1. Introduction
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) is a primary inflammatory choriocapillaropathy (PICCP) that was first described in 1968, by John Donald Macintyre Gass [
1]. As its name indicates, the main lesion process was attributed to the retinal pigment epithelium (RPE). This pathophysiological explanation was, however, reoriented towards the preponderant role of inflammatory choriocapillaris hypo or non-perfusion at the origin of the disease process. Analysing fluorescein angiographic (FA) images showing early geographic hypofluorescence, Deutman came up with a more appropriate denomination of the disease by calling it Acute Multifocal Ischaemic Choriocapillaritis (AMIC) [
2,
3]. Indocyanine green angiography (ICGA) later showed that the FA hypofluorescent areas corresponded not only to a choriocapillaris perfusion delay but to non-perfusion throughout the angiographic sequence causing ischaemia to the outer retina-RPE complex [
4].
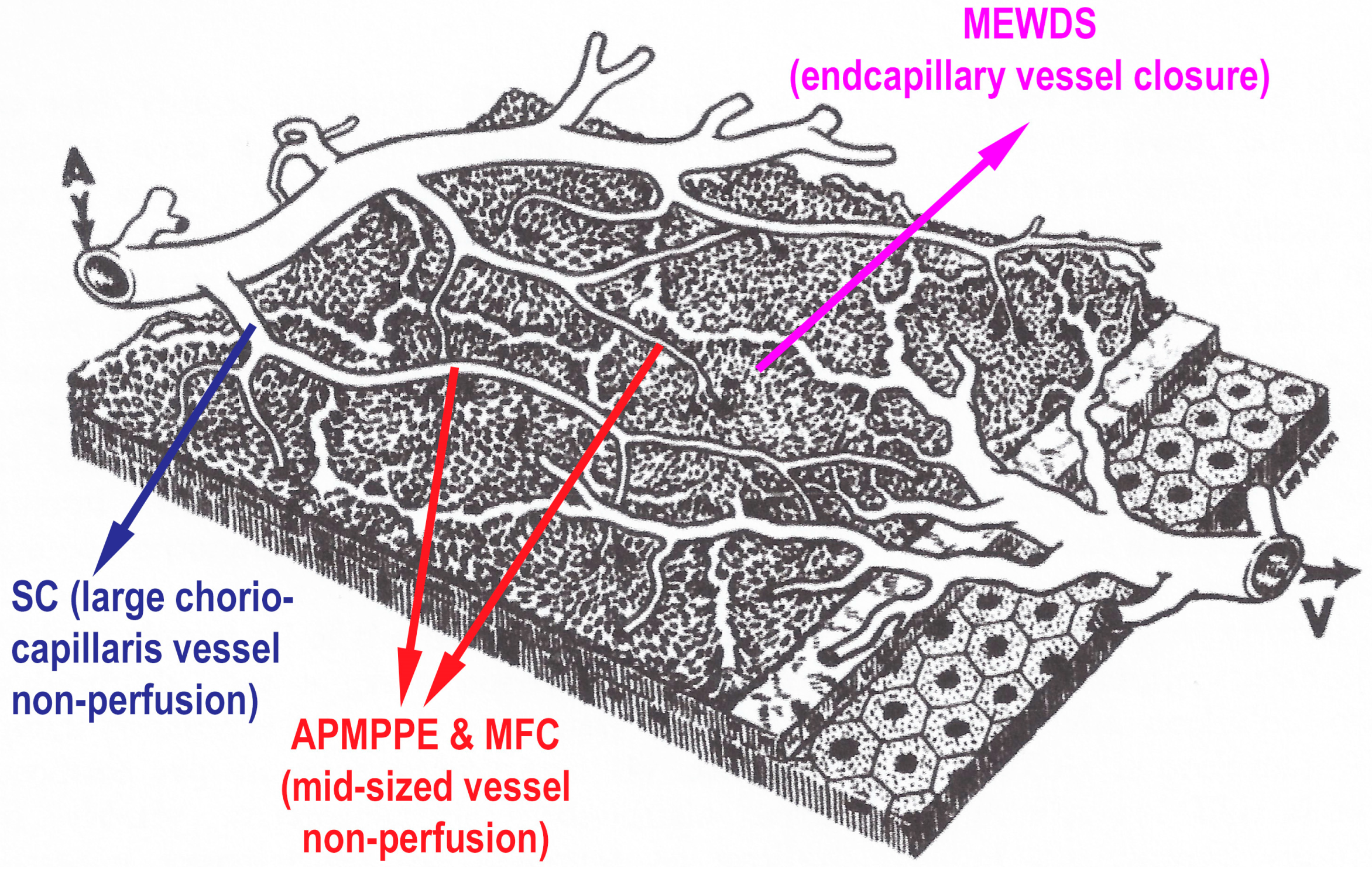
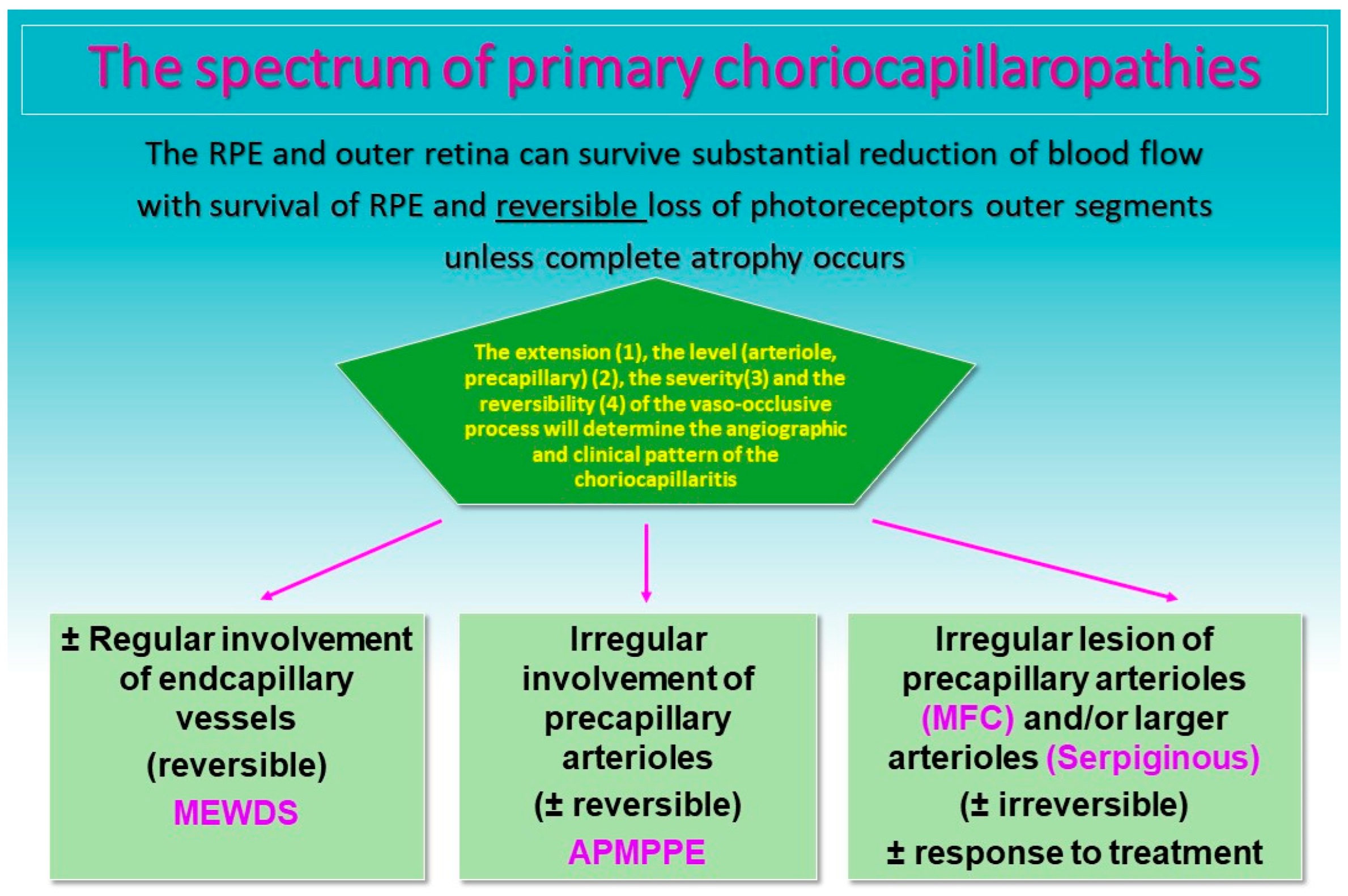
The common denominator of PICCPs is a dysfunction of the choriocapillaris perfusion of diverse severity and extension with grading from small end-capillary nonperfusion in Multiple Evanescent White Dot Syndrome (MEWDS) to progressively more proximal capillary involvement in Idiopathic Multifocal Choroiditis (MFC), APMPPE/AMIC and Serpiginous Choroiditis (SC) [
5,
6] (
Figure 1 and
Figure 2). It is called primary because there is no known cause at the origin of these entities.
In APMPPE/AMIC, inflammatory non-perfusion affects larger choriocapillaris or pre-choriocapillaris vessels (
Figure 1), with more or less widespread involvement. Its particularity is the variability of involvement from limited non-perfused ICGA hypofluorescent areas to more severe and extensive lesions. It is therefore not possible to attribute a mean expected severity level of involvement to cases of this condition. Indeed, cases may resolve without treatment as it is usually reported in textbooks. It is, however, far from rare to have severe and extended ischaemic areas with potentially deleterious consequences, especially when the central macula is involved. In the latter cases, systemic corticosteroid therapy, with or without immunosuppressants, seems to be absolutely required [
8,
9,
10,
11]. APMPPE/AMIC develops preponderantly in young adults, being possibly slightly more frequent in men [
12]. A viral or febrile episode or other systemic manifestation has been reported to precede the ocular disease in approximately half of the patients, an occurrence similar to other PICCPs. Sometimes the presumed virus can be tracked [
13]. As in other PICCPs, vaccination can be the trigger at the origin of APMPPE/AMIC [
14,
15,
16]. Bilaterality, concomitant or with a slight delay from one eye to the other, and limitation to a single episode determine the characteristic clinical pattern. In case of unilaterality and/or recurrences the diagnosis should be questioned and can be considered either as atypical APMPPE/AMIC or as a non-classifiable choriocapillaritis [
5]. Mixed forms between APMPPE/AMIC and SC (Ampiginous choroiditis) can also occur and should be differentiated [
17].
Symptoms include a decrease in visual acuity depending on the localisation of lesions in the macula, subjective scotomas and photopsias [
12]. Anterior segment involvement, exceptionally including synechiae, is not a rare occurrence and subclinical anterior segment inflammation can be looked for with laser flare photometry [
18].
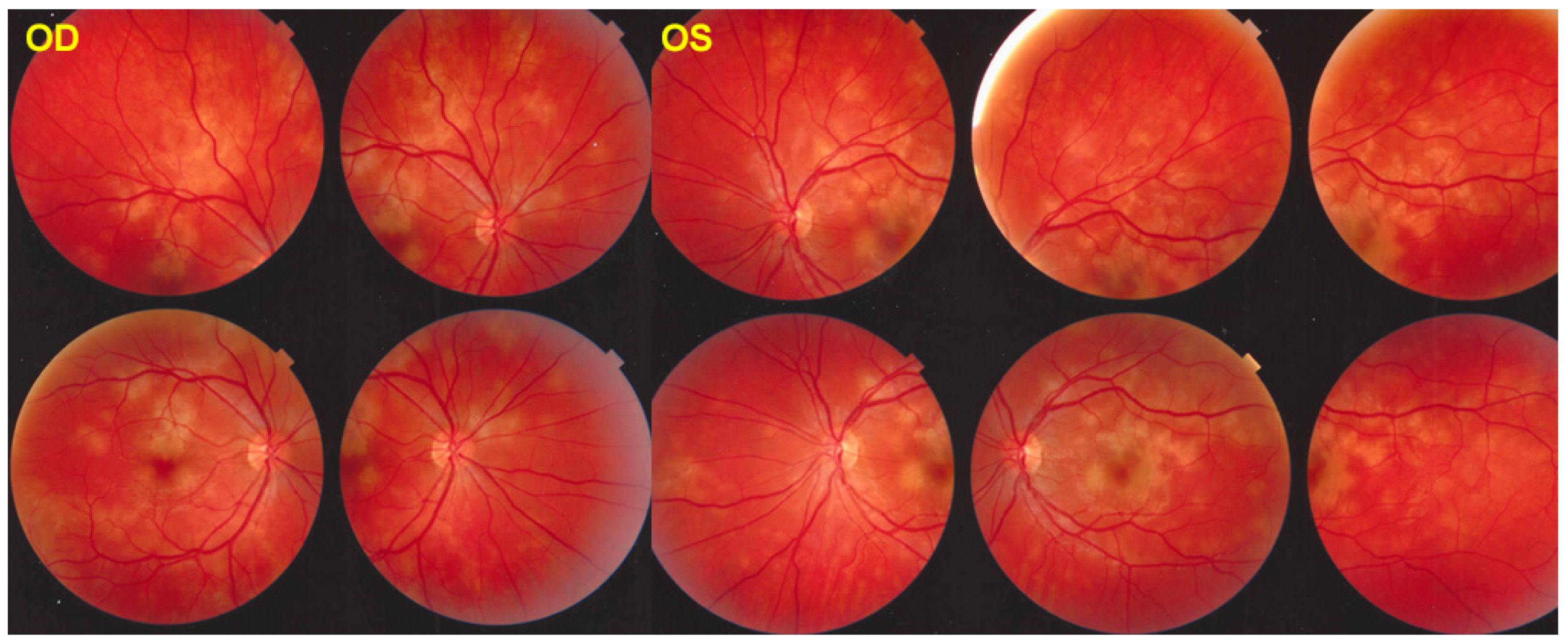
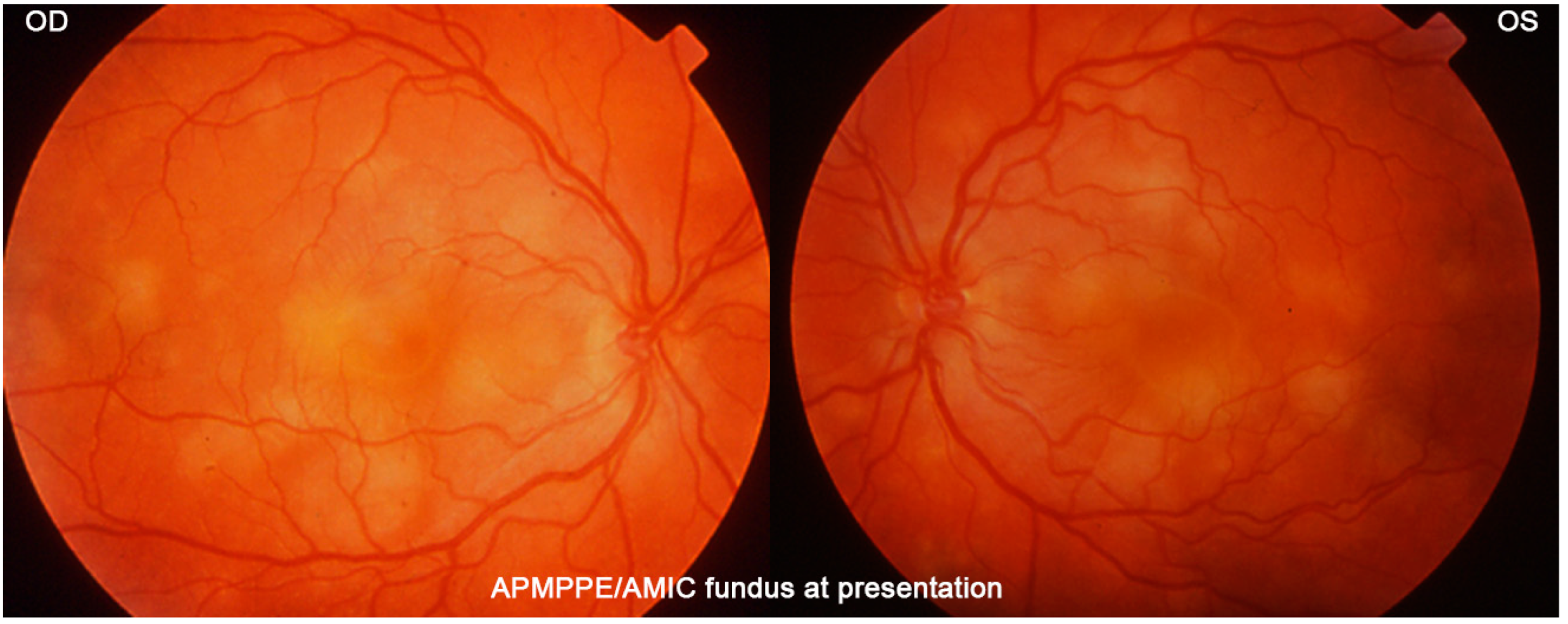
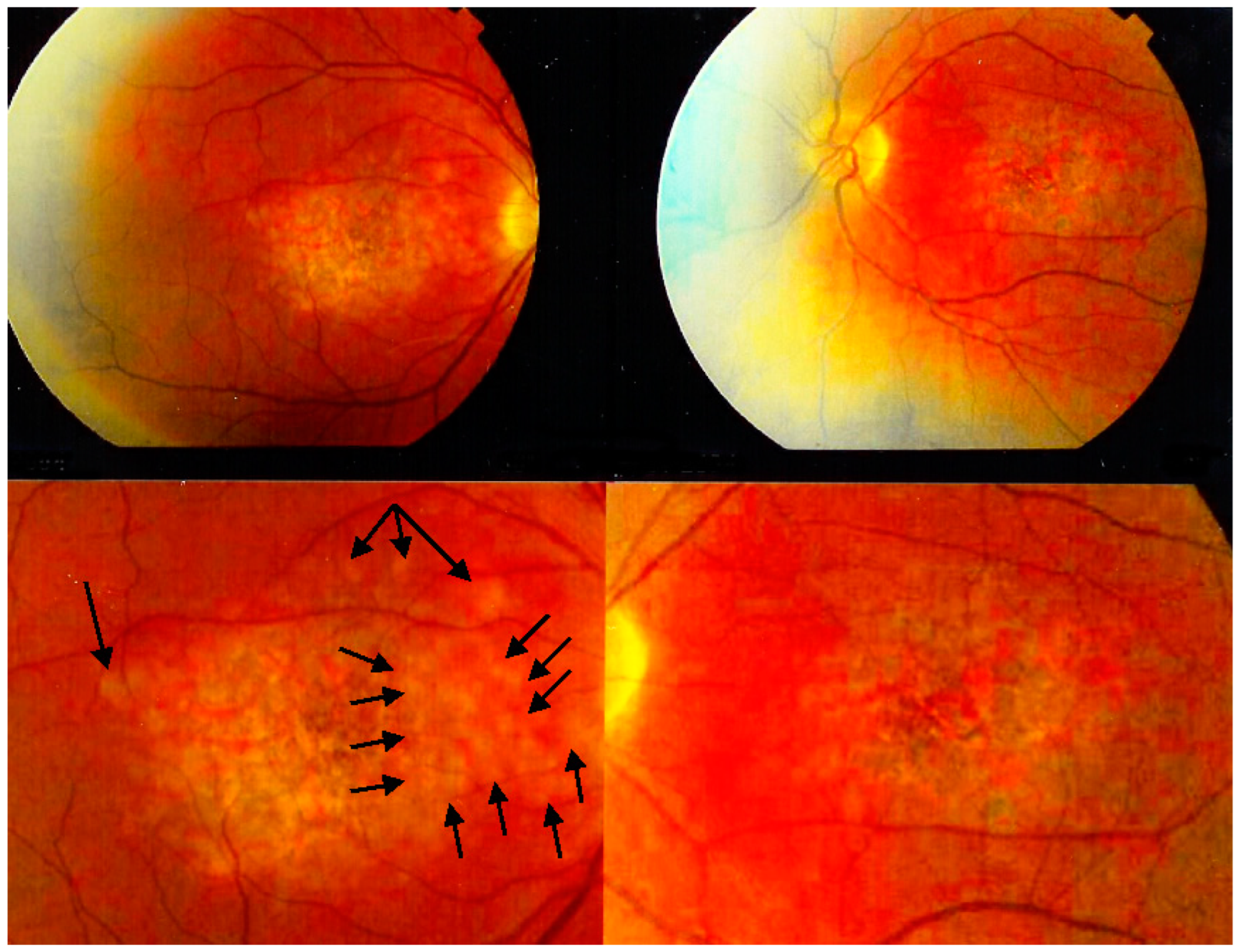
The fundus appearance consists of the characteristic bilateral yellow-white discoloured “placoid” lesions in the posterior pole and mid-periphery after which the disease was originally named (
Figure 3). Involvement may sometimes be at a slightly different stage in one and the contralateral eye [
1,
6]. In the differential diagnosis, infection-related conditions, such as acute syphilitic posterior placoid chorioretinitis (ASPPC) or tuberculosis-related serpiginoïd choroiditis (TB-SC), which can have similar presentations, should be considered [
19,
20].
Multimodal imaging contributes substantially to the diagnosis, the different imaging modalities pointing towards choriocapillaris non-perfusion and its consequences [
5,
21,
22].
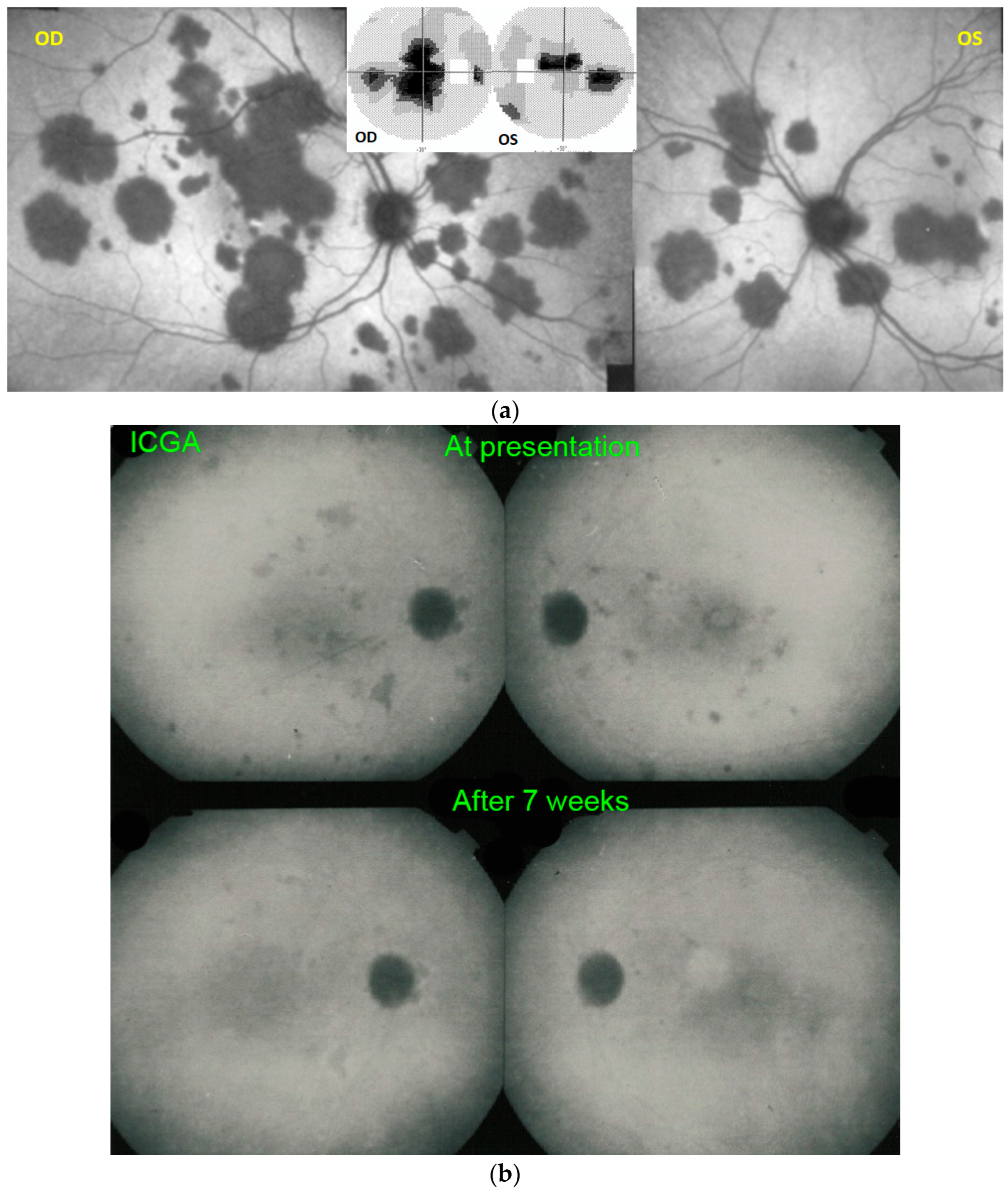
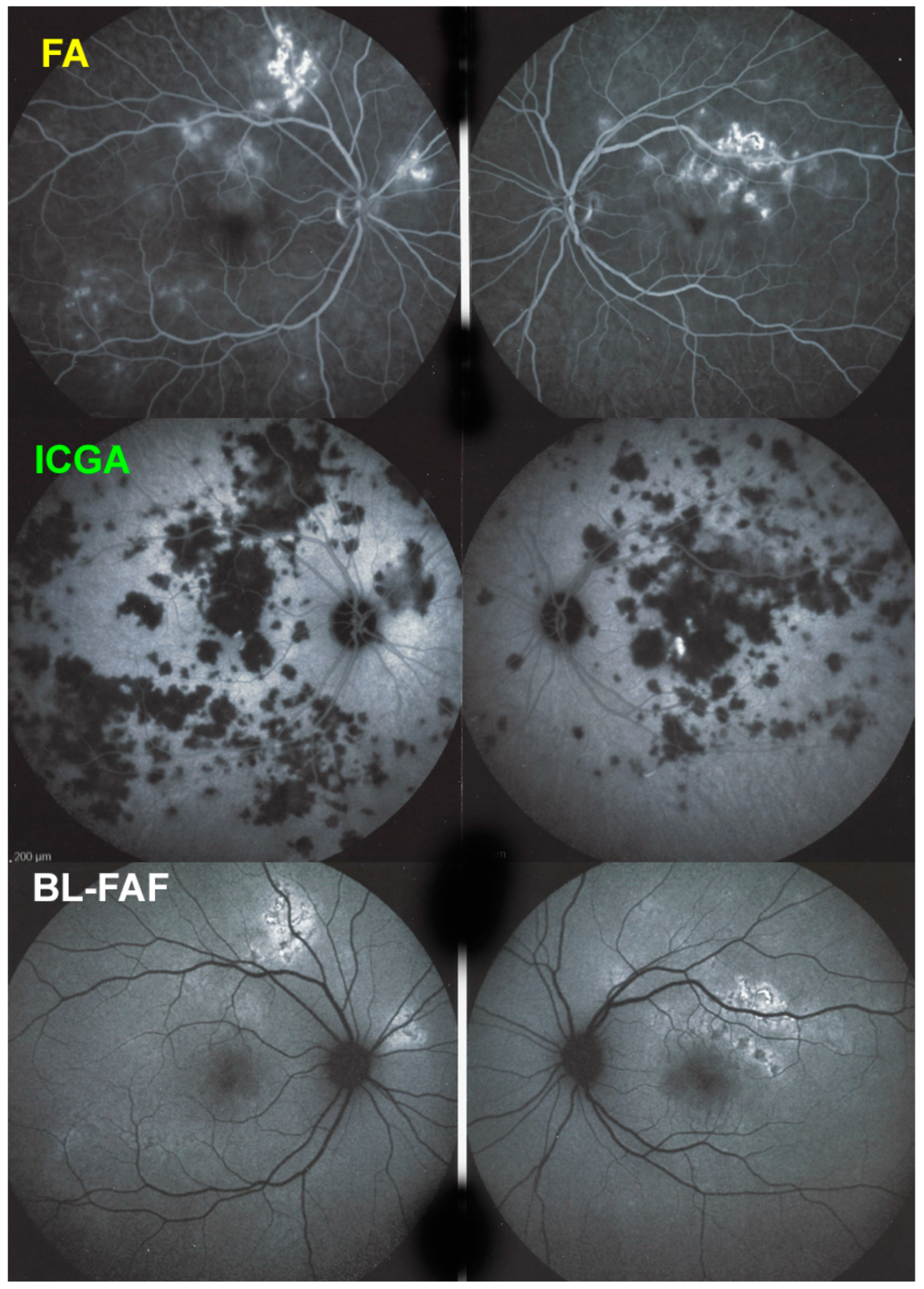
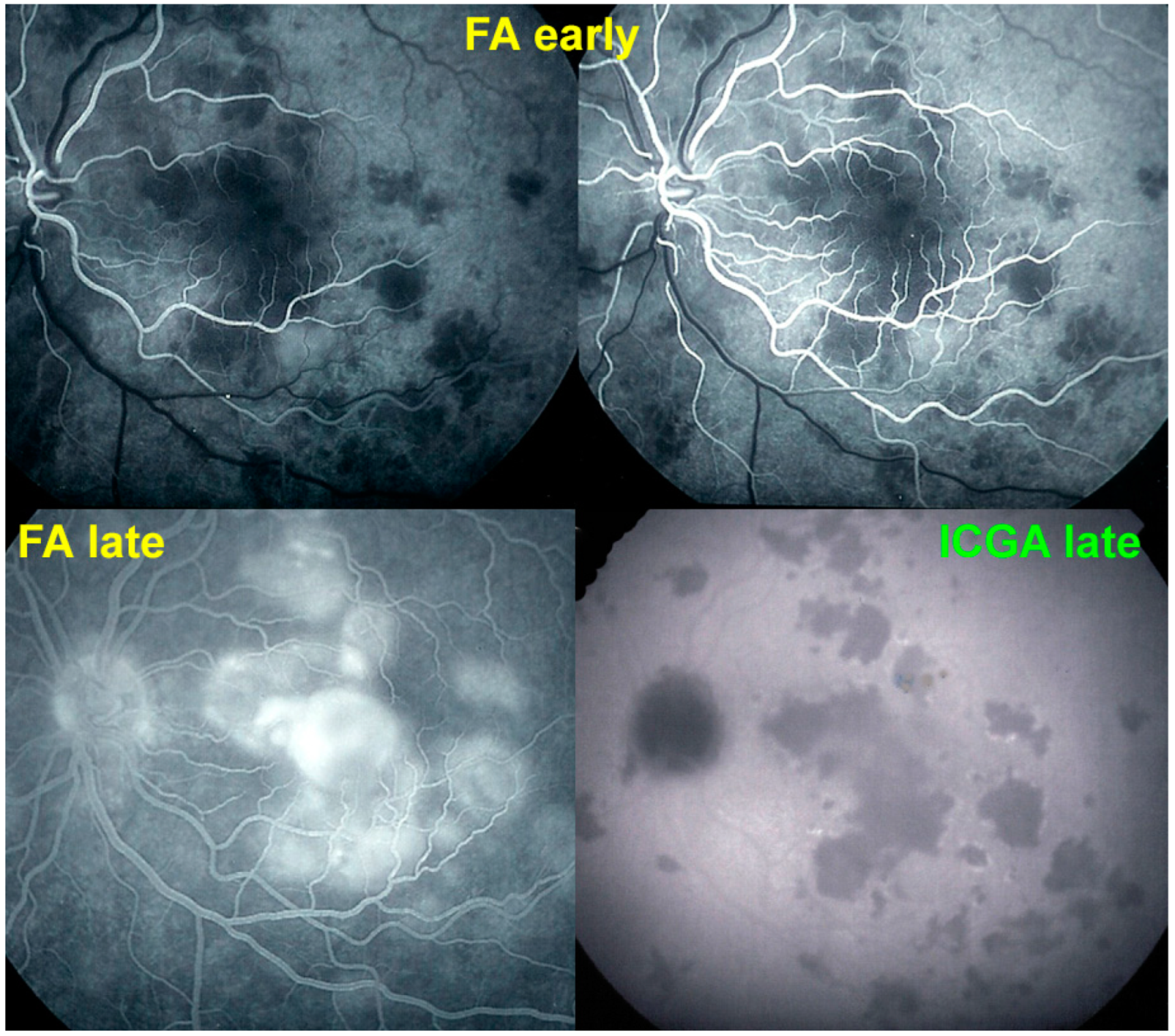
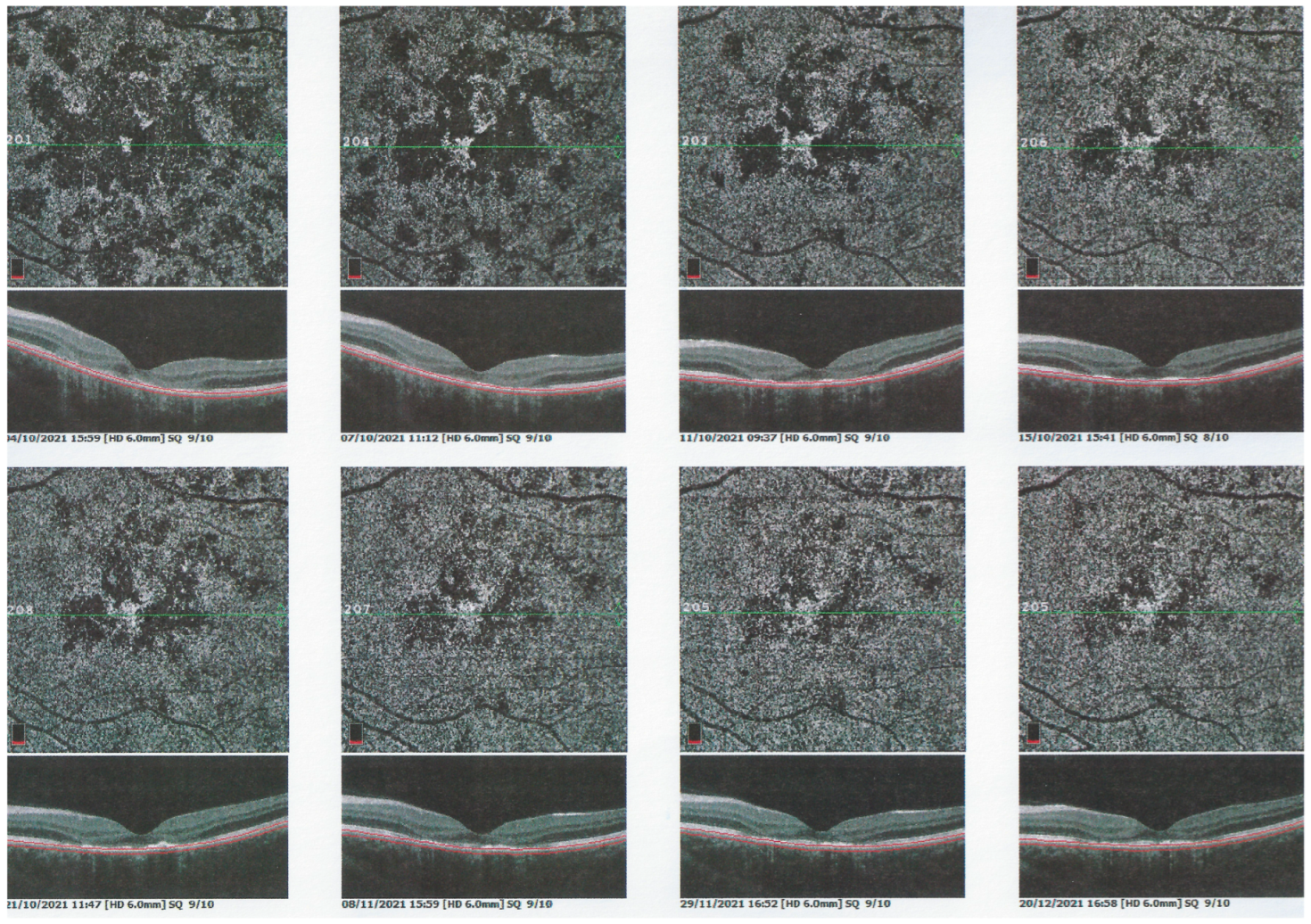
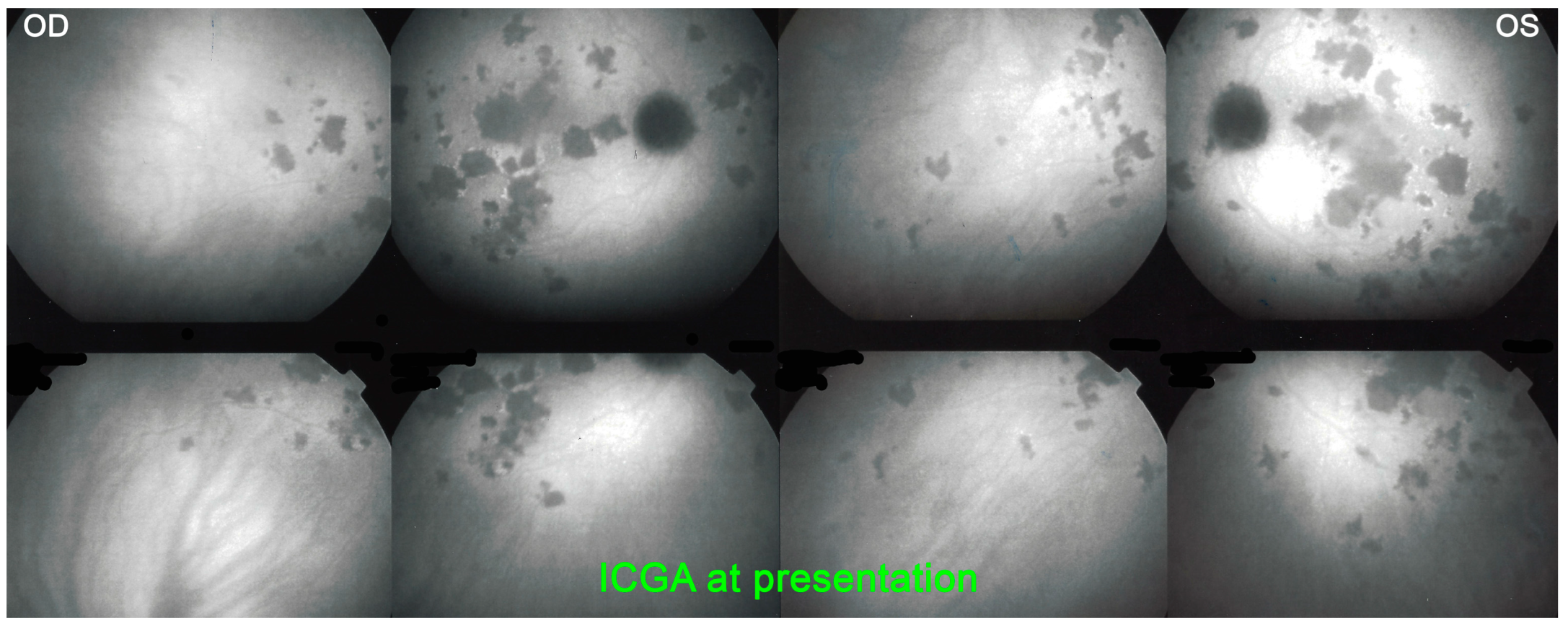
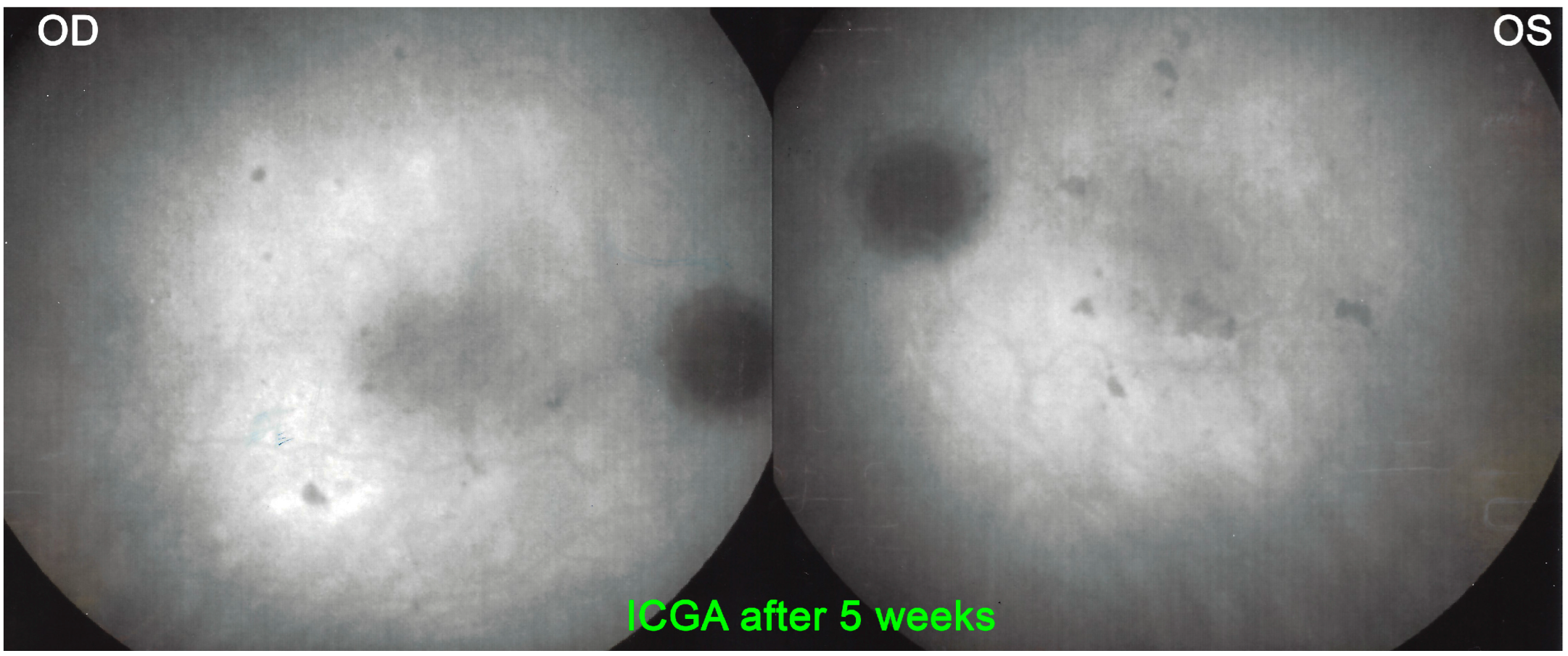
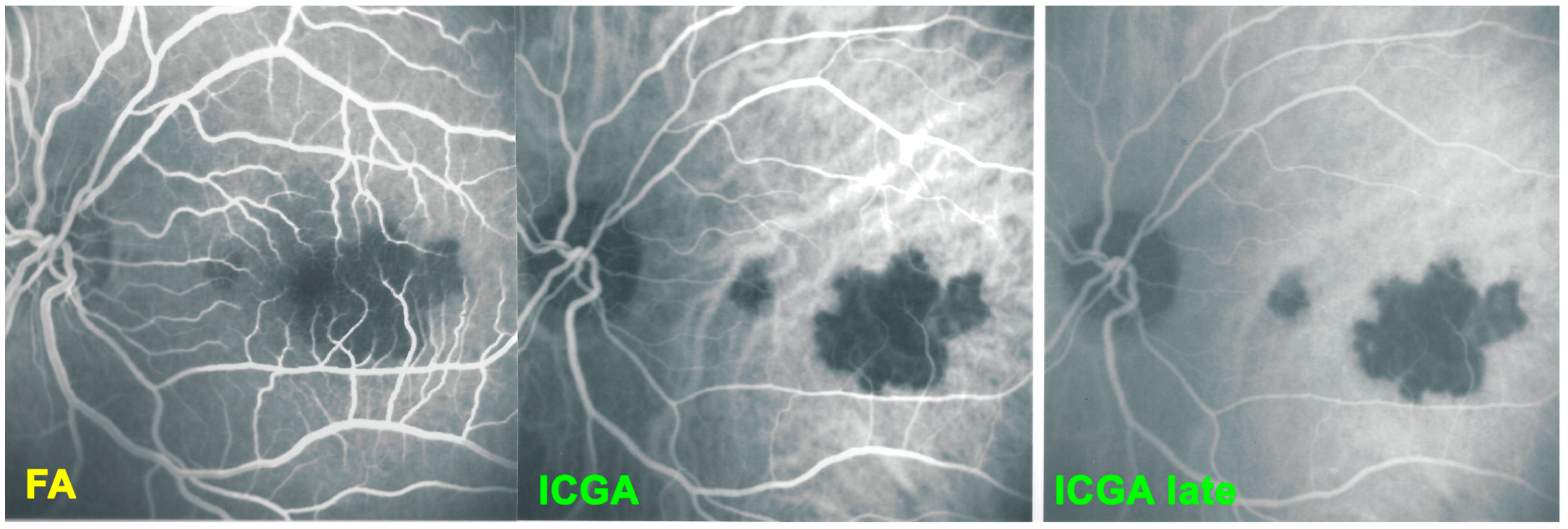
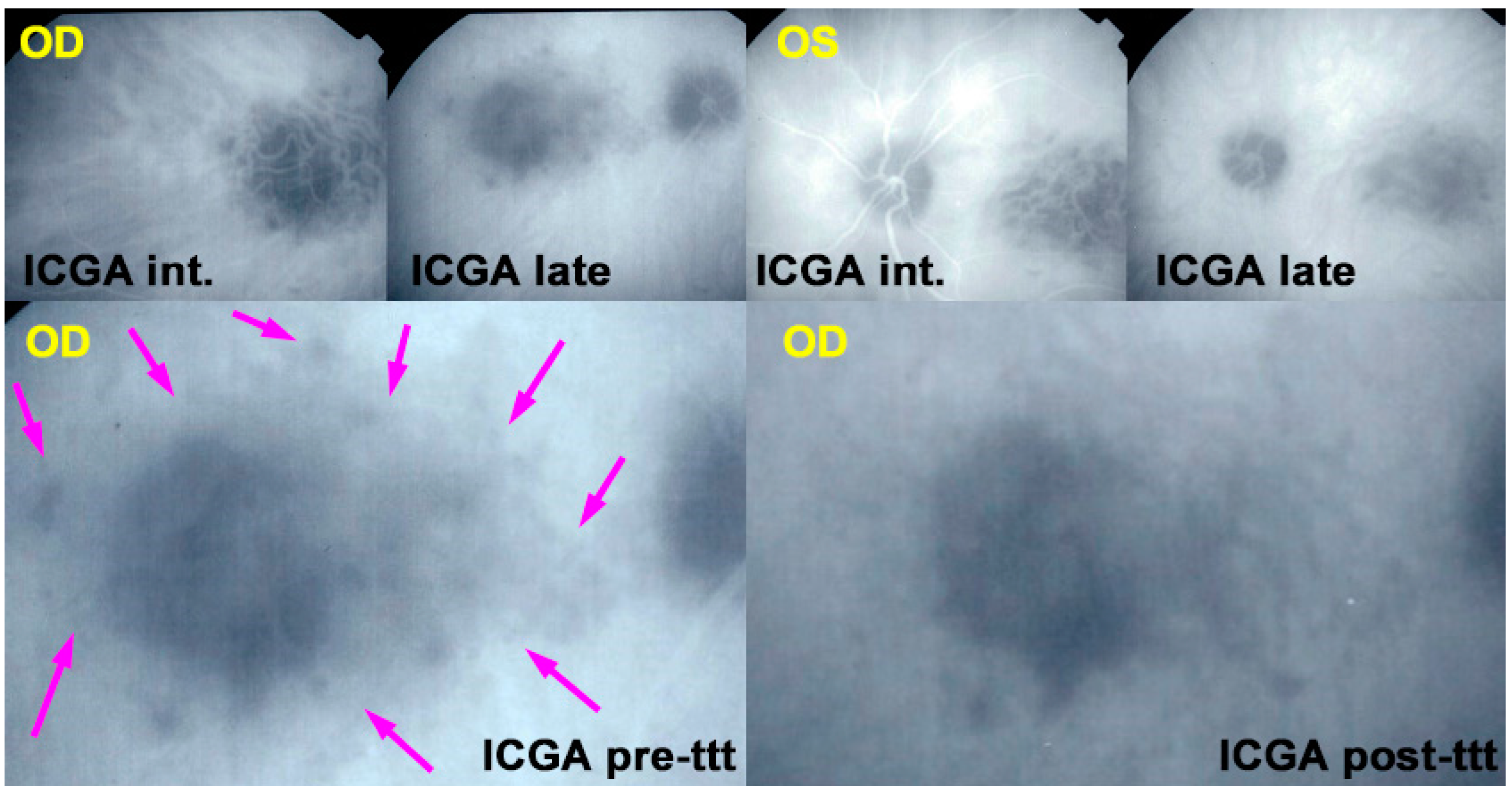
For precise evaluation of areas of choriocapillaris non-perfusion and their follow-up, ICGA is by far the most valuable modality, revealing discrete to large areas of scattered or confluent hypofluorescent dark areas present from early to late angiographic frames [
22,
23,
24] (
Figure 4a,b and
Figure 5).
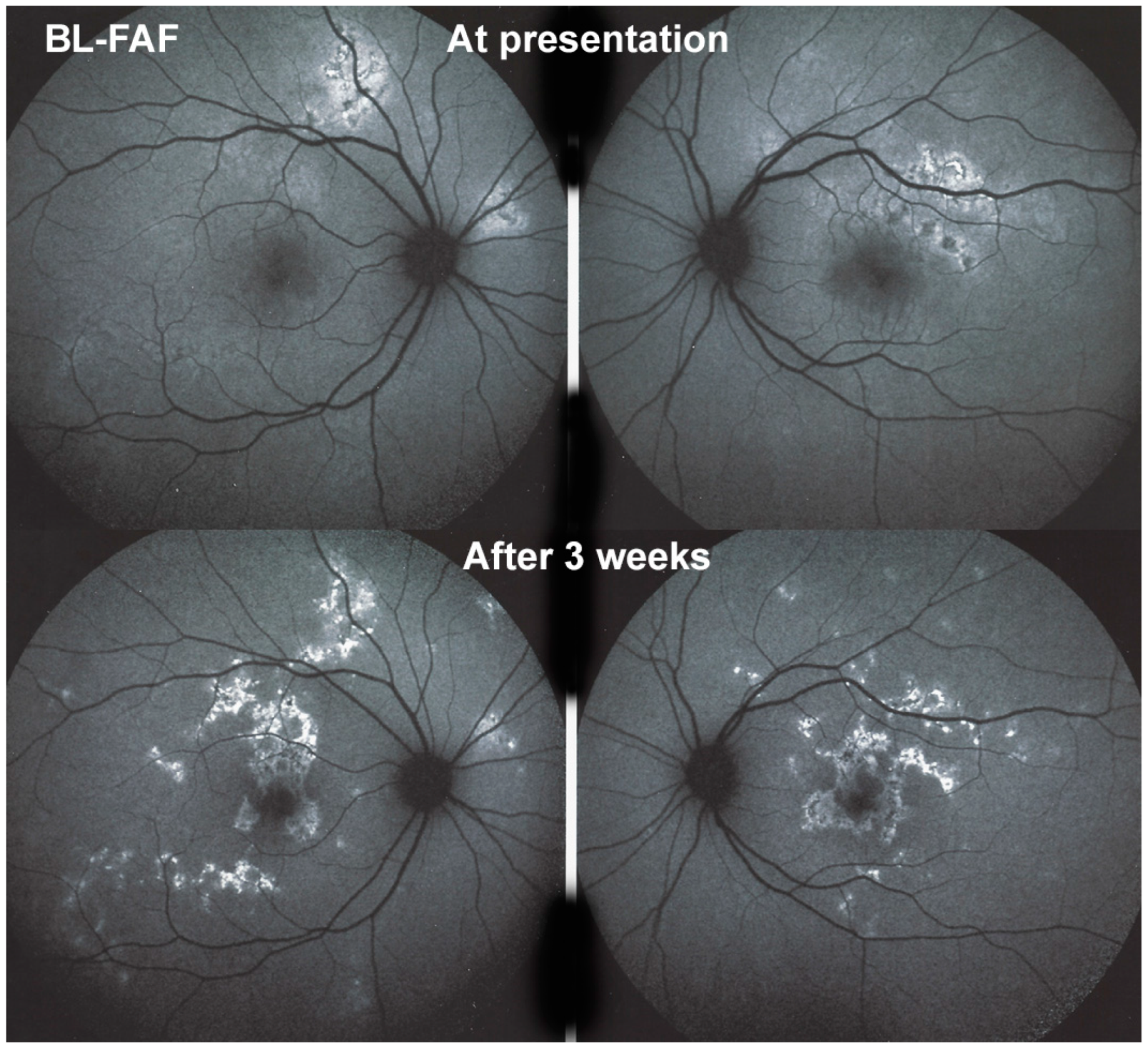
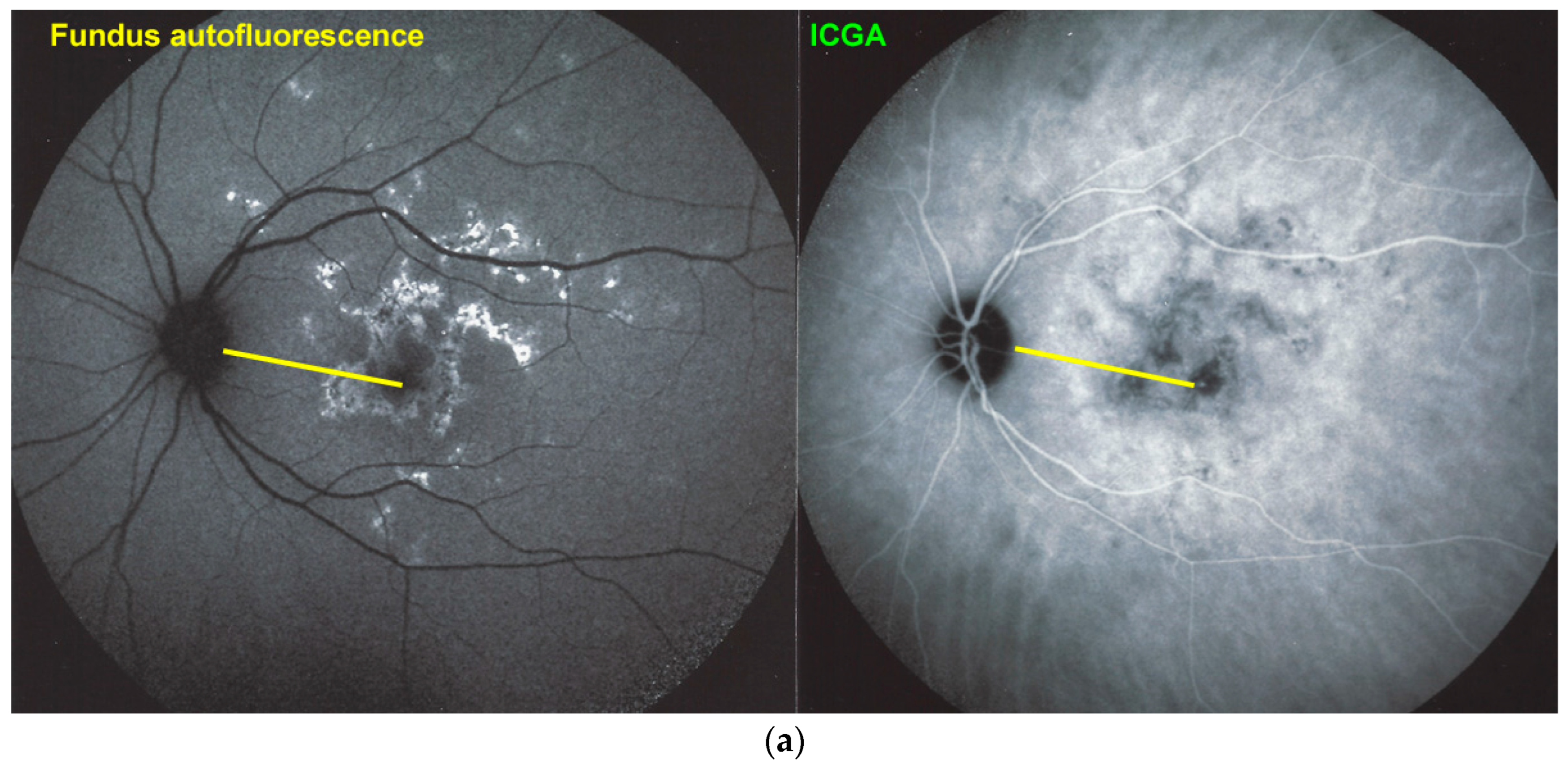
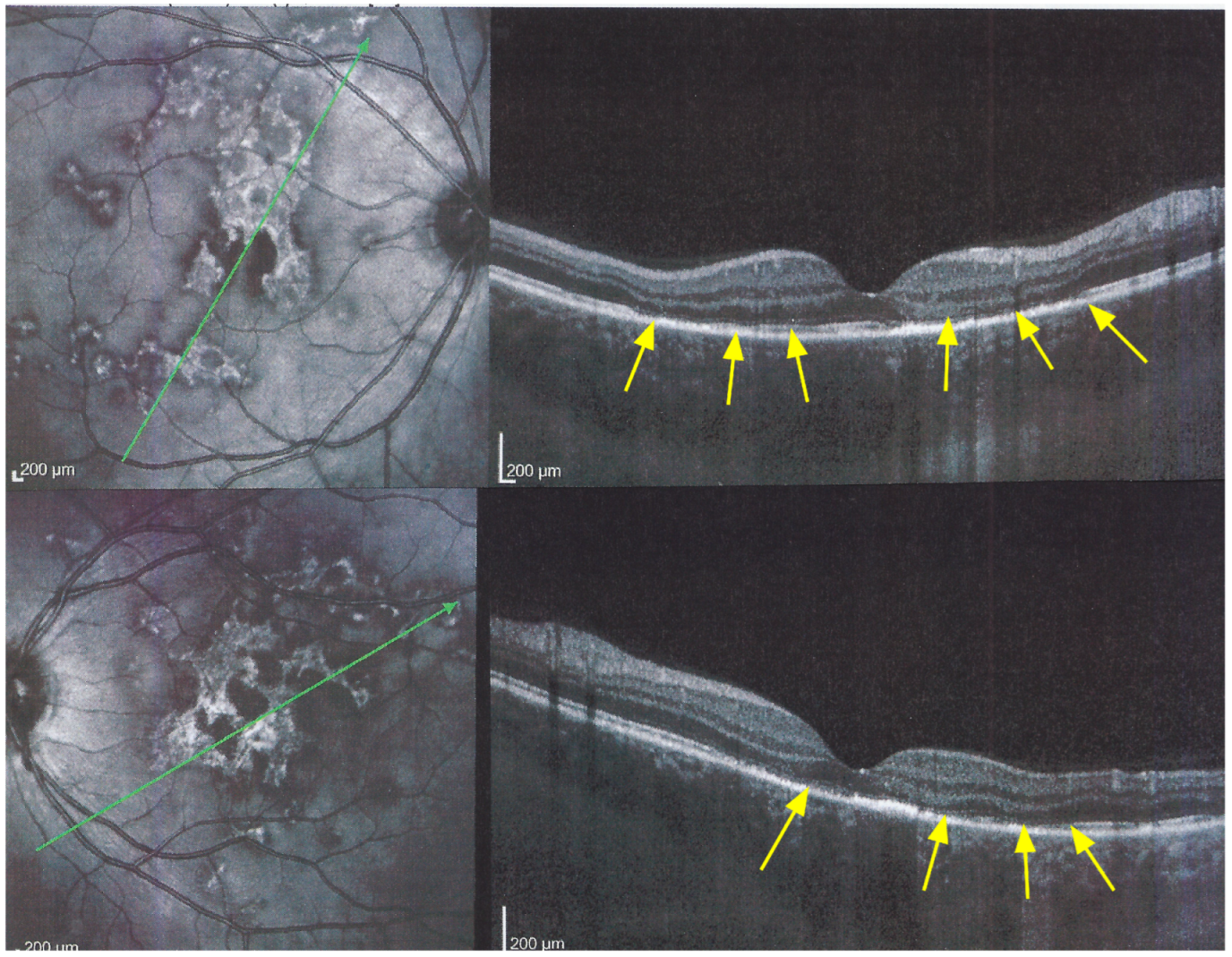
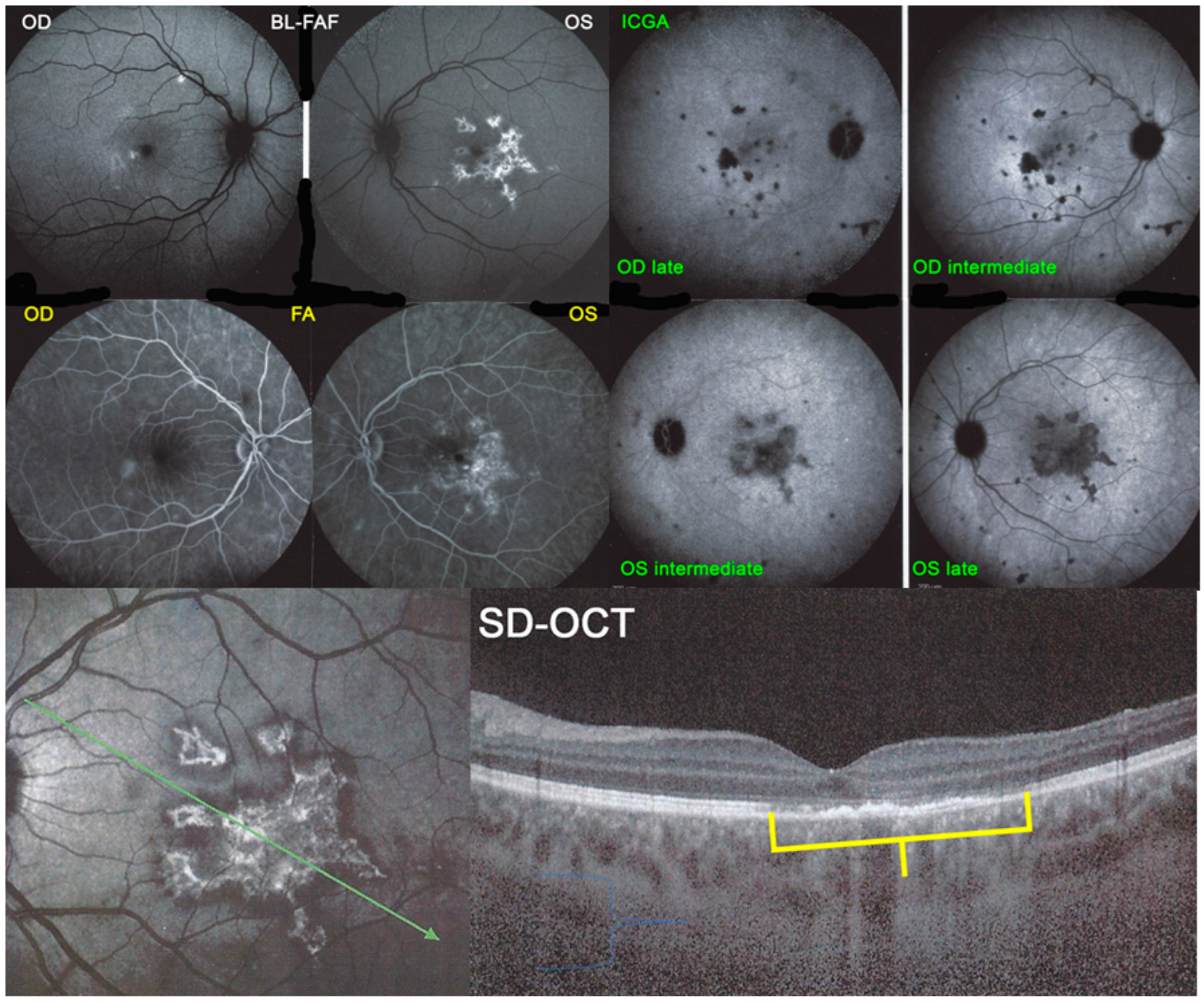
Blue light fundus autofluorescence (BL-FAF) shows both hyperautofluorescence and hypoautofluorescence depending on the stage of evolution of the disease and is sometimes difficult to interpret. In the very acute stage, there are still limited areas of hyperautofluorescence following the loss of photoreceptor outer segments due to non-perfusion which has caused ischaemia without chorioretinal atrophy. In the subacute stage, there is an alternation of hyperautofluorescence either due to loss of photoreceptor outer segments or damage of RPE cells with accumulation of fluorophore debris, or hypoautofluorescence due to chorioretinal atrophy with loss of RPE [
5,
7,
25] (
Figure 6 and
Figure 7a,b). In progressing lesions hypoautofluorescence is central (atrophy) and hyperautofluorescence is located in the periphery of lesions where ischaemia causes photoreceptor outer segment loss with still conserved RPE and/or RPE cells with accumulation of cellular fluorophore debris (
Figure 7a,b).
The evolution of BL-FAF findings during the different disease stages is illustrated in
Figure 7b.
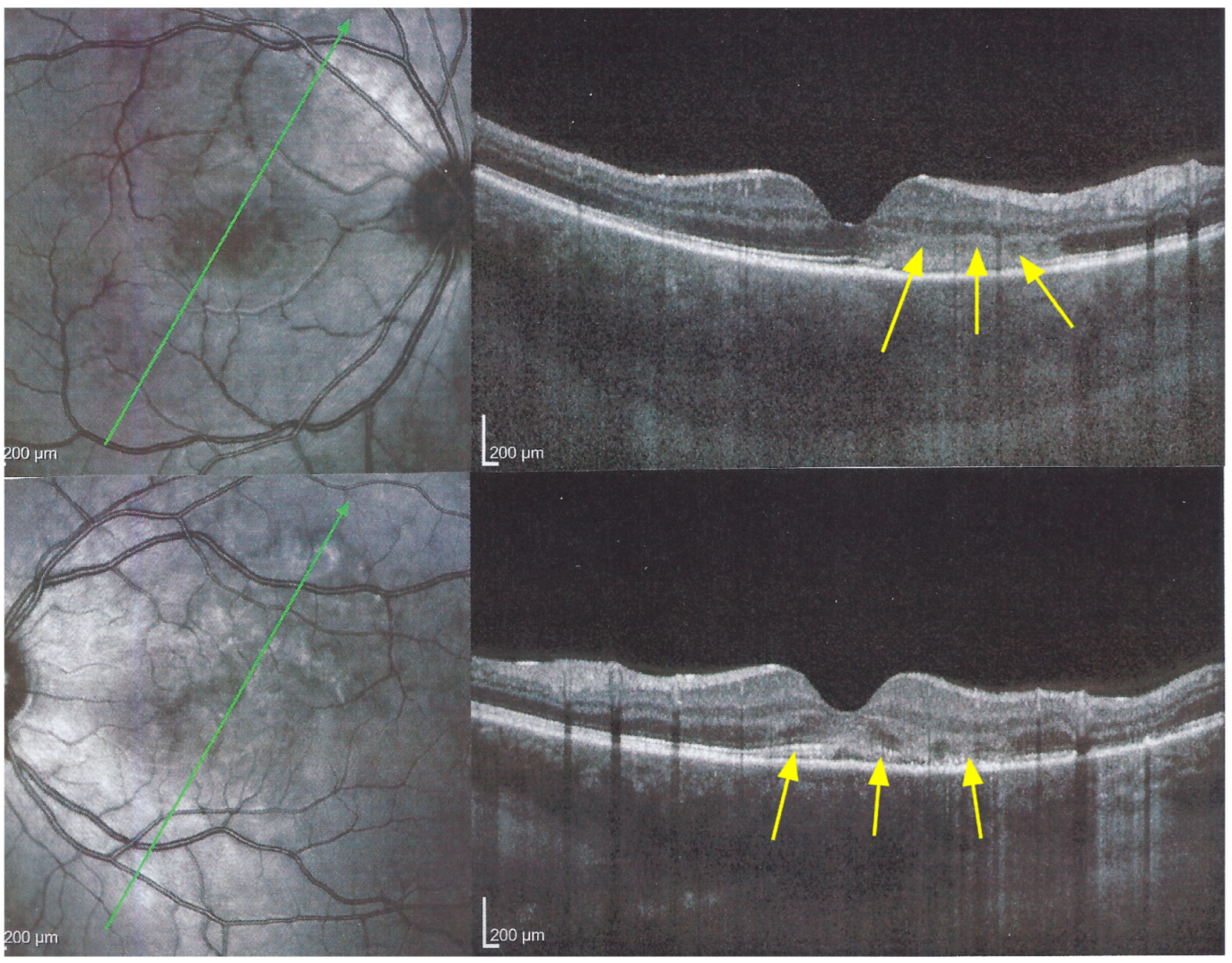
The chorioretinal findings by spectral domain optical coherence tomography (SD-OCT) depend on the degree of involvement and the stage of the disease [
5,
7,
25]. In early-acute disease ischaemia, induced by choriocapillaris non-perfusion, causes thickening of the outer retina including the IS/OS line and beyond (
Figure 8). In later stages, SD-OCT can show either simple loss of photoreceptor outer segments, thickened RPE or atrophy with loss of RPE (
Figure 9).
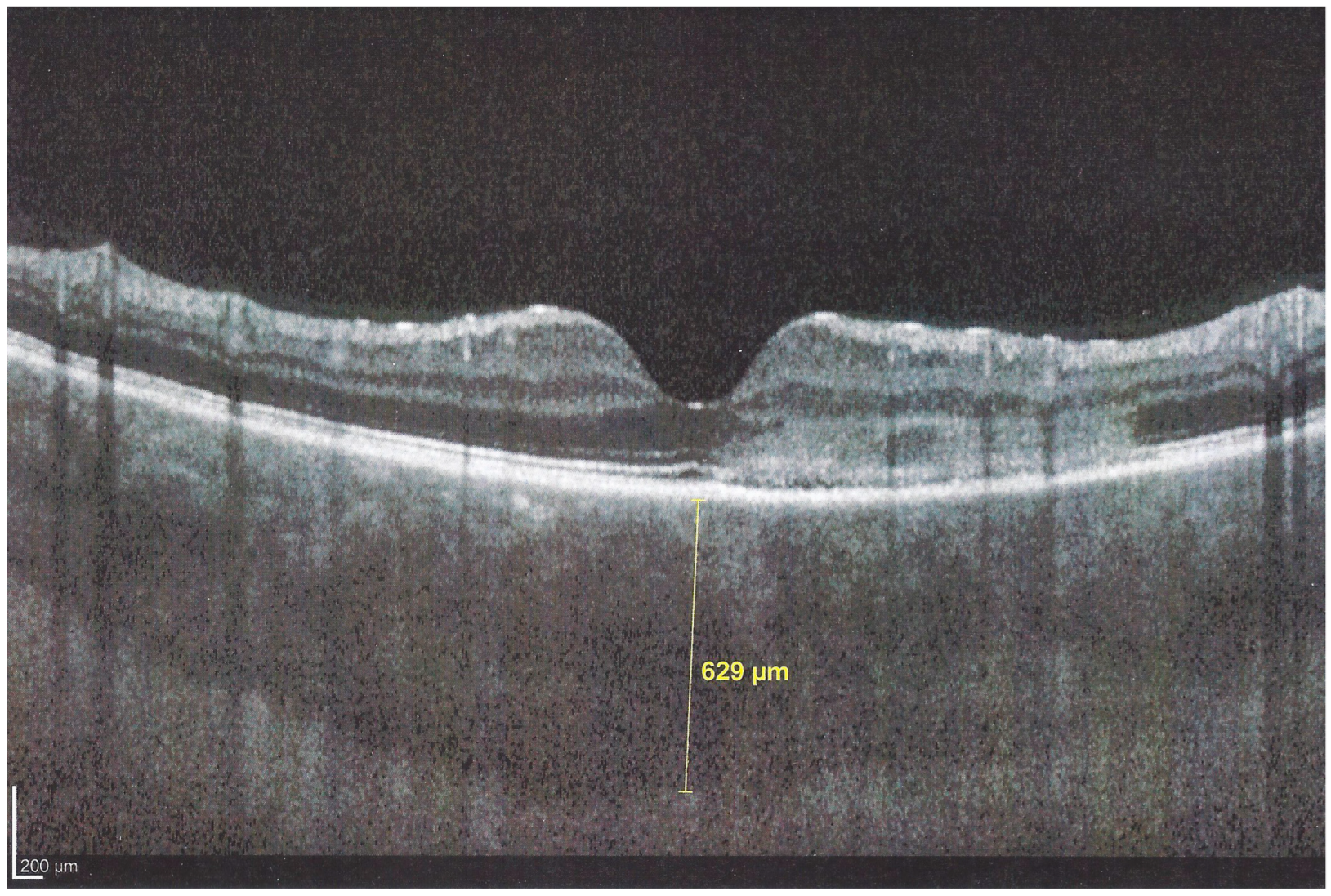
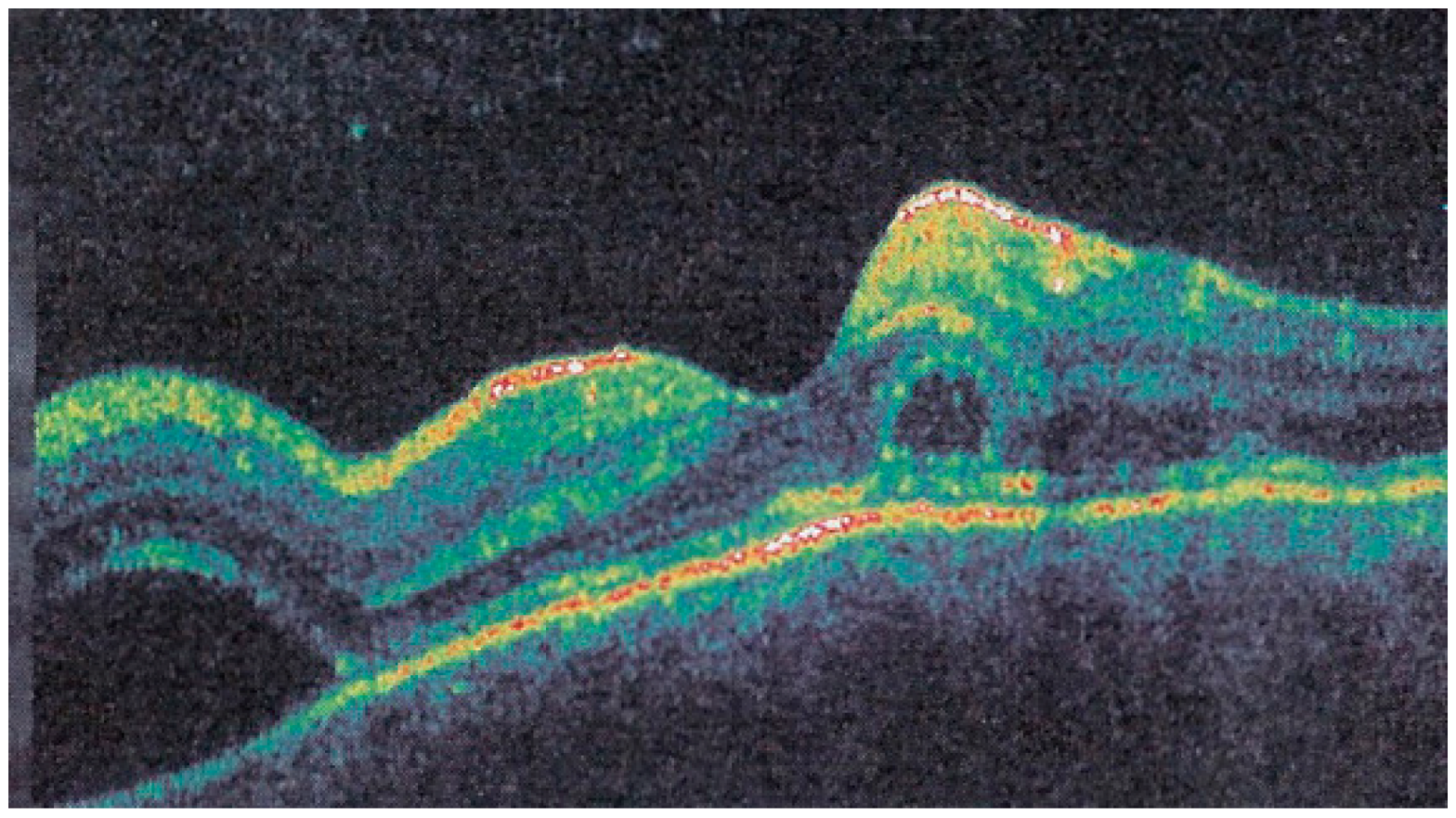
Interestingly, in some cases, the degree and extension of outer retina ischaemia in early-acute disease are such that, similar to diabetic retinal ischaemia, it causes reactionary dilatation and exudation of inner retinal vessels causing pooling visible on FA and visible as serous detachments on SD-OCT [
25] (
Figure 10).
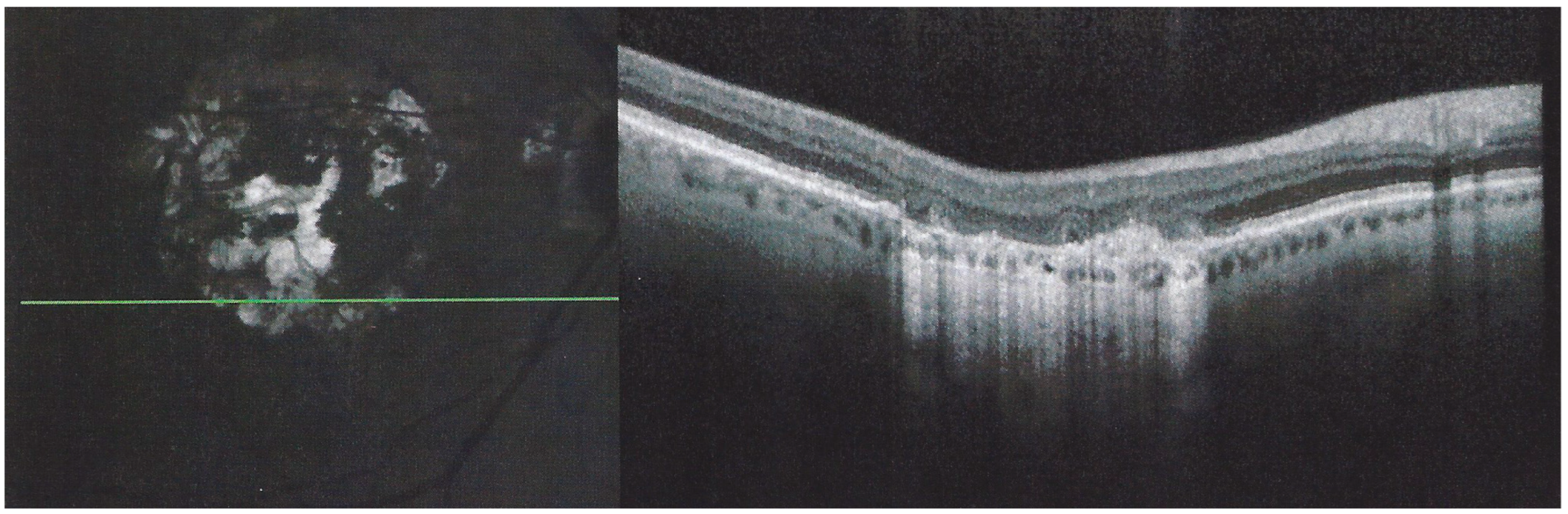
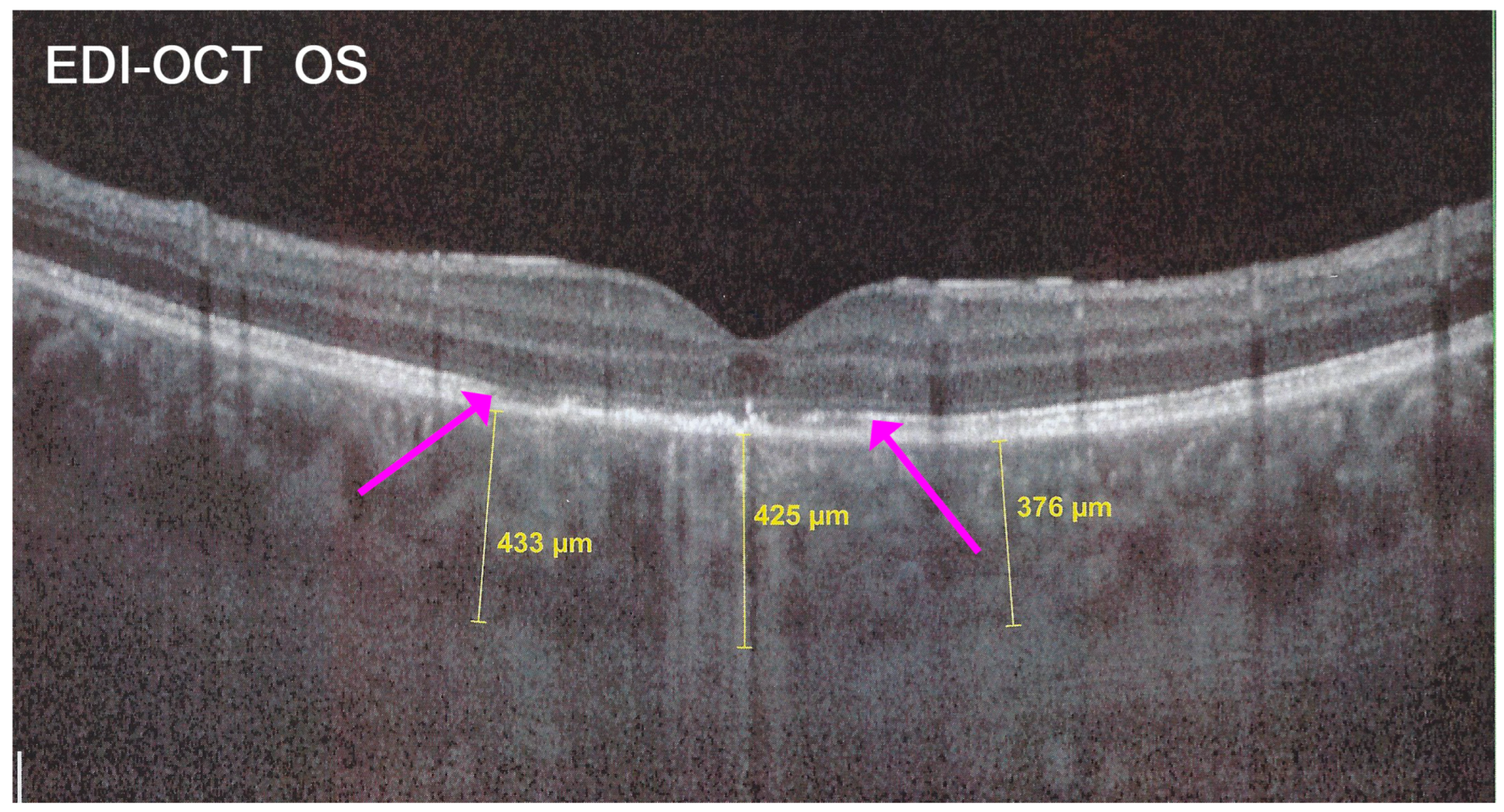
In addition to choriocapillaris non-perfusion, in most acute cases, the entire choroid is thickened, which can be shown by enhanced depth imaging OCT (EDI-OCT) (
Figure 11). In complement to ICGA, OCT angiography (OCT-A) shows areas of choriocapillary drop-out that correlate with the areas of ICGA hypofluorescence the analysed area being however, limited to part of the posterior pole and less precise than ICGA [
5,
7,
25] (
Figure 12).
The prognosis for APMPPE cases varies from one case to the other and depends on the location and severity of involvement. In the literature, it is usually described as a self-limited condition. As indicated here above, there are reports of cases with extensive lesions and marked visual impairment needing systemic corticosteroid treatment. Additional immunosuppression does not seem to be necessary in most cases [
8,
9,
10,
11]. Cerebral vasculitis is a rare complication or co-morbidity that has to be suspected when neurological symptoms are noted possibly confirmed by a positive cerebral magnetic resonance imaging angiography (angio-MRI) [
26,
27]. Systemic vasculitis associated with APMPPE has also been reported [
28]. Most numerous complications include chorioretinal scars and, as is always possible with chorioretinal scars, the development of CNVs needing intravitreal anti-VEGF therapy.
The aim of this study was to retrospectively analyse a relatively large series of APMPPE/AMIC patients seen in our centre, determine their clinical and imaging characteristics, and establish the proportion of patients with a more pronounced disease where systemic prednisone therapy was deemed necessary and compare these findings to the literature. In patients that were followed in our centre, the evolution of cases and outcomes were documented.
2. Patients and Methods
This retrospective case series was performed in the Centre for Ophthalmic Specialised Care (COS), Lausanne, Switzerland. Patients diagnosed from 2000 to 2021 with APMPPE/AMIC were included. Patients having a positive QuantiFERON test and/or positive VDRL-TPHA test were excluded. Imaging analysis included spectral domain optical coherence tomography (SD-OCT) and enhanced depth imaging OCT (EDI-OCT) (Heidelberg Engineering GmbH, Heidelberg, Germany), OCT angiography (OCT-A) (AngioVue®, Optovue, Fremont, CA, USA) Fluorescein and Indocyanine angiography (FA, ICGA) (Heidelberg Engineering GmbH, Heidelberg, Germany). Best corrected visual acuity (BCVA) and routine ocular examination, as well as laser flare photometry (LFP), were performed at presentation and during the follow-up of patients. The presence or not of a prodromal febrile and/or viral episode or other systemic symptoms was noted. Eyes were divided into 2 groups, depending on the localisation of the lesions by ICGA and/or SD-OCT. The lesions were qualified as foveal when the hypofluorescent areas included the fovea on ICGA and/or when alterations of the outer retina and/or RPE were noted in the fovea by SD-OCT. The lesions were characterised as perifoveal or parafoveal when hypofluorescent areas were spotted outside the fovea by ICGA and/or when the outer retina was untouched in the fovea by SD-OCT.
3. Results
3.1. Demographics
Nineteen out of 1664 new patients (35 eyes) (1.14%) were diagnosed with APMPPE/AMIC in the uveitis clinic of the Centre for Ophthalmic Specialised Care (COS) during the period from 2000 to 2021 and were included in our study. 13 (68%) were male and 6 (32%) were female. The mean age was 33.1 ± 9.2 years. 16 (84%) patients mentioned a viral prodromal episode, or other systemic symptoms and 3 (16%) did not mention any episode before the onset of the ocular symptoms.
3.2. Clinical Findings
The series was divided into 2 groups depending on whether lesions were foveal or extra-foveal. 15 of 38 eyes (38%) had a foveal location of lesions while 20/38 (52.6%) eyes had parafoveal or peri-foveal lesions and 3 eyes were normal in the 3 unilateral cases (15%).
The mean BCVA for the whole series at presentation was 0.83 ± 0.24. The mean flare at presentation was 10.52 ± 7.7, indicating minimal anterior segment involvement. One patient had bilateral non-granulomatous uveitis with synechiae. 3 eyes presented choroidal neovascular membranes that were treated by 4 intravitreal injections of anti-VEGF agents.
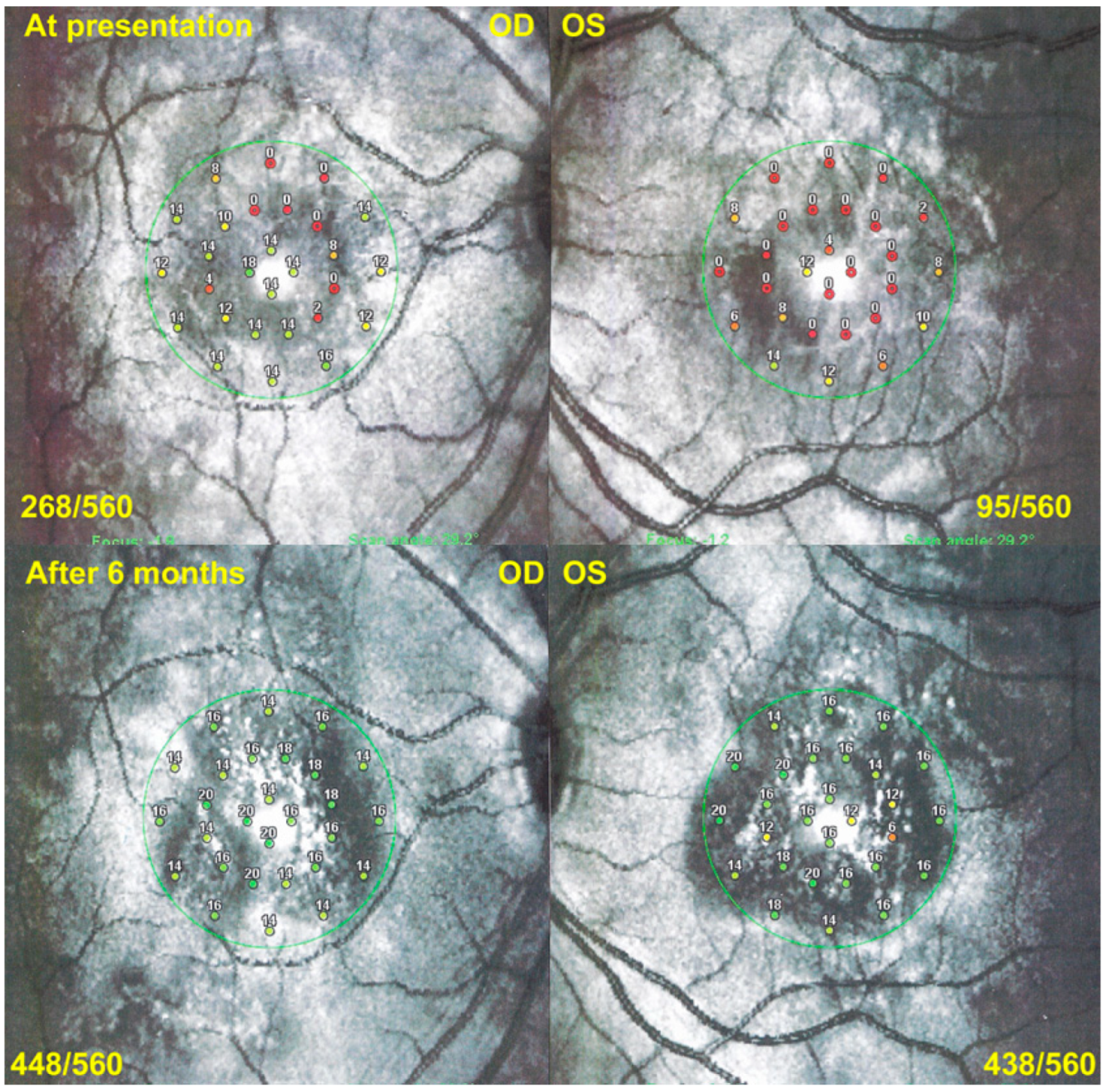
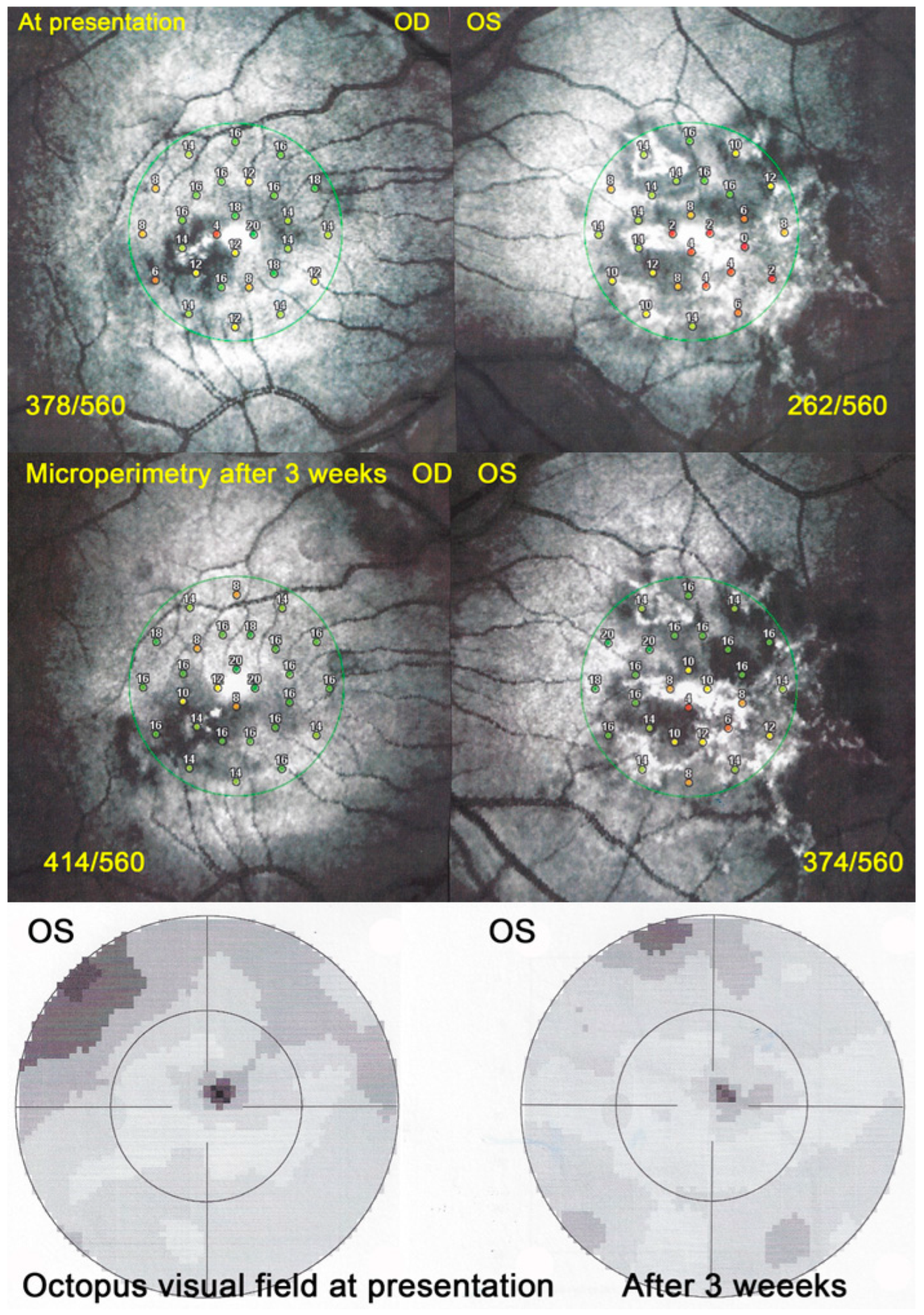
Visual field (VF) showed central and/or paracentral scotomas with a mean MD at a presentation of 5 ± 3.6 db. However, microperimetry, when available, showed a much more precise topography of the decreased retinal sensitivity and a numerical measurement at presentation and on follow-up (
Figure 13).
3.3. Imaging Findings
The most important and constant imaging modality was
ICGA which showed early to late hypofluorescent dots/areas scattered or confluent taking a geographic pattern. This finding was present in absolutely all 35/35 eyes and is the hallmark of the disease. Lesions were hypofluorescent from early to late angiographic frames. (
Figure 4a,b,
Figure 5,
Figure 7 and
Figure 10).
SD-OCT findings included thickened hyperreflective areas of the outer retina in the very early disease phase (10/35 eyes) (
Figure 8 and
Figure 11), photoreceptor outer segment loss and/or ellipsoid zone disruption-RPE alterations (22/35 eyes) (
Figure 9), subretinal fluid (serous retinal detachment—SRD) (4/35 eyes), (
Figure 14) and atrophy (3/35 eyes) in the late stage of the disease (
Figure 15).
FA consistently showed early hypofluorescence with late hyperfluorescence in 19/35 eyes, (
Figure 10) early hypofluorescence with late iso-fluorescence in 4/35 eyes, and disc hyperfluorescence in 6/35 eyes while in 2 eyes there was early to late hypofluorescence due to extended chorioretinal atrophy including the RPE.
In hyperacute cases, there was late subretinal pooling corresponding to the SDR on SD-OCT found in 4/35 eyes (
Figure 10).
BL-FAF: 3 pathological eyes had normal FAF, while hyperautofluorescence mixed with hypoautofluorescence was detected in 5/23 eyes; (
Figure 7) hyperautofluorescence alone was detected in 12/23 eyes and hypoautofluorescence in 3/23 eyes. There were no FAF data available in 6 patients.
OCT-A showed patchy choriocapillaris drop-out in acute phases and absence of signal in atrophic lesions, but it was not available for 12 patients (
Figure 16).
3.4. Localisation of Lesions and Visual Outcomes
While BCVA for the whole series at presentation amounted to 0.83 ± 0.24, in the 15 eyes of 12 patients that had foveal lesions, BCVA at presentation was 0.58 ± 0.28, increasing to 0.97 ± 0.13 at final follow-up, a difference that was statistically significant (p = 0.0028). For the 20 eyes of 15 patients that presented with peri/parafoveal localisation, BCVA at presentation was 0.94 ± 0.18 while at the last follow-up it was 1.18 ± 0.10, also statistically significant (p = 0.0039).
3.5. Corticosteroid/Immunosuppressive Treatment and Visual Outcome
13/19(68%). patients had received systemic prednisone therapy and 2 patients (11%) had received additionally at least one immunosuppressive agent. 4 (21%) patients had received no treatment. No information was available for 2 patients (11%). Treatment was either already given by the referring ophthalmologist or by us. 11 out of 12 patients with foveal involvement of at least 1 eye had received systemic prednisone treatment (3 patients with bilateral foveal lesions, 9 patients with unilateral foveal lesions). One referred patient with bilateral foveal involvement had not received any treatment and was seen in our centre with severe bilateral foveal atrophy as a consequence and visual acuities of 0.1 bilaterally. Among the patients with peri/parafoveal localization, 12 eyes (10 patients) had received treatment, 5 eyes (3 patients) had not received treatment, and for 3 eyes (2 patients), no information was available. 3/35 eyes (8%) had received intraocular anti-VEGF injections because of choroidal neovascularisation.
25/32(78%). eyes had received oral corticosteroid/immunosuppressive therapy of which follow-up data were available for 19 eyes. BCVA at presentation was 0.74 ± 0.28 versus 1.06 ± 0.15 at the last follow-up. (p < 0.0001).
We have data of follow-up from only 5 eyes that received no treatment. BCVA at presentation was 0.64 ± 0.22 versus 0.66 ± 0.4 at last follow, not significantly increased (p = 0.17). This indicates that the eyes were probably doing better functionally when treated.
5. Discussion
We report on 19 APMPPE/AMIC cases seen over a period of 22 years at the COS uveitis clinic, being among the largest series published when excluding meta-analysis series. This represented a percentage of 1.14% of all new patients seen during this period and corresponding to approximately 1 patient per year. Although the clinical presentation is usually characteristic, there is a variability of presentations from discrete to very severe cases which are reflected in our series. The evolution usually described in textbooks, series and case reports is that of a self-limited disease not necessarily needing systemic corticosteroid treatment. Therefore, astonishingly, in more than two-thirds of our patients, systemic corticosteroid therapy with or without additional non-steroidal immunosuppression was deemed necessary. Indeed, one non-treated referred patient lost both maculae to atrophy. We found a higher than reported rate of prodromal systemic illnesses including symptoms of viral disease or other amounting to close to 85%. Men were more frequent in our series than generally reported, representing a little over two-thirds; although as reported in other series, there does not seem to be a significant gender difference in APMPPE/AMIC [
29]. Due to the lack of predictability of the evolution of the disease, close monitoring of non-treated patients should be the standard of management in order to intervene promptly with corticosteroid treatment in case of deleterious evolution. Close monitoring has become easier with the multimodal imaging modalities at our disposal nowadays. The gold standard to establish and closely follow lesions is without contest ICGA. The latter is also the modality that is able to show early lesions not yet demonstrated by the other modalities, indicating that the “primum movens” is indeed choriocapillaris non-perfusion at the origin of all other imaging signs. OCT-A represented a precise and non-invasive method to follow non-perfusion areas, able to show progression in non-treated cases and subsequent regression after the introduction of prednisone therapy. BL-FAF is more difficult to interpret in APMPPE/AMIC when compared to MEWDS where it represents almost exclusively loss of IS/OS line and can therefore be used for the follow-up. In APMPPE/AMIC BL-FAF shows both losses of photoreceptor outer segments together with accumulation of fluorophore debris in metabolically damaged RPE cells in more hardly hit areas, producing hyperautofluorescence of different patterns and proportions. When RPE cells are lost altogether hypoautofluorescence is noted. Initial SD-OCT signs, in the acute phase of the disease, showed thickening of the outer retina which became hyperreflective and, later, loss of IS/OS line of photoreceptor outer segments with a focal thickness of the RPE in more hardly hit areas. The most characteristic FA sign in severe disease was late pooling explained by the exudation from retinal vessels in reaction to outer retina ischaemia, sometimes producing SRD in SD-OCT.
Outcomes were favourable without treatment in eyes with extrafoveal lesions. Visual outcome in eyes with and without foveal involvement was better in eyes treated with corticosteroids when compared to non-treated eyes, with a good visual acuity and good recovery of visual fields. This was in contrast to the one non-treated patient with severe lesions the location of which included the fovea leading to a deleterious evolution. Despite the publication of cases and a series reporting favourable outcomes without treatment [
30], abstention may be harmful. In case of potentially dangerous location of lesions and view of the unpredictability of the disease together with the numerous publications indicating that the notion of the self-limited character of the disease is incorrect [
9,
10,
11,
31,
32], a short course of corticosteroids represents a limited risk. Therefore, the decision to introduce prednisone therapy should be taken liberally, unless lesions are far from the central macula and not potentially vision-threatening.
Choroidal neovascular membranes occurred in 3 of 35 eyes but no central nervous system complications such as cerebral vasculitis were noted in our collective.
Many articles in the literature report cases resembling APMPPE/AMIC; however, they are lacking the classical criteria. These cases should better be termed as APMPPE/AMIC-like cases or undefined choriocapillaritis cases.
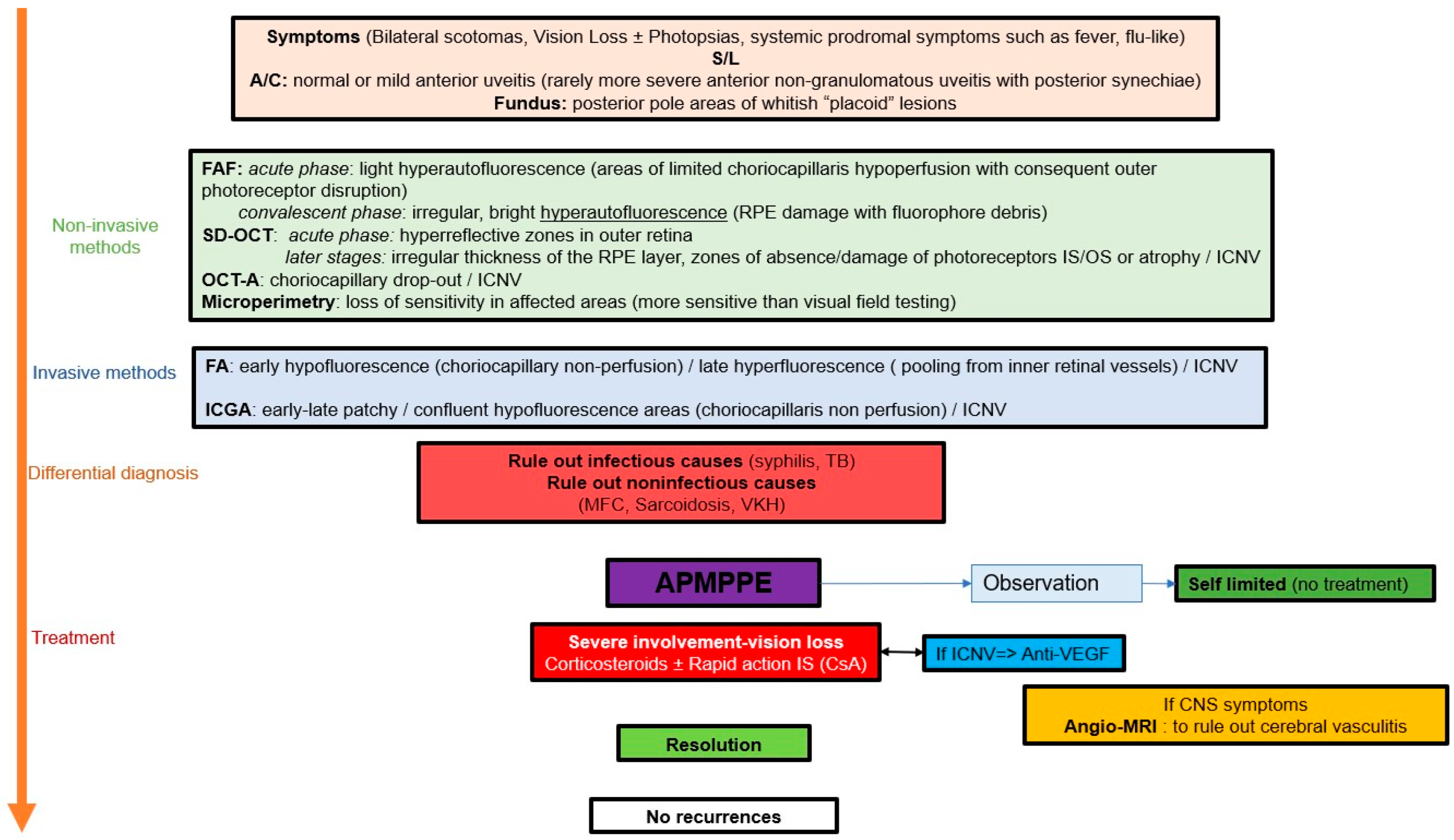
The key to responsible management of APMPPE/AMIC is the establishment of a clear delineation of lesions at presentation obtained thanks to ICGA, followed by precise and close monitoring of lesions which is very much helped by non-invasive imaging modalities at our disposal such as SD-OCT, BL-FAF and OCT-A, as well as functional investigations with a high grade of precision such as microperimetry (
Scheme 1).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



























