google Scholar] [CrossRef] [PubMed]
- Grosser, T.; Naji, A.; FitzGerald, G.A. Urinary Prostaglandin Metabolites: An Incomplete Reckoning and a Flush to Judgment. Circ. Res. 2018, 122, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, G.; Grosser, T. Prostanoids and inflammatory pain. Prostaglandins Other Lipid Mediat. 2013, 104–105, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.G.; Padilla, J.; Koumas, L.; Ray, D.; Phipps, R.P. Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23, 144–150. [Google Scholar] [CrossRef]
- Smyth, E.M.; Grosser, T.; Wang, M.; Yu, Y.; FitzGerald, G.A. Prostanoids in health and disease. J. Lipid Res. 2009, 50, S423–S428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Yang, V.W.; Geiman, D.E.; Hubbard, W.C.; Spannhake, E.W.; Hylind, L.M.; Hamilton, S.R.; Giardiello, F.M. Tissue prostanoids as biomarkers for chemoprevention of colorectal neoplasia: Correlation between prostanoid synthesis and clinical response in familial adenomatous polyposis. Prostaglandins Other Lipid Mediat. 2000, 60, 83–96. [Google Scholar] [CrossRef]
- Almer, G.; Teismann, P.; Stevic, Z.; Halaschek-Wiener, J.; Deecke, L.; Kostic, V.; Przedborski, S. Increased levels of the pro-inflammatory prostaglandin PGE2 in CSF from ALS patients. Neurology 2002, 58, 1277–1279. [Google Scholar] [CrossRef]
- Björk, L.; Leifsdottir, K.; Saha, S.; Herlenius, E. PGE2—Metabolite levels in CSF correlate to HIE score and outcome after perinatal asphyxia. Acta Paediatr. 2013, 102, 1041–1047. [Google Scholar] [CrossRef]
- Mattsson, N.; Yaong, M.; Rosengren, L.; Blennow, K.; Månsson, J.E.; Andersen, O.; Zetterberg, H.; Haghighi, S.; Zho, I.; Pratico, D. Elevated cerebrospinal fluid levels of prostaglandin E2 and 15-(S)-hydroxyeicosatetraenoic acid in multiple sclerosis. J. Intern. Med. 2009, 265, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Matsuura, T.; Matsuura, M.; Fujiwara, M.; Okayasu, I.; Ito, S.; Arihiro, S. Prostaglandin E-Major Urinary Metabolite as a Biomarker for Inflammation in Ulcerative Colitis: Prostaglandins Revisited. Digestion 2016, 93, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Campbell, J.; Yoo, W.; Taylor, J.A.; Sandler, D.P. Systemic levels of estrogens and PGE2 synthesis in relation to postmenopausal breast cancer risk. Cancer Epidemiol. Biomark. Prev. 2017, 26, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.C.; Schmidt, C.R.; Shrubsole, M.J.; Billheimer, D.D.; Joshi, P.R.; Morrow, J.D.; Heslin, M.J.; Washington, M.K.; Ness, R.M.; Zheng, W.; et al. Urine PGE-M: A metabolite of prostaglandin E2 as a potential biomarker of advanced colorectal neoplasia. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2006, 4, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Jabr, S.; Gartner, S.; Milne, G.L.; Roca-Ferrer, J.; Casas, J.; Moreno, A.; Gelpí, E.; Picado, C. Quantification of major urinary metabolites of PGE2 and PGD2 in cystic fibrosis: Correlation with disease severity. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 121–126. [Google Scholar] [CrossRef]
- Labat, C.; Temmar, M.; Nagy, E.; Bean, K.; Brink, C.; Benetos, A.; Bäck, M. Inflammatory mediators in saliva associated with arterial stiffness and subclinical atherosclerosis. J. Hypertens. 2013, 31, 2251–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anyona, S.B.; Kempaiah, P.; Davenport, G.C.; Vulule, J.M.; Hittner, J.B.; Ong’echa, J.M.; Perkins, D.J. Suppressed circulating bicyclo-PGE2 levels and leukocyte COX-2 transcripts in children co-infected with P. falciparum malaria and HIV-1 or bacteremia. Biochem. Biophys. Res. Commun. 2013, 436, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Nakamura, T.; Harada, H.; Tachibana, Y.; Aritake, K.; Shimosawa, T.; Yatomi, Y.; Murata, T. Prostaglandin D2 metabolite in urine is an index of food allergy. Sci. Rep. 2017, 7, 17687. [Google Scholar] [CrossRef]
- Higashi, N.; Taniguchi, M.; Mita, H.; Yamaguchi, H.; Ono, E.; Akiyama, K. Aspirin-Intolerant Asthma (AIA) Assessment Using the Urinary Biomarkers, Leukotriene E4 (LTE4) and Prostaglandin D2 (PGD2) Metabolites. Allergol. Int. 2012, 61, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Kolmert, J.; Gómez, C.; Balgoma, D.; Sjödin, M.; Bood, J.; Konradsen, J.R.; Ericsson, M.; Thörngren, J.-O.; James, A.; Mikus, M.; et al. Urinary Leukotriene E4 and Prostaglandin D2 Metabolites Increase in Adult and Childhood Severe Asthma Characterized by Type-2 Inflammation. Am. J. Respir. Crit. Care Med. 2020, 203, 37–53. [Google Scholar] [CrossRef]
- Takeshita, E.; Komaki, H.; Tachimori, H.; Miyoshi, K.; Yamamiya, I.; Shimizu-Motohashi, Y.; Ishiyama, A.; Saito, T.; Nakagawa, E.; Sugai, K.; et al. Urinary prostaglandin metabolites as Duchenne muscular dystrophy progression markers. Brain Dev. 2018, 40, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Buchheit, K.M.M.D.; Cahill, K.N.M.D.; Katz, H.P.; Murphy, K.; Feng, C.M.D.; Lee-Sarwar, K.; Lai, J.; Bhattacharyya, N.M.D.; Israel, E.M.D.F.; Boyce, J.A.M.D.F.; et al. Thymic Stromal Lymphopoietin Controls Prostaglandin D2 Generation in Aspirin-Exacerbated Respiratory Disease. J. Allergy Clin. Immunol. 2016, 137, AB198. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Quehenberger, O.; Armando, A.; Dennis, E.A. Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for noninvasive diagnosis of nonalcoholic steatohepatitis1. J. Lipid Res. 2015, 56, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassiri, M.; Eckermann, O.; Babina, M.; Edenharter, G.; Worm, M. Serum levels of 9α,11β-PGF 2 and cysteinyl leukotrienes are useful biomarkers of anaphylaxis. J. Allergy Clin. Immunol. 2016, 137, 312–314.e7. [Google Scholar] [CrossRef] [Green Version]
- Coras, R.; Kavanaugh, A.; Kluzniak, A.; Holt, D.; Weilgosz, A.; Aaron, A.; Quehenberger, O.; Ritchlin, C.; Guma, M. Differences in oxylipin profile in psoriasis versus psoriatic arthritis. Arthritis Res. Ther. 2021, 23, 200. [Google Scholar] [CrossRef]
- Vazzana, N.; Santilli, F.; Lattanzio, S.; Liani, M.; Giacci, L.; Del Rosso, G.; Salvati, F.; Boccatonda, A.; Ferroni, P.; Davì, G. Determinants of thromboxane biosynthesis in patients with moderate to severe chronic kidney disease. Eur. J. Intern. Med. 2016, 33, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Szczuko, M.; Koziol, I.; Kotlega, D.; Brodowski, J.; Drozd, A. The Role of Thromboxane in the Course and Treatment of Ischemic Stroke: Review. Int. J. Mol. Sci. 2021, 22, 11644. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Vendrov, K.C.; Simmons, B.P.; Schuck, R.N.; Stouffer, G.A.; Lee, C.R. Urinary 11-dehydro-thromboxane B2 levels are associated with vascular inflammation and prognosis in atherosclerotic cardiovascular disease. Prostaglandins Other Lipid Mediat. 2018, 134, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Hishinuma, T.; Koseki, Y.; Murai, Y.; Yamazaki, T.; Suzuki, K.; Mizugaki, M. Urinary thromboxane A2/prostacyclin balance reflects the pathological state of a diabetic. Prostaglandins Other Lipid Mediat. 1999, 58, 263–271. [Google Scholar] [CrossRef]
- Ujike-Omori, H.; Maeshima, Y.; Kinomura, M.; Tanabe, K.; Mori, K.; Watatani, H.; Hinamoto, N.; Sugiyama, H.; Sakai, Y.; Morimatsu, H.; et al. The urinary levels of prostanoid metabolites predict acute kidney injury in heterogeneous adult Japanese ICU patients: A prospective observational study. Clin. Exp. Nephrol. 2015, 19, 1024–1036. [Google Scholar] [CrossRef]
- Thorén, S.; Jakobsson, P.J. Coordinate up- and down-regulation of glutathione-dependent prostaglandin E synthase and cyclooxygenase-2 in A549 cells. Inhibition by NS-398 and leukotriene C4. Eur. J. Biochem. 2000, 267, 6428–6434. [Google Scholar] [CrossRef]
- Stichtenoth, D.O.; Thorén, S.; Bian, H.; Peters-Golden, M.; Jakobsson, P.J.; Crofford, L.J. Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J. Immunol. 2001, 167, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qasem, H.; Al-Ayadhi, L.; Bjørklund, G.; Chirumbolo, S.; El-Ansary, A. Impaired lipid metabolism markers to assess the risk of neuroinflammation in autism spectrum disorder. Metab. Brain Dis. 2018, 33, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Varedi, A.; Rahman, H.; Kumar, D.; Catrow, J.L.; Cox, J.E.; Liu, T.; Florell, S.R.; Boucher, K.M.; Okwundu, N.; Burnett, W.J.; et al. ASA Suppresses PGE2 in Plasma and Melanocytic Nevi of Human Subjects at Increased Risk for Melanoma. Pharmaceuticals 2020, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Bäck, M.; Hlawaty, H.; Labat, C.; Michel, J.B.; Brink, C. The oral cavity and age: A site of chronic inflammation? PLoS ONE 2007, 2, e1351. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimzadeh, T.; Kuprasertkul, A.; Neugent, M.L.; Lutz, K.C.; Fuentes, J.L.; Gadhvi, J.; Khan, F.; Zhang, C.; Sharon, B.M.; Orth, K.; et al. Urinary prostaglandin E2 as a biomarker for recurrent UTI in postmenopausal women. Life Sci. Alliance 2021, 4, e202000948. [Google Scholar] [CrossRef]
- Doña, I.; Pérez-Sánchez, N.; Eguiluz-Gracia, I.; Muñoz-Cano, R.; Bartra, J.; Torres, M.J.; Cornejo-García, J.A. Progress in understanding hypersensitivity reactions to nonsteroidal anti-inflammatory drugs. Allergy 2020, 75, 561–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, S.; Nakamura, T.; Hamasaki, Y.; Yamamoto-Hanada, K.; Fukuie, T.; Narita, M.; Shimosawa, T.; Murata, T.; Ohya, Y. Prostaglandin D(2) metabolite is not a useful clinical indicator for assessing atopic dermatitis. Clin. Exp. Dermatol. 2021, 46, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Nakamura, T.; Takenouchi, S.; Hayashi, A.; Omori, K.; Murata, T. Urinary 8-iso PGF2α and 2,3-dinor-8-iso PGF2α can be indexes of colitis-associated colorectal cancer in mice. PLoS ONE 2021, 16, e0245292. [Google Scholar] [CrossRef]
- Noort, W.A.; van Bulck, B.; Vereecken, A.; de Zwart, F.A.; Keirse, M.J.N.C. Changes in plasma of PGF2α and PGI2 metabolites at and after delivery at term. Prostaglandins 1989, 37, 3–12. [Google Scholar] [CrossRef]
- Martini, D.; Dominguez-Perles, R.; Rosi, A.; Tassotti, M.; Angelino, D.; Medina, S.; Ricci, C.; Guy, A.; Oger, C.; Gigliotti, L.; et al. Effect of Coffee and Cocoa-Based Confectionery Containing Coffee on Markers of DNA Damage and Lipid Peroxidation Products: Results from a Human Intervention Study. Nutrients 2021, 13, 2399. [Google Scholar] [CrossRef] [PubMed]
- Cangemi, R.M.D.; Casciaro, M.M.D.; Rossi, E.M.D.; Calvieri, C.M.D.; Bucci, T.M.D.; Calabrese, C.M.M.D.; Taliani, G.M.D.; Falcone, M.M.D.; Palange, P.M.D.; Bertazzoni, G.M.D.; et al. Platelet Activation Is Associated with Myocardial Infarction in Patients with Pneumonia. J. Am. Coll. Cardiol. 2014, 64, 1917–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.L.; Lawson, J.A.; Wang, M.; Zou, H.; Fitzgerald, G.A. Noninvasive Assessment of the Role of Cyclooxygenases in Cardiovascular Health: A Detailed HPLC/MS/MS Method. Methods Enzymol. 2007, 433, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.M.D.; Pignatelli, P.M.D.; Farcomeni, A.P.; Cangemi, R.M.D.; Hiatt, W.R.M.D.; Bartimoccia, S.P.; Nocella, C.P.; Vicario, T.M.D.; Bucci, T.M.D.; Carnevale, R.P.; et al. Urinary 11-dehydro-thromboxane B2 is associated with cardiovascular events and mortality in patients with atrial fibrillation. Am. Heart J. 2015, 170, 490–497.e1. [Google Scholar] [CrossRef]
- Van der Weiden, R.M.F.; Helmerhorst, F.M.; Keirse, M.J.N.C. Which prostanoid metabolites should be determined for the study of reproductive processes? Prostaglandins Leukot. Essent. Fat. Acids 1998, 58, 205–207. [Google Scholar] [CrossRef]
Figure 1.
Schematic overview of the prostanoid pathway with the major metabolites that can be detected in human plasma or urine, respectively. The individual metabolites are described in detail in the text. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites. Red arrows indicate reduction, green, blue, or yellow arrows oxidation, and grey arrows non-enzymatic reactions. 1, Cyclooxygenase-1/2 (COX, EC 1.14.99.1); 2, Prostacyclin Synthase (PTGIS, EC 5.3.99.4); 3, Thromboxane A Synthase (TBXAS1, EC 5.3.99.5); 4, Hematopoietic Prostaglandin D Synthase (HPGDS, EC 2.5.1.18); 5, Lipocalin-Type Prostaglandin D Synthase (L-PGDS, EC 5.3.99.2); 6, Microsomal Prostaglandin E Synthase-1 (MPGES1, EC 5.3.99.3); 7, Microsomal Prostaglandin E Synthase-2 (MPGES2, EC 5.3.99.3); 8, Cytosolic Prostaglandin E Synthase (CPGES, EC 5.3.99.3); 9, Prostaglandin F Synthase (PGFS; EC 1.1.1.188); 10, non-enzymatic degradation; 11, albumin-mediated degradation; 12, 11-hydroxy Thromboxane Dehydrogenase (EC 1.2.1.3); 13, 15-hydroxyprostaglandin Dehydrogenase (15-PGDH, EC 1.1.1.141); 14, 15-oxo-prostaglandin Δ13-reductase (EC 1.3.1.48); 15, NADPH-dependent PGD2 11-ketoreductase (EC 1.1.1.188); 16, enzymatic β-oxidation; 17, enzymatic ω-oxidation.
Figure 1.
Schematic overview of the prostanoid pathway with the major metabolites that can be detected in human plasma or urine, respectively. The individual metabolites are described in detail in the text. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites. Red arrows indicate reduction, green, blue, or yellow arrows oxidation, and grey arrows non-enzymatic reactions. 1, Cyclooxygenase-1/2 (COX, EC 1.14.99.1); 2, Prostacyclin Synthase (PTGIS, EC 5.3.99.4); 3, Thromboxane A Synthase (TBXAS1, EC 5.3.99.5); 4, Hematopoietic Prostaglandin D Synthase (HPGDS, EC 2.5.1.18); 5, Lipocalin-Type Prostaglandin D Synthase (L-PGDS, EC 5.3.99.2); 6, Microsomal Prostaglandin E Synthase-1 (MPGES1, EC 5.3.99.3); 7, Microsomal Prostaglandin E Synthase-2 (MPGES2, EC 5.3.99.3); 8, Cytosolic Prostaglandin E Synthase (CPGES, EC 5.3.99.3); 9, Prostaglandin F Synthase (PGFS; EC 1.1.1.188); 10, non-enzymatic degradation; 11, albumin-mediated degradation; 12, 11-hydroxy Thromboxane Dehydrogenase (EC 1.2.1.3); 13, 15-hydroxyprostaglandin Dehydrogenase (15-PGDH, EC 1.1.1.141); 14, 15-oxo-prostaglandin Δ13-reductase (EC 1.3.1.48); 15, NADPH-dependent PGD2 11-ketoreductase (EC 1.1.1.188); 16, enzymatic β-oxidation; 17, enzymatic ω-oxidation.

Figure 2.
Major metabolic pathway of PGE2. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites. The red arrow indicates reduction, the green, blue, or yellow arrows oxidation, and the grey arrows non-enzymatic/albumin mediated reactions. 1, 15-hydroxyprostaglandin Dehydrogenase (15-PGDH, EC 1.1.1.141); 2, 15-oxo-prostaglandin Δ13-reductase (EC 1.3.1.48); 3, enzymatic β-oxidation; 4, enzymatic ω-oxidation; 5, non-enzymatic degradation; 6, albumin-mediated degradation.
Figure 2.
Major metabolic pathway of PGE2. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites. The red arrow indicates reduction, the green, blue, or yellow arrows oxidation, and the grey arrows non-enzymatic/albumin mediated reactions. 1, 15-hydroxyprostaglandin Dehydrogenase (15-PGDH, EC 1.1.1.141); 2, 15-oxo-prostaglandin Δ13-reductase (EC 1.3.1.48); 3, enzymatic β-oxidation; 4, enzymatic ω-oxidation; 5, non-enzymatic degradation; 6, albumin-mediated degradation.

Figure 3.
Major metabolic pathways of PGD2. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites; metabolites on a grey background are cyclopentenone derivatives of PGD2. Red arrows indicate reduction, green, blue, or yellow arrows oxidation, and grey arrows non-enzymatic/albumin mediated reactions. The letters in the lower left corner denote the ring structure of the respective metabolite. 1, 15-hydroxyprostaglandin Dehydrogenase (15-PGDH, EC 1.1.1.141); 2, 15-oxo-prostaglandin Δ13-reductase (EC 1.3.1.48); 3, enzymatic β-oxidation; 4, enzymatic ω-oxidation; 5, NADPH-dependent PGD2 11-ketoreductase (EC 1.1.1.188); 6, albumin-mediated degradation; 7, non-enzymatic degradation.
Figure 3.
Major metabolic pathways of PGD2. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites; metabolites on a grey background are cyclopentenone derivatives of PGD2. Red arrows indicate reduction, green, blue, or yellow arrows oxidation, and grey arrows non-enzymatic/albumin mediated reactions. The letters in the lower left corner denote the ring structure of the respective metabolite. 1, 15-hydroxyprostaglandin Dehydrogenase (15-PGDH, EC 1.1.1.141); 2, 15-oxo-prostaglandin Δ13-reductase (EC 1.3.1.48); 3, enzymatic β-oxidation; 4, enzymatic ω-oxidation; 5, NADPH-dependent PGD2 11-ketoreductase (EC 1.1.1.188); 6, albumin-mediated degradation; 7, non-enzymatic degradation.

Figure 4.
Major metabolic pathway of PGF2α. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites. The red arrow indicates reduction, and the green, blue, or yellow arrows oxidation reactions. 1, 15-hydroxyprostaglandin Dehydrogenase (15-PGDH, EC 1.1.1.141); 2, 15-oxo-prostaglandin Δ13-reductase (EC 1.3.1.48); 3, enzymatic β-oxidation; 4, enzymatic ω-oxidation.
Figure 4.
Major metabolic pathway of PGF2α. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites. The red arrow indicates reduction, and the green, blue, or yellow arrows oxidation reactions. 1, 15-hydroxyprostaglandin Dehydrogenase (15-PGDH, EC 1.1.1.141); 2, 15-oxo-prostaglandin Δ13-reductase (EC 1.3.1.48); 3, enzymatic β-oxidation; 4, enzymatic ω-oxidation.

Figure 5.
Major metabolic pathway of TXA2. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites. The grey arrow indicates non-enzymatic decomposition, the green and blue arrows oxidation reactions. 1, non-enzymatic degradation; 2, 11-hydroxy Thromboxane Dehydrogenase (EC 1.2.1.3); 3, enzymatic β-oxidation.
Figure 5.
Major metabolic pathway of TXA2. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites. The grey arrow indicates non-enzymatic decomposition, the green and blue arrows oxidation reactions. 1, non-enzymatic degradation; 2, 11-hydroxy Thromboxane Dehydrogenase (EC 1.2.1.3); 3, enzymatic β-oxidation.

Figure 6.
Major metabolic pathway of PGI2. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites The grey arrow indicates non-enzymatic decomposition and the blue arrow oxidation reactions. 1, non-enzymatic degradation; 2, enzymatic β-oxidation.
Figure 6.
Major metabolic pathway of PGI2. Metabolites on a red background are regarded as major plasma metabolites; metabolites on a yellow background are major urinary metabolites The grey arrow indicates non-enzymatic decomposition and the blue arrow oxidation reactions. 1, non-enzymatic degradation; 2, enzymatic β-oxidation.


{kind=link}
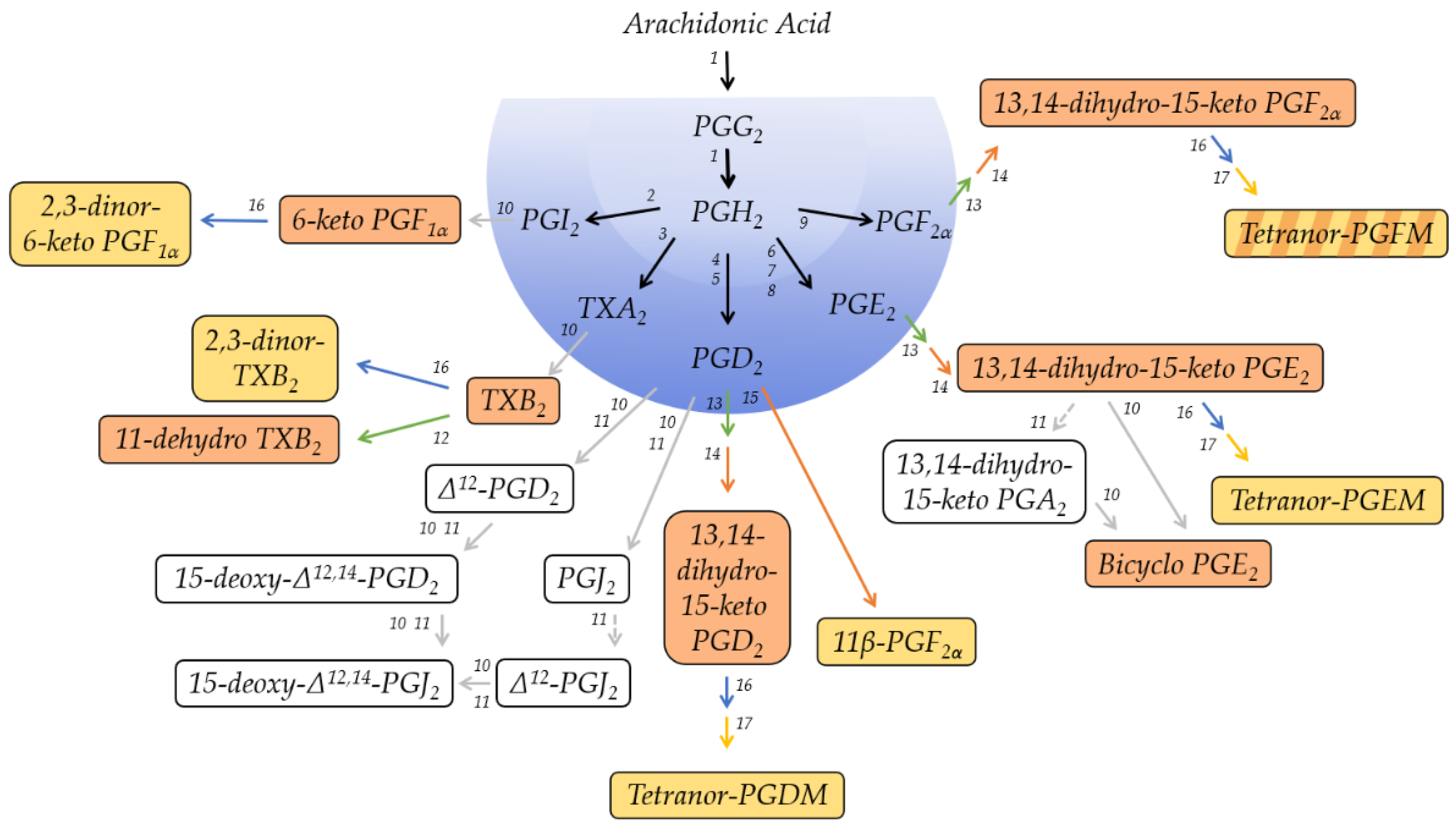{kind=link}
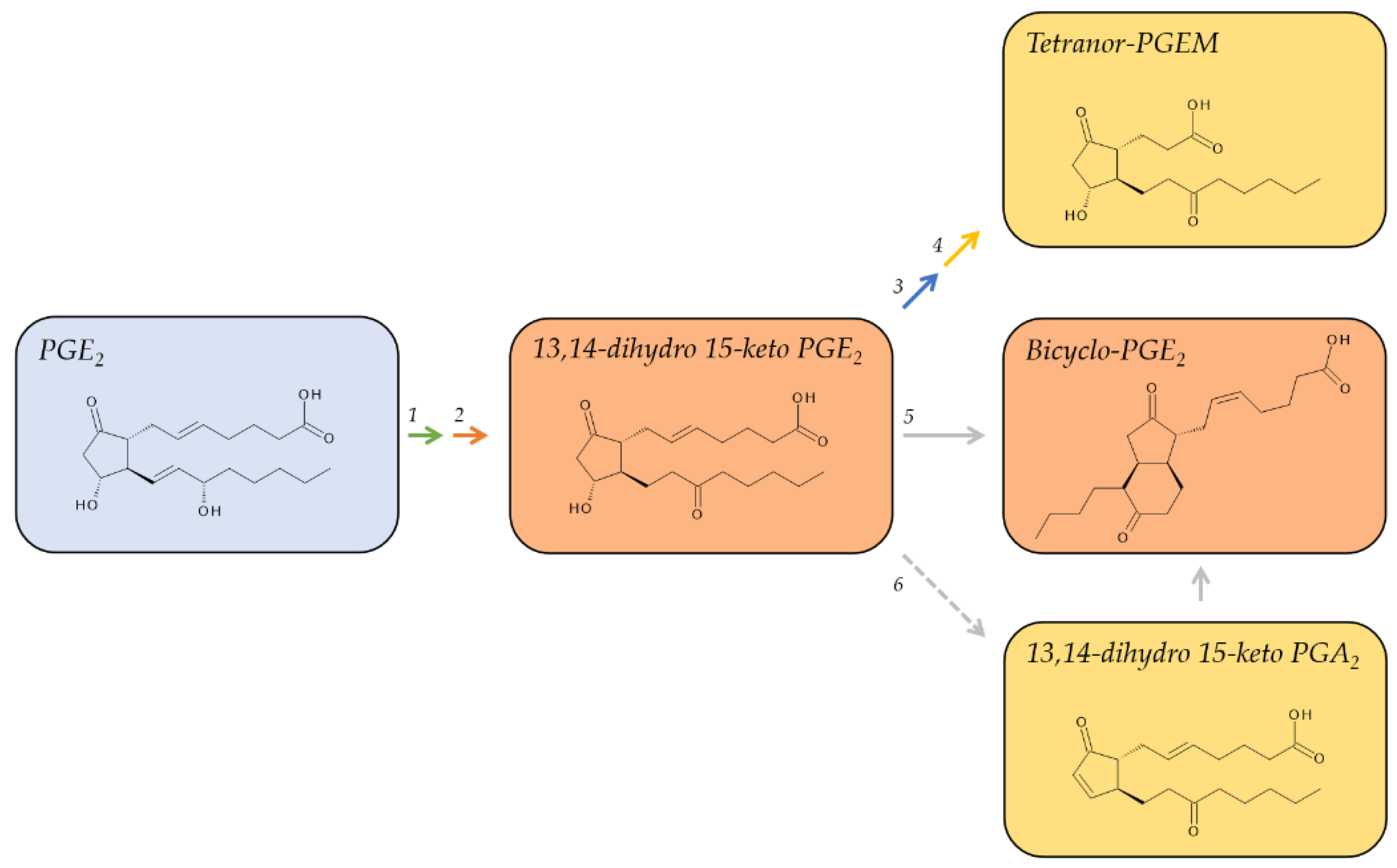{kind=link}
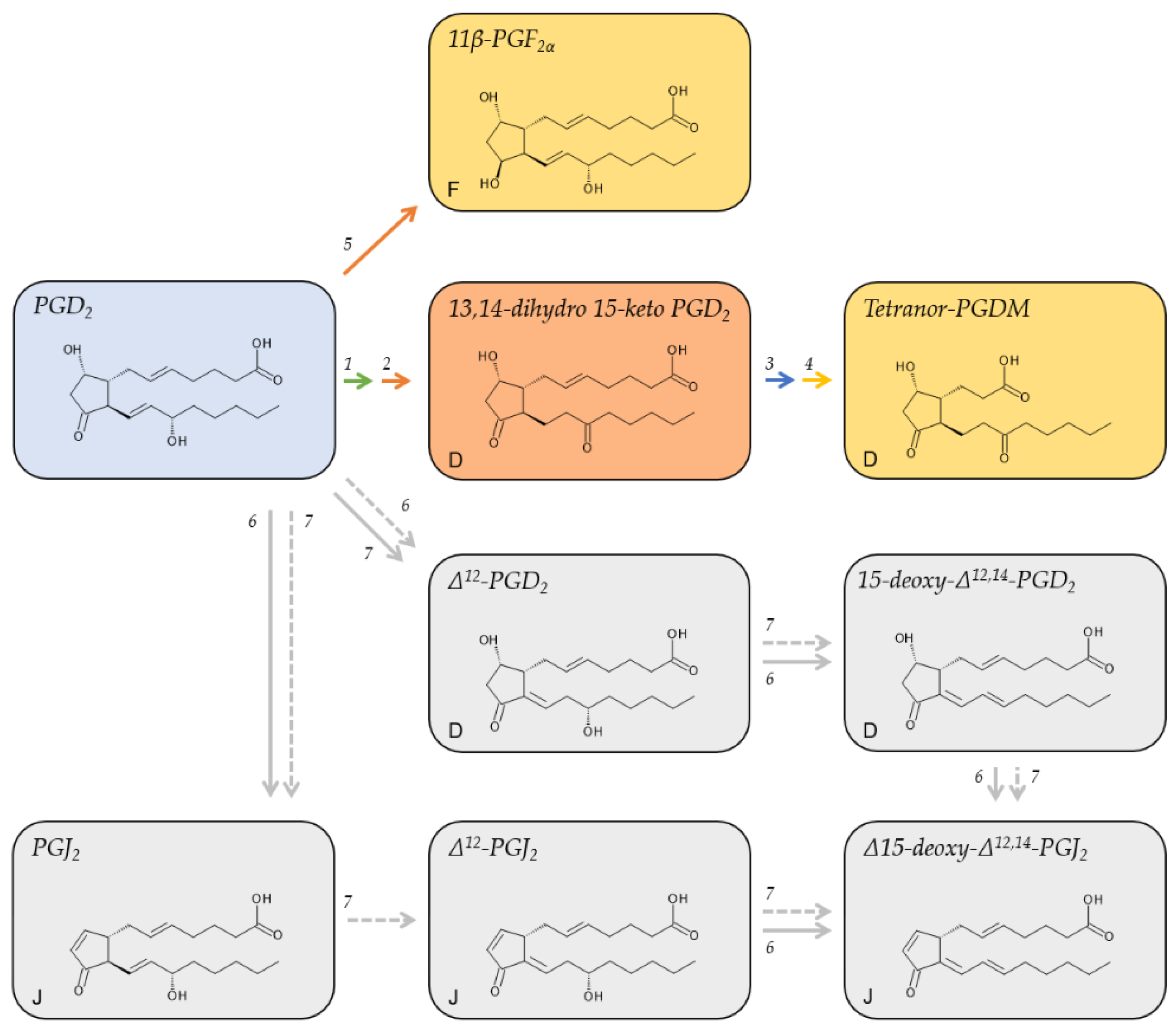{kind=link}
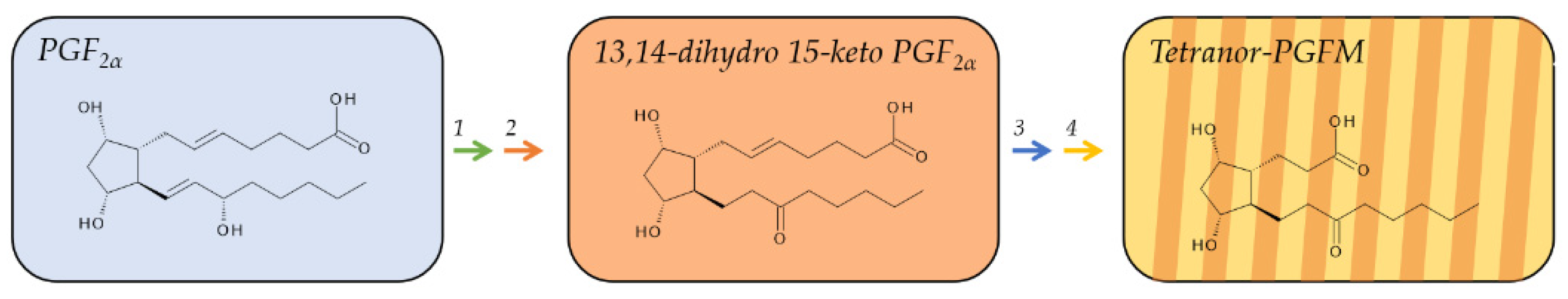{kind=link}
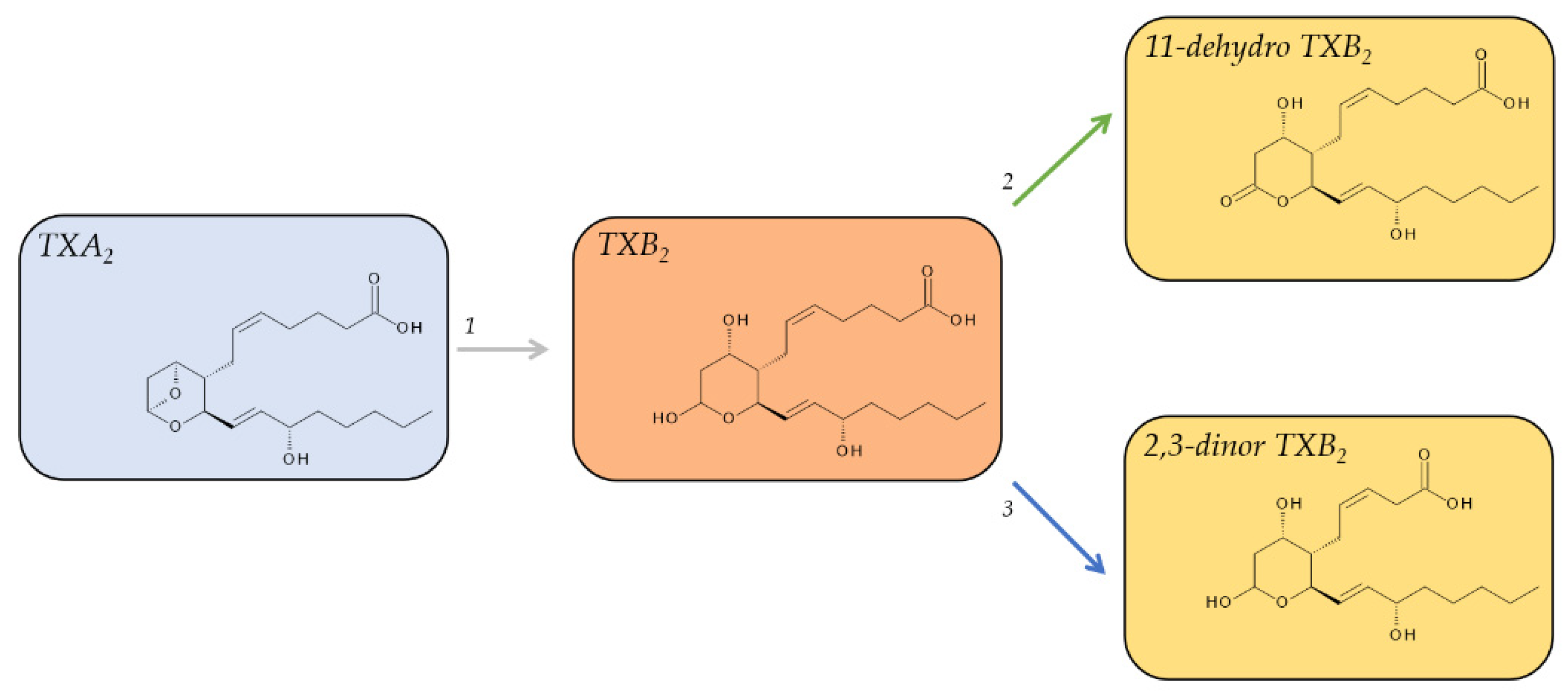{kind=link}
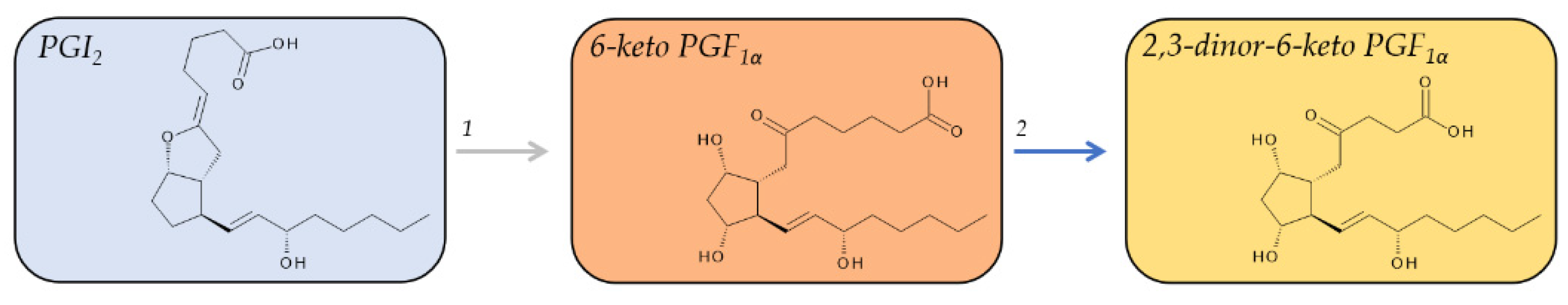{kind=link}
Table 1.
Prostanoid metabolites and their concentrations in biofluids.
| Primary Prostanoid | Metabolite | Concentration 1 | Comment |
|---|---|---|---|
| PGE2 | Plasma: 3–12 pg/mL | t1/2 < 1min in circulation [27]. | |
| 13,14-dihydro-15-keto PGE2 | Plasma: 10–100 pg/mL [28] | t1/2 = 9 min (in vivo in dogs) [29]. t1/2 = 7 h in protein-free buffer [30]. t1/2 = 3 h in diluted plasma [30]. | |
| tetranor-PGEM | Urine: 7–40 µg per 24 h urine collection (µg/day) [31] | Major urinary metabolite of PGE2 in humans. Marker for systemic PGE2 biosynthesis in vivo. Considered a stable metabolite. | |
| bicyclo PGE2 | Plasma: 20–25 pg/mL [28,32] | Non-enzymatically formed, but base-catalyzed. Considered a stable metabolite. | |
| 13,14-dihydro-15-keto PGA2 | n.d. | Non-enzymatically formed. Considered a stable metabolite. | |
| PGD2 | t1/2 = 0.9 min [33]. | ||
| tetranor-PGDM | Urine: 1.5 ± 0.3 ng/mg creatinine [31] | Major urinary metabolite of PGD2 in humans. Marker for systemic PGD2 biosynthesis in vivo. | |
| 11β-PGF2α | n.d. | ||
| 15d-PGD2 | n.d. | Bioactive metabolite of PGD2; activates PPARγ. | |
| 15-deoxy-Δ12,14-PGJ2 | Plasma: 2 to 350 pg/mL [34] Urine: 6.3 ± 2.7 pg/mg creatinine [35] | ||
| 13,14-dihydro-15-keto PGD2 | n.d. | Not regularly measured as PGD2 metabolite in plasma or urine. | |
| Δ12-PGJ2 | n.d. | Not regularly measured as PGD2 metabolite in plasma or urine. | |
| PGJ2 | n.d. | Not regularly measured as PGD2 metabolite in plasma or urine. | |
| PGF2α | |||
| 13,14-dihydro-15-keto PGF2α | Plasma: 0.08–20 pmol/mL (basal levels) [36]; 40–60 pg/mL (late pregnancy); 1200–4100 pg/mL (parturition) [37] | ||
| tetranor-PGFM | Plasma: 60–100 pg/mL (late pregnancy); 1000 and 2000 pg/mL (parturition); 100–300 pg/mL (24 h after parturition) [37] Urine: 11–59 µg/day (♂), 7–13 µg/day (♀), 2- to 5-fold increase during pregnancy [38] | Major urinary metabolite of PGF2α in humans. | |
| TXA2 | t1/2 = 30 s in circulation [39]. | ||
| TXB2 | 1–2 pg/mL (3–6 fmol/mL) in uninduced blood 300–400 ng/mL (0.8–1.0 nmol/mL) in fully coagulated blood [40] | t1/2 = 7 min [41]. Non-enzymatically formed. Detectable in plasma. Stable metabolite ex vivo. Urinary TXB2 may reflect intrarenal TXA2 production. Serum TXB2 reflects capacity of platelets to synthesize TXA2 in vitro. | |
| 11-dehydro TXB2 | Plasma: 0.9–1.8 pg/mL [42] Urine: 0.9–4.3 pg/mL, 30–70 ng/mmol creatinine, 80–190 pmol/mmol creatinine [43] | Detectable in plasma. t1/2 = 45 min in circulation [42]. Major urinary metabolite of TXA2 in mice. | |
| 2,3-dinor TXB2 | Urine: 10.3 ng/h (138 pg/mg creatine) 45 pmol/mmol creatinine [43] | Urinary 2,3-dinor TXB2 is a marker for systemic TXA2 synthesis in vivo. | |
| PGI2 | t1/2 = 30 s (in vivo). t1/2 = 1.29–1.52 min (in vivo in cats) [44], t1/2 = 6–10 min (in vitro) [45,46,47]. | ||
| 6-keto PGF1α | n.d. | Non-enzymatically formed. Major plasma metabolite of PGI2. Urinary 6-keto PGF1α may reflect intrarenal PGI2 production. | |
| 2,3-dinor-6-keto PGF1α | Urine: 100 pg/mg creatinine [48,49] | Major urinary metabolite of PGI2 in humans. Urinary 2,3-dinor-6-keto PGF1α is a marker for systemic PGI2 synthesis in vivo. |
1 Concentrations are reported as found in the literature; therefore, units and normalization approaches may differ. When available, data on prostanoid metabolites in healthy human subjects are reported; however, some information comes from other species. n.d., no data.
Table 2.
Prostanoid metabolites as urinary/plasma biomarkers in human disease.
| Prostanoid Metabolite | Biological Fluid | Association to Disease |
|---|---|---|
| PGE2 | ||
| tetranor-PGEM | Urine | Ulcerative colitis [122]; Viral infections in infants [8]; Breast cancer [123]; Colorectal cancers [124]; Abdominal obesity/pre-diabetes [31]; Cystic fibrosis [125] |
| combined PGE2 and PGE2 metabolites | Plasma/serum/saliva | Obesity [31]; Arterial stiffness [126]; Malaria pathogenesis [127] |
| PGD2 | ||
| tetranor-PGDM | Urine | Cystic fibrosis [125]; Food allergy [128]; Aspirin-intolerant asthma [129]; Astma [130]; Obesity [31]; Duchenne muscular dystrophy [131]; Aspirin-exacerbated respiratory disease [132] |
| 2,3-dinor-11β-PGF2α | Urine | Asthma [130] |
| 13,14-dihydro-15-keto-prostaglandin D2 | Serum | Nonalcoholic fatty liver (NASH) [133] |
| 11β-PGF2α | Serum | Anaphylaxis [134] |
| 15-deoxy-Δ12,14-PGJ2 | Plasma | Diabetes [34] |
| PGF2α | ||
| tetranor-PGFM | Urine | Psoriasis [135] |
| TXA2 | ||
| 2,3-dinor-TXB2 | Urine | Acute coronary syndromes [40] |
| 11-dhydro-TXB2 | Urine | Cardiovascular risk/Acute coronary syndromes [40]; Chronic kidney disease [136]; Cardiovascular disease [137]; Vascular inflammation [138]; Diabetes [139] |
| PGI2 | ||
| 2,3-dinor-6-keto PGF1α | Urine | Diabetes [139]; Acute kidney injury [140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Idborg, H.; Pawelzik, S.-C. Prostanoid Metabolites as Biomarkers in Human Disease. Metabolites 2022, 12, 721. https://doi.org/10.3390/metabo12080721
AMA Style
Idborg H, Pawelzik S-C. Prostanoid Metabolites as Biomarkers in Human Disease. Metabolites. 2022; 12(8):721. https://doi.org/10.3390/metabo12080721
Chicago/Turabian StyleIdborg, Helena, and Sven-Christian Pawelzik. 2022. "Prostanoid Metabolites as Biomarkers in Human Disease" Metabolites 12, no. 8: 721. https://doi.org/10.3390/metabo12080721
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.