
Preclinical Development of Seriniquinones as Selective Dermcidin Modulators for the Treatment of Melanoma

 , and
, and
Abstract
:
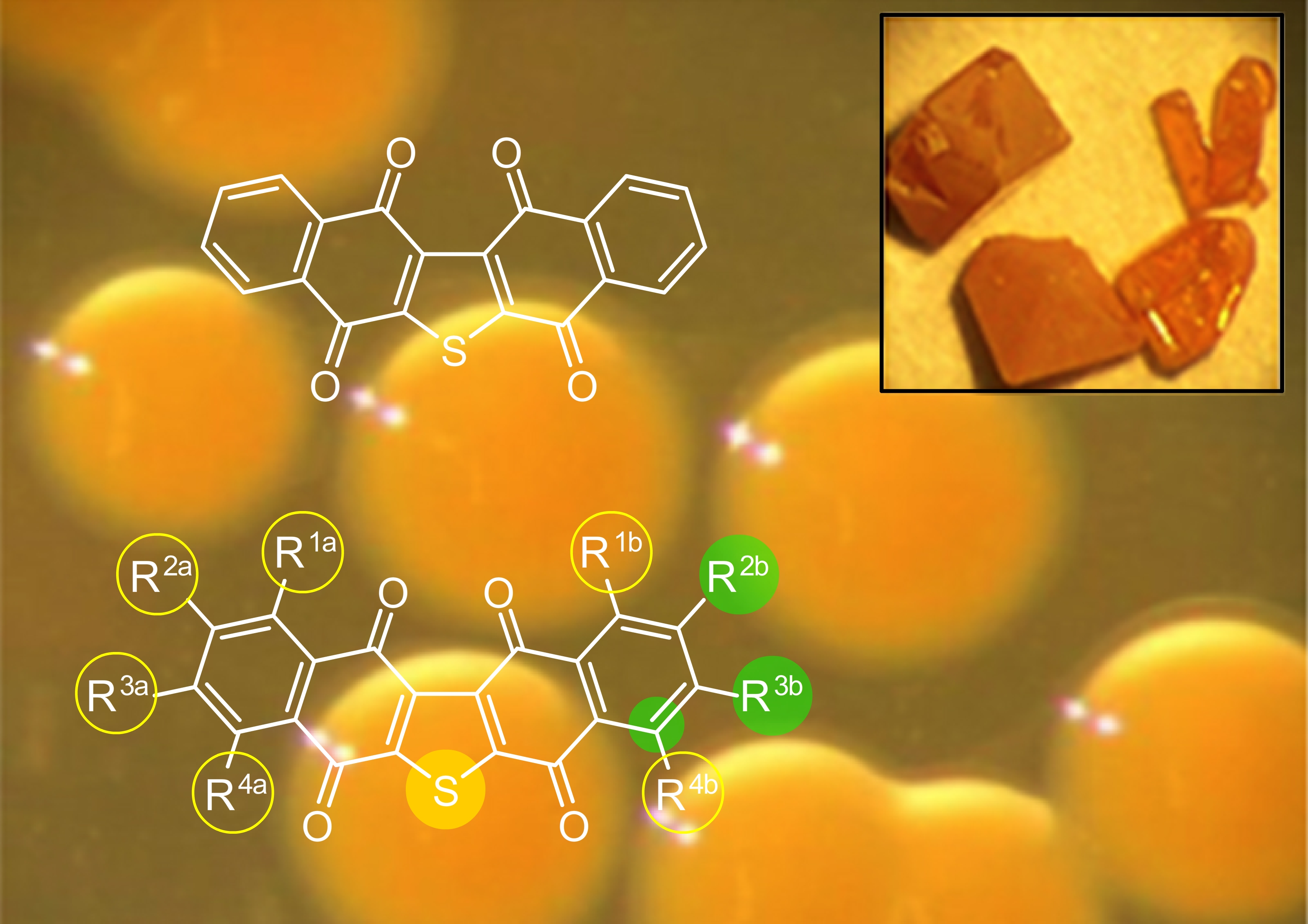{kind=link}
{kind=link}
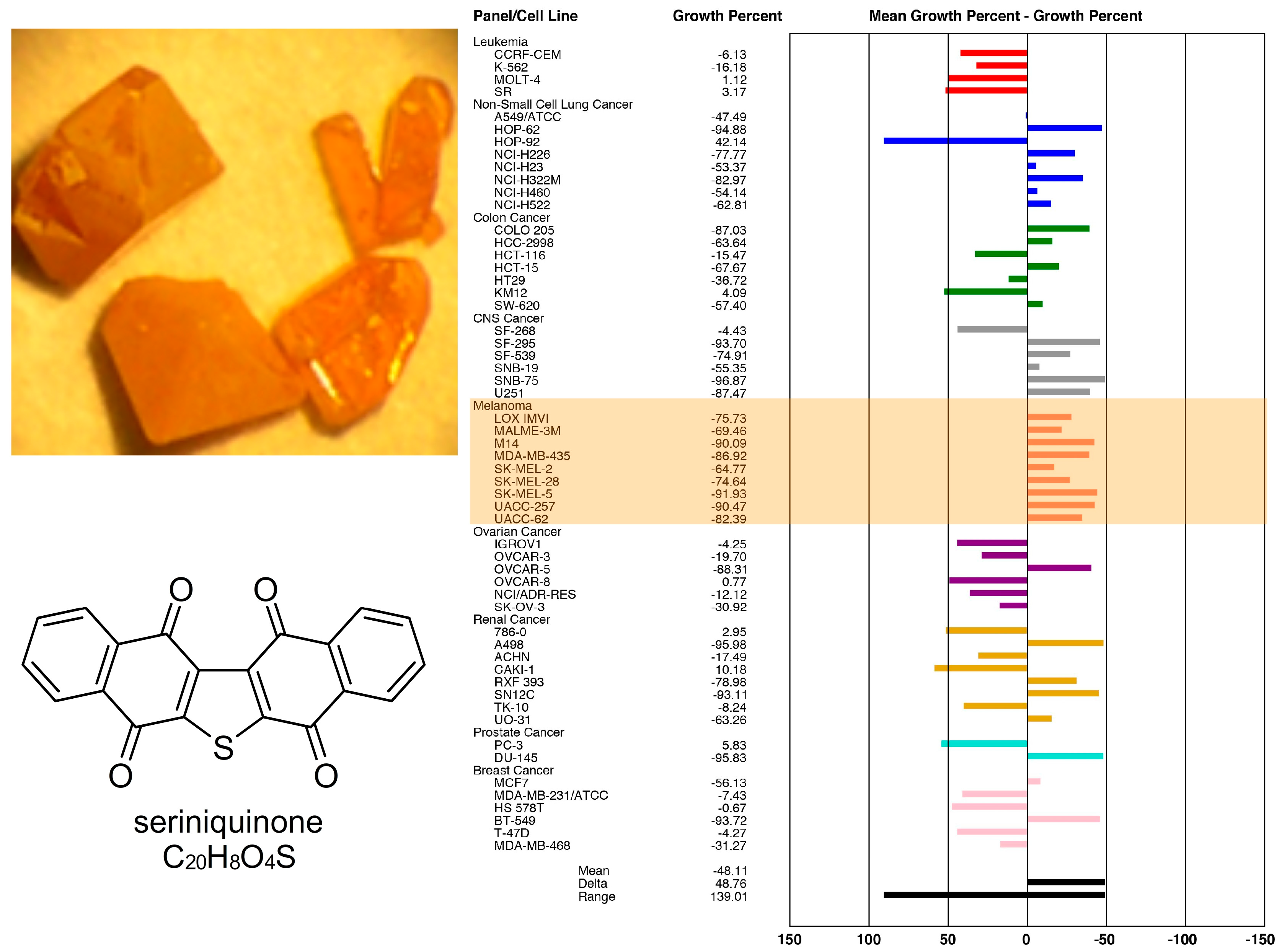{kind=link}
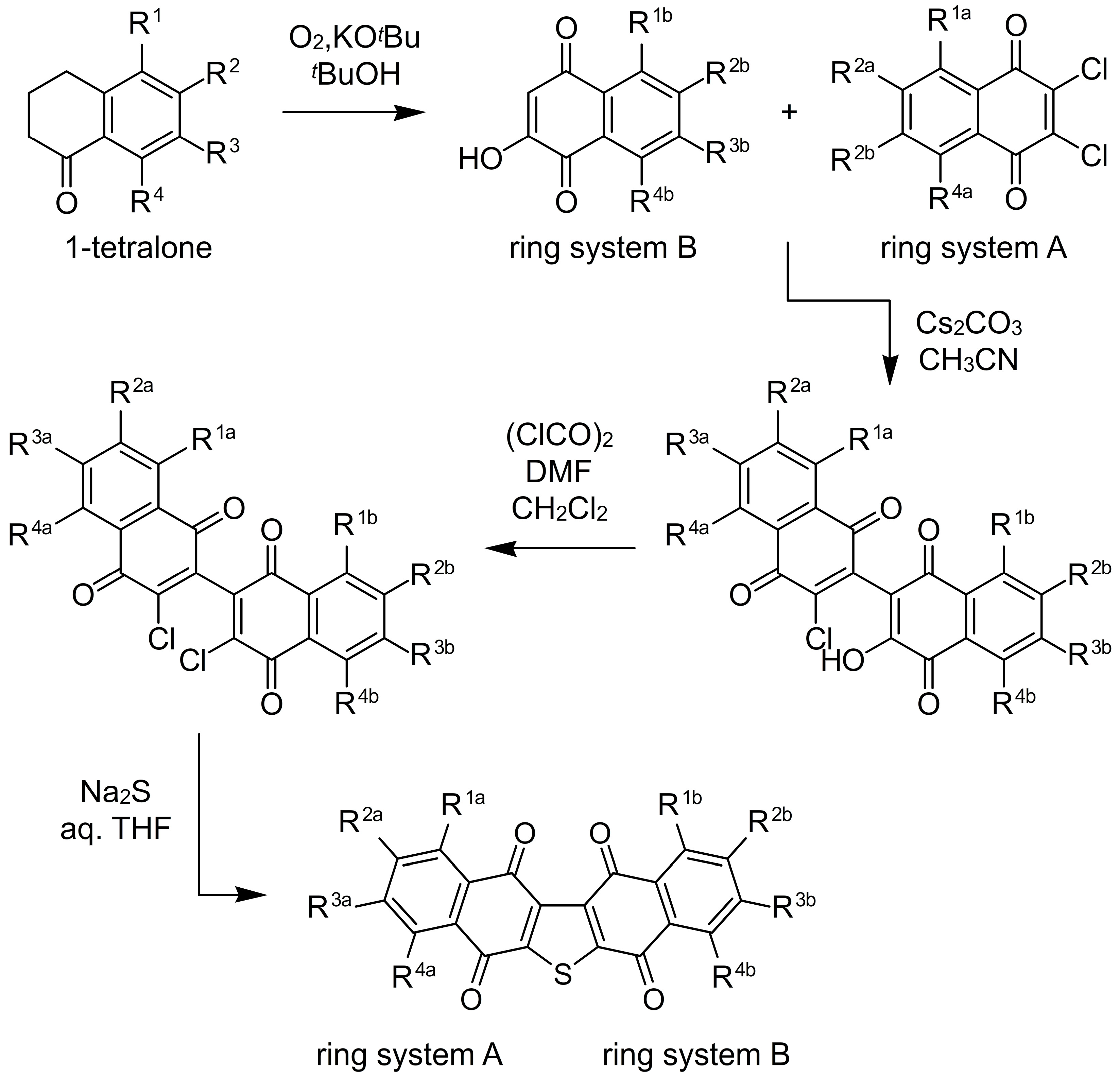{kind=link}
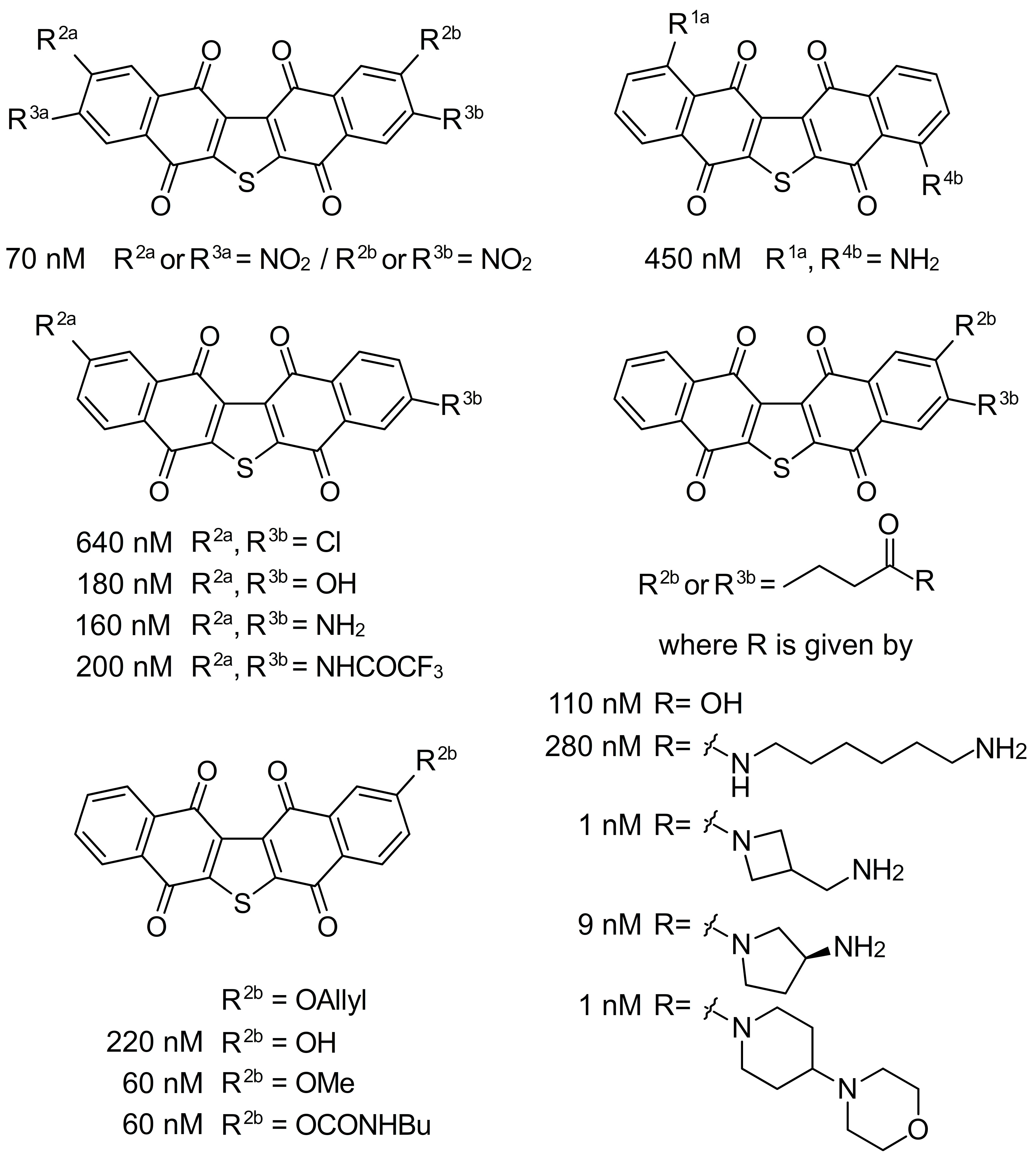{kind=link}
{kind=link}
{kind=link}
{kind=link}
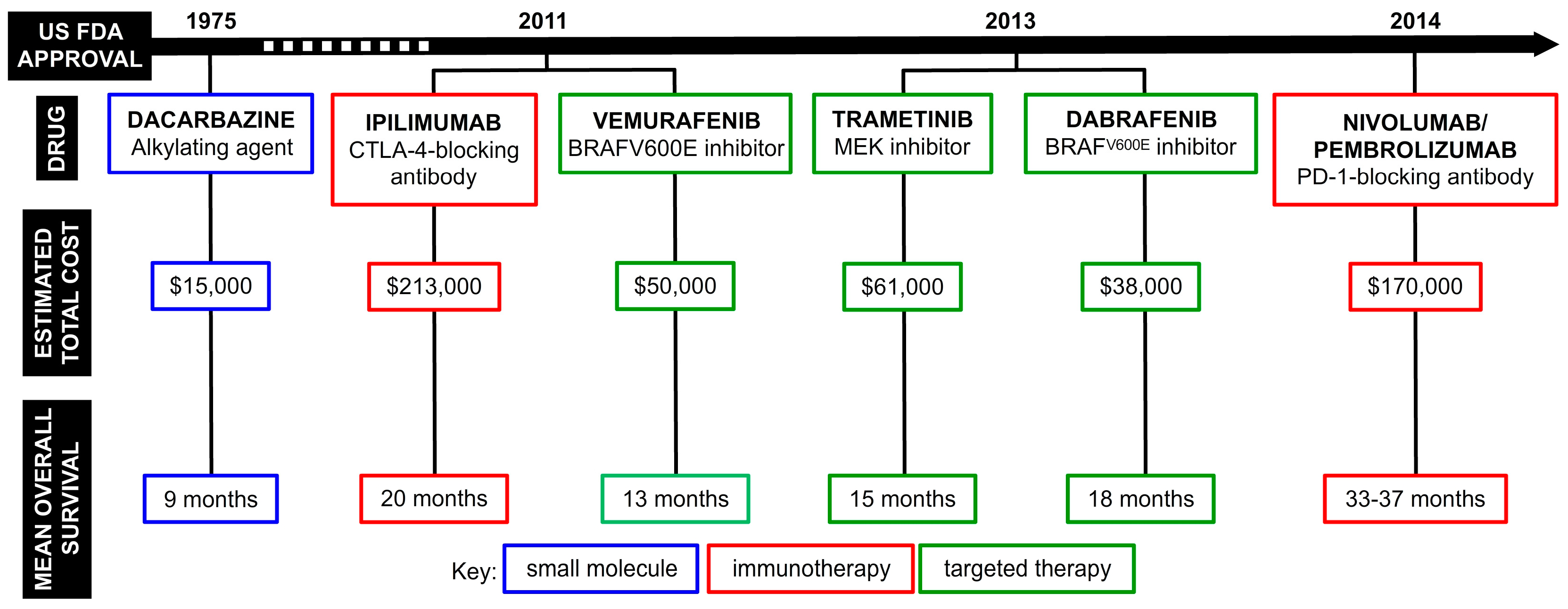
1. The Discovery of Seriniquinone and its Early Bioactivity



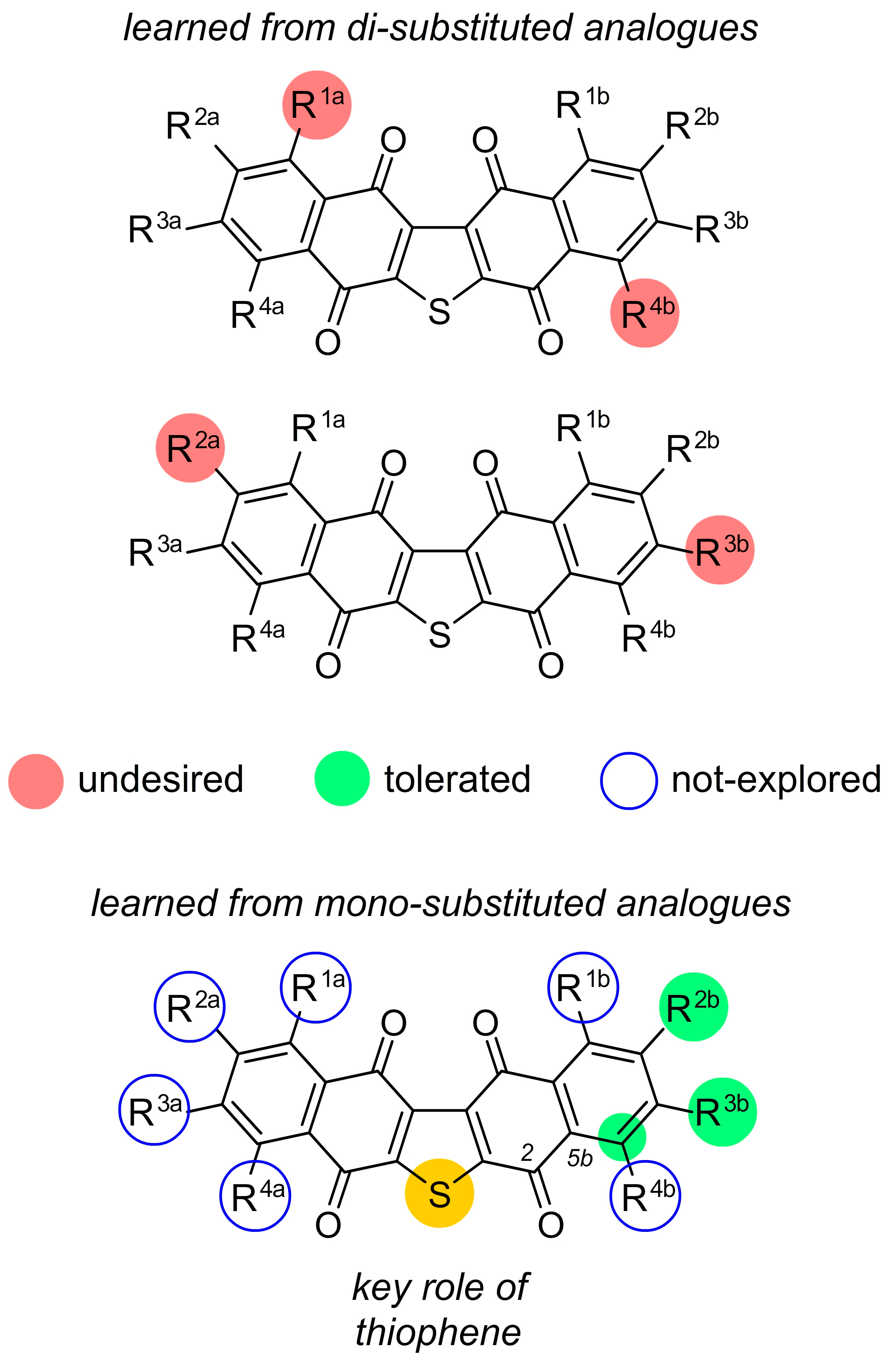
2. Facile Synthetic Access Enables Detailed Structure–Activity Relationship (SAR) Studies


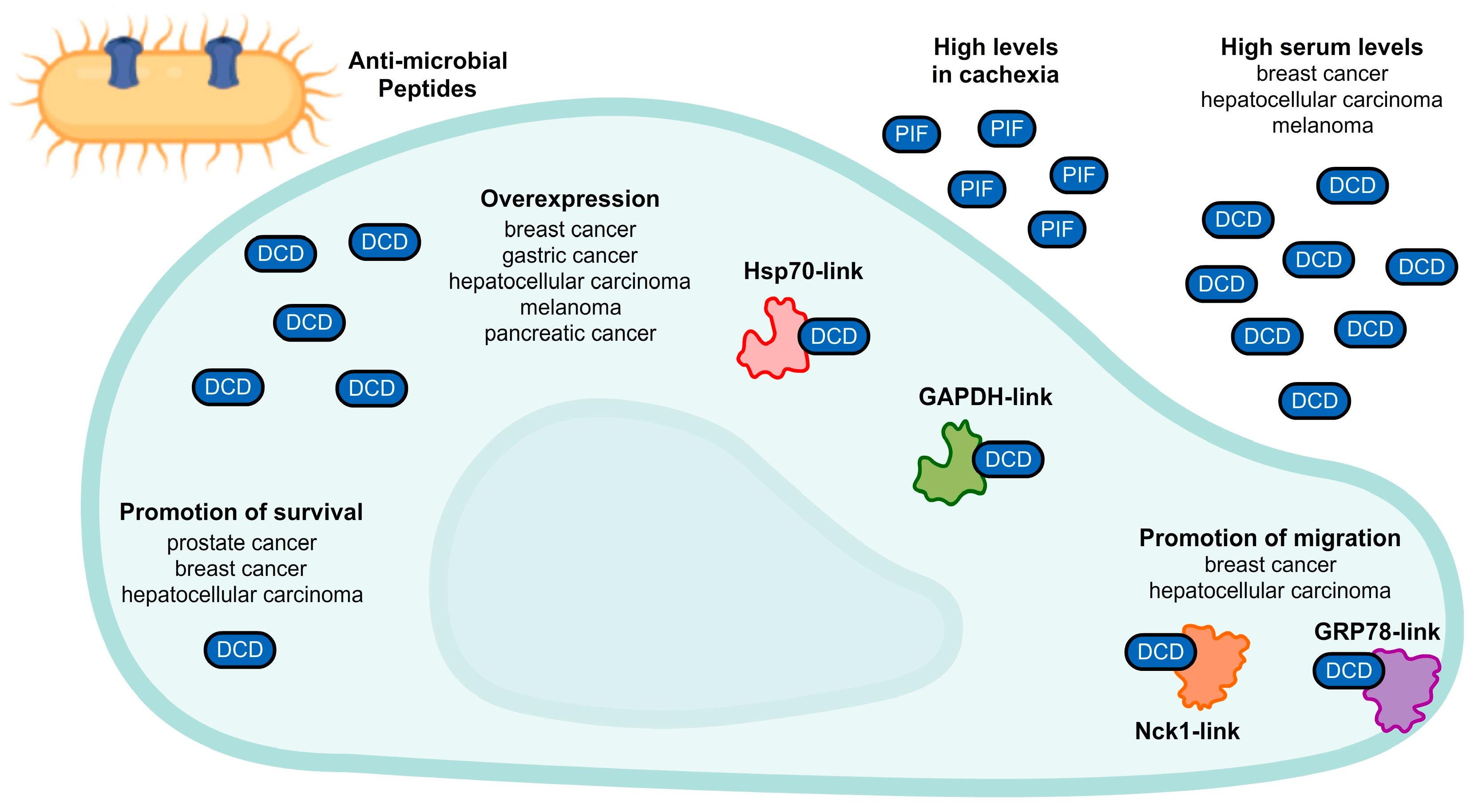
3. A Unique Mode of Action: Dermcidin
4. Complex Pharmacology
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Hammons, J.C.; Trzoss, L.; Jimenez, P.C.; Hirata, A.S.; Costa-Lotufo, L.V.; La Clair, J.J.; Fenical, W. Advance of Seriniquinone Analogues as Melanoma Agents. ACS Med. Chem. Lett. 2019, 10, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Trzoss, L.; Fukuda, T.; Costa-Lotufo, L.V.; Jimenez, P.; La Clair, J.J.; Fenical, W. Seriniquinone, a selective anticancer agent, induces cell death by autophagocytosis, targeting the cancer-protective protein dermcidin. Proc. Natl. Acad. Sci. USA 2014, 111, 14687–14692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, K.; Tanaka, T.; Nagai, K.; Furuichi, Y.; Terahara, T.; Anda, M.; Tsukamasa, Y.; Fukuda, T. New dihydronaphthothiophene derivatives by the biological transformation of seriniquinone using marine-derived actinomycete Streptomyces albogriseolus OM27-12. J. Antibiot. 2022, 65, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.D.; Burkart, M.D.; Leonard, M.S.; Portonovo, P.; Liang, B.; Ding, X.; Joullié, M.M.; Gulledge, B.M.; Aggen, J.B.; Chamberlin, A.R.; et al. A Central Strategy for Converting Natural Products into Fluorescent Probes. ChemBioChem 2006, 7, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.C.; MacMillan, J.B.; Gaudêncio, S.P.; Fenical, W.; La Clair, J.J. Ammosamides A and B Target Myosin. Angew. Chemie Int. Ed. 2009, 48, 728–732. [Google Scholar] [CrossRef] [Green Version]
- IARC. The Global Cancer Observatory (Globocan) 2020 Database. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/16-Melanoma-of-skin-fact-sheet.pdf (accessed on 13 December 2021).
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Collichio, F.; Ollila, D.; Moschos, S. Historical review of melanoma treatment and outcomes. Clin. Dermatol. 2013, 31, 141–147. [Google Scholar] [CrossRef]
- Wagner, D.E.; Ramirez, G.; Weiss, A.J.; Hill, G., Jr. Combination Phase 1–II Study of Imidazole Carboxamide (NCS 45388). Oncology 1972, 26, 310–316. [Google Scholar] [CrossRef]
- Hill, G.J.; Ruess, R.; Berris, R.; Philpott, G.W.; Parkin, P. Chemotherapy of Malignant Melanoma with Dimethyl Triazeno lmidazole Carboxamide (DITC) and Nitrosourea Derivatives (BCN U, CCNU). Ann. Surg. 1974, 180, 167–174. [Google Scholar] [CrossRef]
- Schadendorf, D.; Vaubel, J.; Livingstone, E.; Zimmer, L. Advances and perspectives in immunotherapy of melanoma. Ann. Oncol. 2012, 23, x104–x108. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Mishra, H.; Mishra, P.K.; Ekielski, A.; Jaggi, M.; Iqbal, Z.; Talegaonkar, S. Melanoma treatment: From conventional to nanotechnology. J. Cancer Res. Clin. Oncol. 2018, 144, 2283–2302. [Google Scholar] [CrossRef]
- Curl, P.; Vujic, I.; van ‘t Veer, L.J.; Ortiz-Urda, S.; Kahn, J.G. Cost-Effectiveness of Treatment Strategies for BRAF-Mutated Metastatic Melanoma. PLoS ONE 2014, 9, e107255. [Google Scholar] [CrossRef]
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of metastatic melanoma: An overview. Oncology (Williston Park) 2009, 23, 488–496. [Google Scholar]
- Oh, A.; Tran, D.M.; McDowell, L.C.; Keyvani, D.; Barcelon, J.A.; Merino, O.; Wilson, L. Cost-Effectiveness of Nivolumab-Ipilimumab Combination Therapy Compared with Monotherapy for First-Line Treatment of Metastatic Melanoma in the United States. J. Manag. Care Spec. Pharm. 2017, 23, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- da Rocha Dias, S.; Salmonson, T.; van Zwieten-Boot, B.; Jonsson, B.; Marchetti, S.; Schellens, J.H.M.; Giuliani, R.; Pignatti, F. The European Medicines Agency review of vemurafenib (Zelboraf®) for the treatment of adult patients with BRAF V600 mutation-positive unresectable or metastatic melanoma: Summary of the scientific assessment of the Committee for Medicinal Products for Huma. Eur. J. Cancer 2013, 49, 1654–1661. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef]
- Lugowska, I.; Kosela-Paterczyk, H.; Kozak, K.; Rutkowski, P. Trametinib: A MEK inhibitor for management of metastatic melanoma. Onco. Targets. Ther. 2015, 8, 2251–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, V.; Ten Ham, R.M.; Bui, C.T.; Tran, D.N.; Ting, J.; Wilson, L. Targeted Therapies Compared to Dacarbazine for Treatment of BRAF(V600E) Metastatic Melanoma: A Cost-Effectiveness Analysis. J. Skin Cancer 2015, 2015, 505302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairlie, W.D.; Tran, S.; Lee, E.F. Crosstalk between apoptosis and autophagy signaling pathways. Int. Rev. Cell Mol. Biol. 2020, 352, 115–158. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Song, X.; Yang, Y.; Wan, X.; Alvarez, A.A.; Sastry, N.; Feng, H.; Hu, B.; Cheng, S.-Y. Autophagy and Hallmarks of Cancer. Crit. Rev. Oncog. 2018, 23, 247–267. [Google Scholar] [CrossRef]
- Rahmati, M.; Ebrahim, S.; Hashemi, S.; Motamedi, M.; Moosavi, M.A. New insights on the role of autophagy in the pathogenesis and treatment of melanoma. Mol. Biol. Rep. 2020, 47, 9021–9032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tang, P.; Zou, L.; Zhang, J.; Chen, J.; Yang, C.; He, G.; Liu, B.; Liu, J.; Chiang, C.M.; et al. Discovery of Novel Dual-Target Inhibitor of Bromodomain-Containing Protein 4/Casein Kinase 2 Inducing Apoptosis and Autophagy-Associated Cell Death for Triple-Negative Breast Cancer Therapy. J. Med. Chem. 2021, 64, 18025–18053. [Google Scholar] [CrossRef]
- Biggers, J.W.; Nguyen, T.; Di, X.; Gupton, J.T.; Henderson, S.C.; Emery, S.M.; Alotaibi, M.; White, K.L.; Brown, R.; Almenara, J.; et al. Autophagy, cell death and sustained senescence arrest in B16/F10 melanoma cells and HCT-116 colon carcinoma cells in response to the novel microtubule poison, JG-03-14. Cancer Chemother. Pharmacol. 2013, 71, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, M.; Wang, D.; Li, X.; Wang, W.; Lou, H.; Yuan, H. Malformin A1 promotes cell death through induction of apoptosis, necrosis and autophagy in prostate cancer cells. Cancer Chemother. Pharmacol. 2016, 77, 63–75. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Hirata, A.S.; Rezende-Teixeira, P.; Machado-Neto, J.A.; Jimenez, P.C.; Clair, J.J.L.; Fenical, W.; Costa-Lotufo, L.V. Seriniquinones as Therapeutic Leads for Treatment of BRAF and NRAS Mutant Melanomas. Molecules 2021, 26, 7362. [Google Scholar] [CrossRef]
- Heppt, M.V.; Siepmann, T.; Engel, J.; Schubert-Fritschle, G.; Eckel, R.; Mirlach, L.; Kirchner, T.; Jung, A.; Gesierich, A.; Ruzicka, T.; et al. Prognostic significance of BRAF and NRAS mutations in melanoma: A German study from routine care. BMC Cancer 2017, 17, 536. [Google Scholar] [CrossRef] [PubMed]
- Ryabaya, O.O.; Inshakov, A.N.; Egorova, A.V.; Emelyanova, M.A.; Nasedkina, T.V.; Zasedatelev, A.S.; Khochenkov, D.A.; Stepanova, E.V. Autophagy inhibitors chloroquine and LY294002 enhance temozolomide cytotoxicity on cutaneous melanoma cell lines in vitro. Anticancer. Drugs 2017, 28, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-H.; Piao, S.-F.; Dey, S.; Mcafee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress–induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Invest. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.-S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [Green Version]
- Strohecker, A.M.; White, E. Targeting Mitochondrial Metabolism by Inhibiting Autophagy in BRAF-Driven Cancers. Cancer Discov. 2014, 4, 766–772. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Koh, J.Y.; Price, S.; White, E.; Mehnert, J.M. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov. 2015, 5, 410–423. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.-Y.; Li, J.; Li, F.; Zhu, Y.; Cui, K.; Wong, S.T.; Chang, E.C.; Liao, Y.-H. Induction of N-Ras degradation by flunarizine-mediated autophagy. Sci. Rep. 2018, 8, 16932. [Google Scholar] [CrossRef]
- Bolton, J.L.; Dunlap, T. Formation and Biological Targets of Quinones: Cytotoxic versus Cytoprotective Effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef]
- Lown, J.W. The mechanism of action of quinone antibiotics. Mol. Cell. Biochem. 1983, 55, 17–40. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Moreira da Silva, R.; Carrão, D.B.; Habenschus, M.D.; Jimenez, P.C.; Lopes, N.P.; Fenical, W.; Costa-Lotufo, L.V.; de Oliveira, A.R.M. Prediction of seriniquinone-drug interactions by in vitro inhibition of human cytochrome P450 enzymes. Toxicol. In Vitro 2020, 65, 104820. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, E.; Cagel, M.; Lagomarsino, E.; Moretton, M.; Chiappetta, D.A. Paclitaxel: What has been done and the challenges remain ahead. Int. J. Pharm. 2017, 526, 474–495. [Google Scholar] [CrossRef]
- Apolinário, A.C.; Hirata, A.S.; Anjos Miguel, R.D.; Costa-Lotufo, L.V.; Pessoa, A.; La Clair, J.J.; Fenical, W.; Lopes, L.B. Exploring the benefits of nanotechnology for cancer drugs in different stages of the drug development pipeline. Nanomedicine 2020, 15, 2539–2542. [Google Scholar] [CrossRef] [PubMed]
- Schittek, B.; Hipfel, R.; Sauer, B.; Bauer, J.; Kalbacher, H.; Stevanovic, S.; Schirle, M.; Schroeder, K.; Blin, N.; Meier, F.; et al. Dermcidin: A novel human antibiotic peptide secreted by sweat glands. Nat. Immunol. 2001, 2, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Hodge, L.; Speicher, D.; Reim, D.; Tyler-Polsz, C.; Levitt, P.; Eagleson, K.; Kennedy, S.; Wang, Y. Identification of a Survival-Promoting Peptide in Medium Conditioned by Oxidatively Stressed Cell Lines of Nervous System Origin. J. Neurosci. 1998, 18, 7047–7060. [Google Scholar] [CrossRef] [Green Version]
- Cariuk, P.; Lorite, M.; Todorov, P.; Field, W.; Wigmore, S.; Tisdale, M. Induction of cachexia in mice by a product isolated from the urine of cachectic cancer patients. Br. J. Cancer 1997, 76, 606–613. [Google Scholar] [CrossRef] [Green Version]
- Lorite, M.; Thompson, M.; Drake, J.; Carling, G.; Tisdale, M. Mechanism of muscle protein degradation induced by a cancer cachectic factor. Br. J. Cancer 1998, 78, 850–856. [Google Scholar] [CrossRef]
- Porter, D.; Weremowicz, S.; Chin, K.; Seth, P.; Keshaviah, A.; Lahti-Domenici, J.; Bae, Y.K.; Monitto, C.L.; Merlos-Suarez, A.; Chan, J.; et al. A neural survival factor is a candidate oncogene in breast cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 10931–10936. [Google Scholar] [CrossRef] [Green Version]
- Brauer, H.A.; D’Arcy, M.; Libby, T.E.; Thompson, H.J.; Yasui, Y.Y.; Hamajima, N.; Li, C.I.; Troester, M.A.; Lampe, P.D. Dermcidin expression is associated with disease progression and survival among breast cancer patients. Breast Cancer Res. Treat. 2014, 144, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deans, D.A.C.; Wigmore, S.J.; Gilmour, H.; Tisdale, M.J.; Fearon, K.C.H.; Ross, J.A. Expression of the proteolysis-inducing factor core peptide mRNA is upregulated in both tumour and adjacent normal tissue in gastro-oesophageal malignancy. Br. J. Cancer 2006, 94, 731–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ding, W.; Kuai, X.; Ji, Y.; Zhu, Z.; Mao, Z.; Wang, Z. Dermcidin as a novel binding protein of lncRNA STCAT3 and its effect on prognosis in gastric cancer. Oncol. Rep. 2018, 40, 2854–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.-L.; Qiu, F.-H.; Dayarathna, T.K.; Wu, J.; Kuang, M.; Li, S.S.C.; Peng, B.-G.; Nie, J. Identification of Dermcidin as a novel binding protein of Nck1 and characterization of its role in promoting cell migration. Biochim. Biophys. Acta 2011, 1812, 703–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, F.; Qiu, F.; Liu, L.; Liu, J.; Xu, J.; Huang, X. The Role of Dermcidin in the Diagnosis and Staging of Hepatocellular Carcinoma. Genet. Test. Mol. Biomark. 2018, 22, 218–223. [Google Scholar] [CrossRef] [Green Version]
- López-Sánchez, L.M.; Jurado-Gámez, B.; Feu-Collado, N.; Valverde, A.; Cañas, A.; Fernández-Rueda, J.L.; Aranda, E.; Rodríguez-Ariza, A. Exhaled breath condensate biomarkers for the early diagnosis of lung cancer using proteomics. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L664–L676. [Google Scholar] [CrossRef] [Green Version]
- Núñez-Naveira, L.; Mariñas-Pardo, L.A.; Montero-Martínez, C. Mass Spectrometry Analysis of the Exhaled Breath Condensate and Proposal of Dermcidin and S100A9 as Possible Markers for Lung Cancer Prognosis. Lung 2019, 197, 523–531. [Google Scholar] [CrossRef]
- Ortega-Martínez, I.; Gardeazabal, J.; Erramuzpe, A.; Sanchez-Diez, A.; Cortés, J.; García-Vázquez, M.D.; Pérez-Yarza, G.; Izu, R.; Luís Díaz-Ramón, J.; de la Fuente, I.M.; et al. Vitronectin and dermcidin serum levels predict the metastatic progression of AJCC I–II early-stage melanoma. Int. J. Cancer 2016, 139, 1598–1607. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, F.; Lage, S.; Rasero, J.; Díaz-Ramón, J.L.; Apraiz, A.; Pérez-Yarza, G.; Ezkurra, P.A.; Penas, C.; Sánchez-Diez, A.; García-Vazquez, M.D.; et al. Serum markers improve current prediction of metastasis development in early-stage melanoma patients: A machine learning-based study. Mol. Oncol. 2020, 14, 1705–1718. [Google Scholar] [CrossRef]
- Stewart, G.D.; Skipworth, R.J.E.; Pennington, C.J.; Lowrie, A.G.; Deans, D.A.C.; Edwards, D.R.; Habib, F.K.; Riddick, A.C.P.; Fearon, K.C.H.; Ross, J.A. Variation in dermcidin expression in a range of primary human tumours and in hypoxic/oxidatively stressed human cell lines. Br. J. Cancer 2008, 99, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.D.; Lowrie, A.G.; Riddick, A.C.P.; Fearon, K.C.H.; Habib, F.K.; Ross, J.A. Dermcidin expression confers a survival advantage in prostate cancer cells subjected to oxidative stress or hypoxia. Prostate 2007, 67, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Bancovik, J.; Moreira, D.F.; Carrasco, D.; Yao, J.; Porter, D.; Moura, R.; Camargo, A.; Fontes-Oliveira, C.C.; Malpartida, M.G.; Carambula, S.; et al. Dermcidin exerts its oncogenic effects in breast cancer via modulation of ERBB signaling. BMC Cancer 2015, 15, 70. [Google Scholar] [CrossRef] [PubMed]
- Lowrie, A.G.; Wigmore, S.J.; Wright, D.J.; Waddell, I.D.; Ross, J.A. Dermcidin expression in hepatic cells improves survival without N-glycosylation, but requires asparagine residues. Br. J. Cancer 2006, 94, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Lager, T.W.; Conner, C.; Keating, C.R.; Warshaw, J.N.; Panopoulos, A.D. Cell surface GRP78 and Dermcidin cooperate to regulate breast cancer cell migration through Wnt signaling. Oncogene 2021, 40, 4050–4059. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirata, A.S.; La Clair, J.J.; Jimenez, P.C.; Costa-Lotufo, L.V.; Fenical, W. Preclinical Development of Seriniquinones as Selective Dermcidin Modulators for the Treatment of Melanoma. Mar. Drugs 2022, 20, 301. https://doi.org/10.3390/md20050301
Hirata AS, La Clair JJ, Jimenez PC, Costa-Lotufo LV, Fenical W. Preclinical Development of Seriniquinones as Selective Dermcidin Modulators for the Treatment of Melanoma. Marine Drugs. 2022; 20(5):301. https://doi.org/10.3390/md20050301
Chicago/Turabian StyleHirata, Amanda S., James J. La Clair, Paula C. Jimenez, Leticia Veras Costa-Lotufo, and William Fenical. 2022. "Preclinical Development of Seriniquinones as Selective Dermcidin Modulators for the Treatment of Melanoma" Marine Drugs 20, no. 5: 301. https://doi.org/10.3390/md20050301