
Biological Activity and Stability of Aeruginosamides from Cyanobacteria

 ,
,  , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
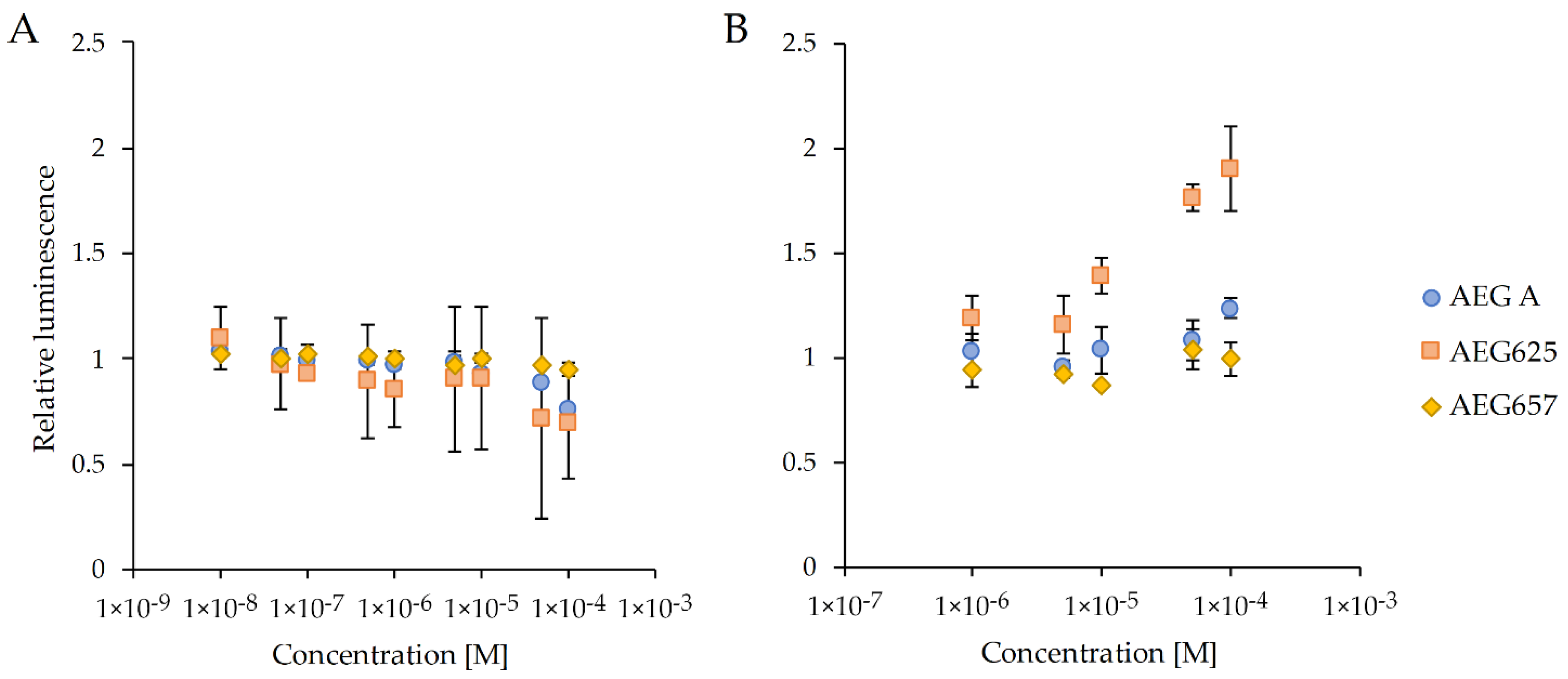
2.1. Biological Activity
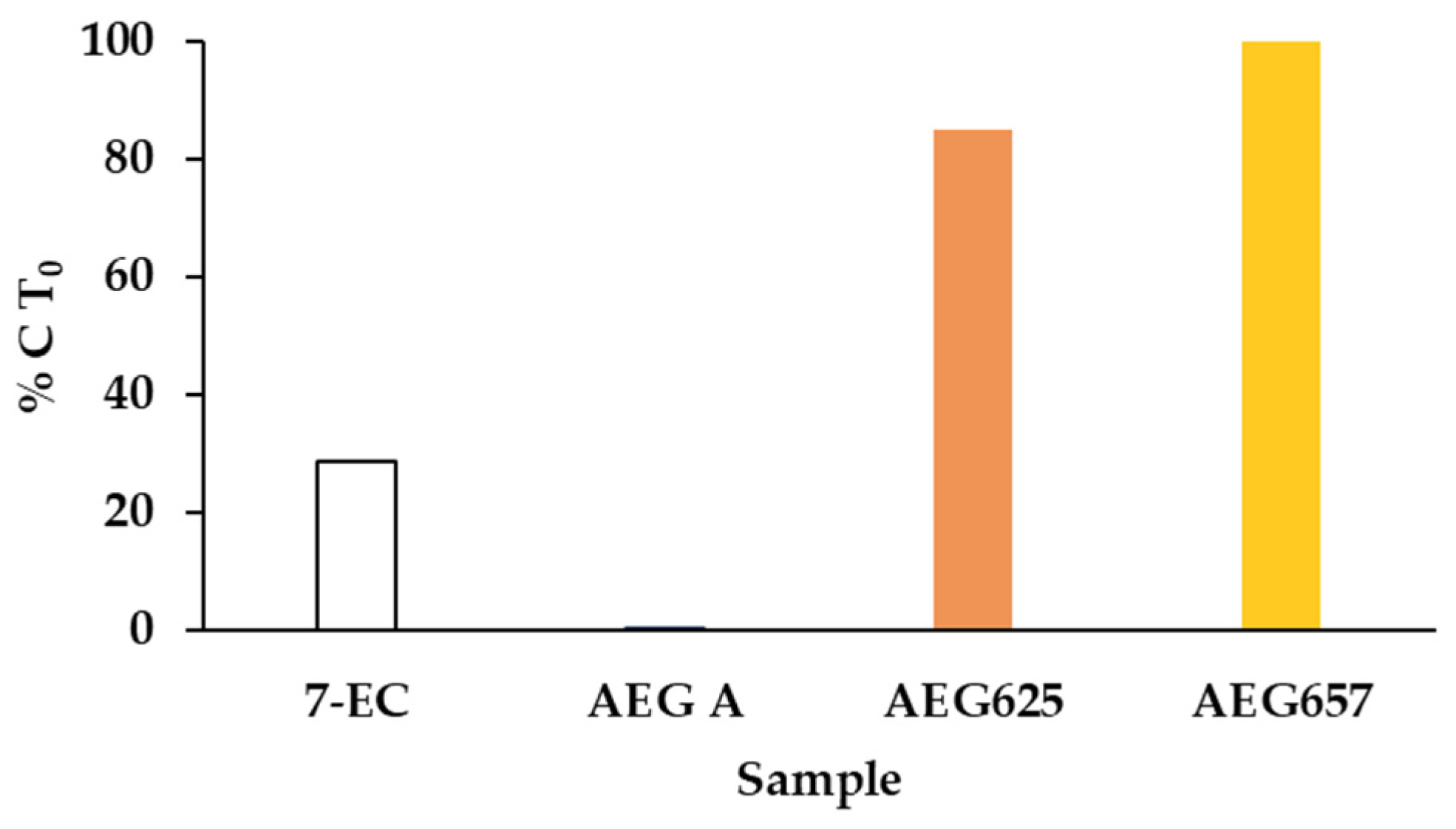
2.2. Metabolic Stability in S9 Fraction
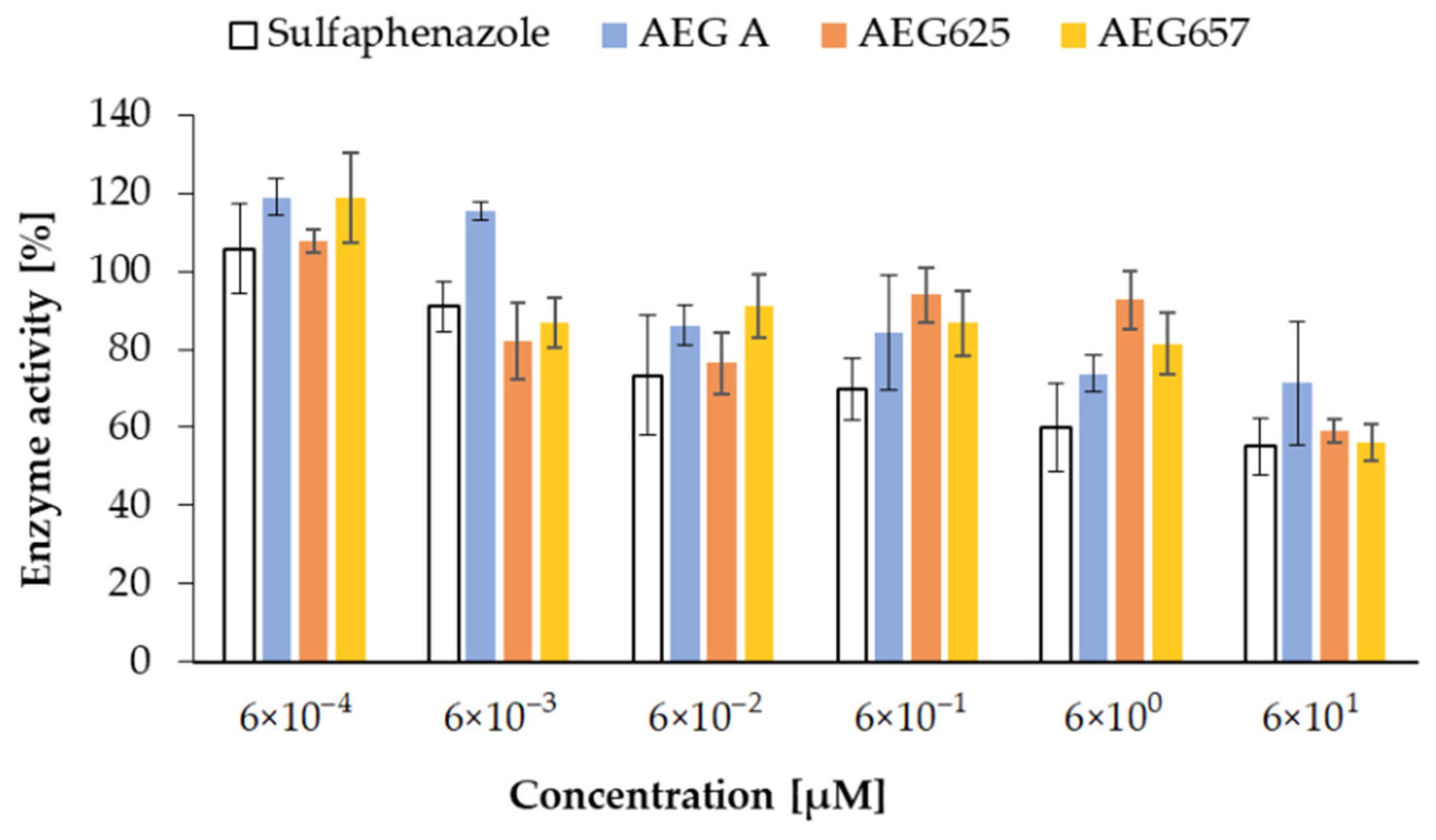
2.3. Inhibition of Cytochrome P450 Enzymes
3. Discussion
4. Materials and Methods
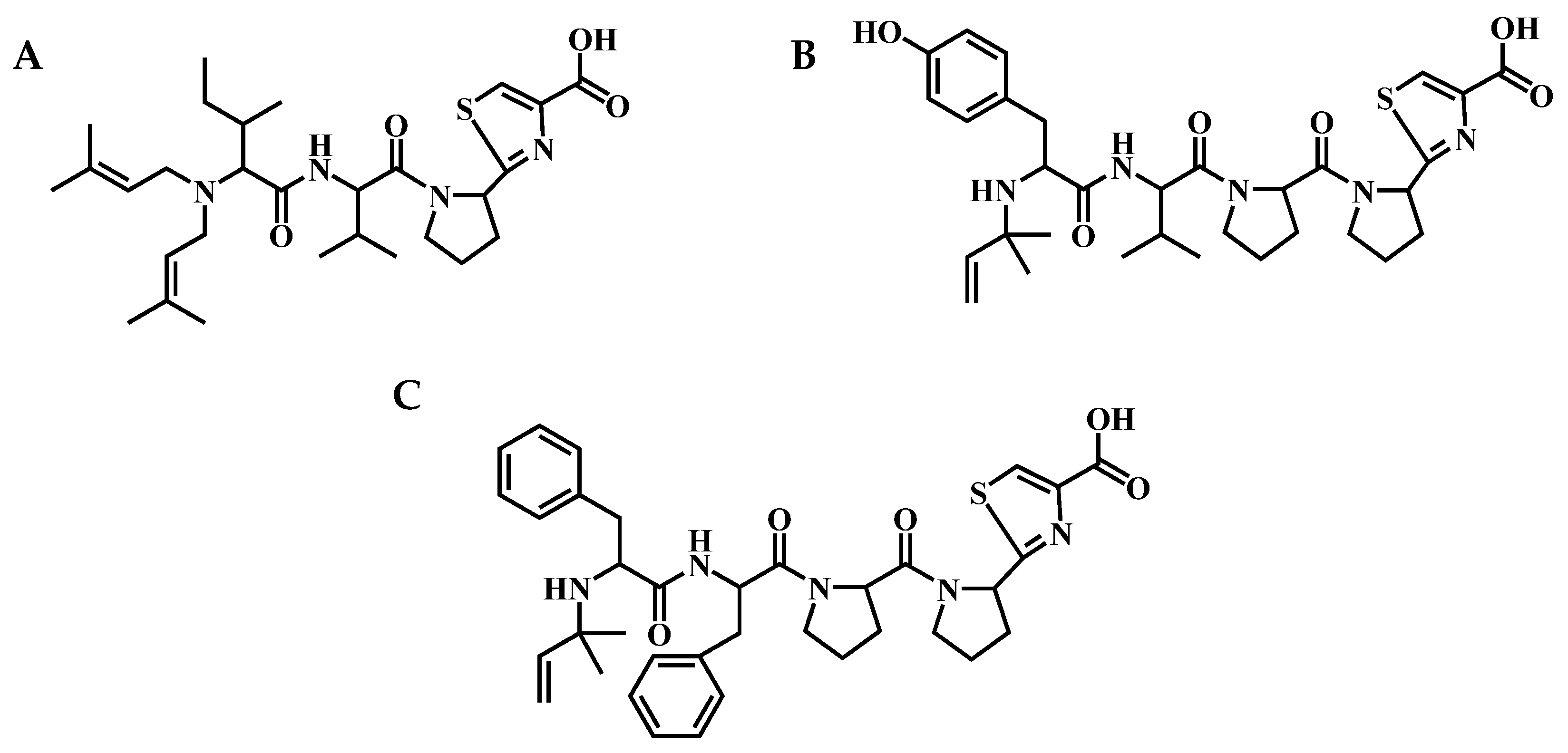
4.1. Extraction and Isolation of Aeruginosamides
4.2. Luciferase Reporter Assays
4.3. MTT Assay
4.4. In Vitro S9 Stability Assay
4.5. Cytochrome P450 Enzyme Inhibition Assays
4.6. LC-MS/MS Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bajpai, V.K.; Shukla, S.; Kang, S.-M.; Hwang, S.K.; Song, X.; Huh, Y.S.; Han, Y.-K. Developments of cyanobacteria for nano-marine drugs: Relevance of nanoformulations in cancer therapies. Mar. Drugs 2018, 16, 179. [Google Scholar] [CrossRef] [Green Version]
- Demay, J.; Bernard, C.; Reinhardt, A.; Marie, B. Natural products from cyanobacteria: Focus on beneficial activities. Mar. Drugs 2019, 17, 320. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, A.K.; Tiwari, B.S. Cyanotherapeutics: An emerging field for future drug discovery. Appl Phyco. 2020, 1, 1–16. [Google Scholar] [CrossRef]
- Khalifa, S.A.M.; Shedid, E.S.; Saied, E.M.; Jassbi, A.R.; Jamebozorgi, F.H.; Rateb, M.E.; Du, M.; Abdel-Daim, M.M.; Kai, G.-Y.; Al-Hammady, M.A.M.; et al. Cyanobacteria-from the oceans to the potential biotechnological and biomedical applications. Mar. Drugs 2021, 19, 241. [Google Scholar] [CrossRef]
- Luesch, H.; Moore, R.E.; Paul, V.K.; Mooberry, S.L.; Corbett, T.H. Isolation of dolastatin 10 from the marine cyanobacterium Symploca species VP642 and total stereochemistry and biological evaluation of its analogue symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Hong, J.; Luesch, H. Largazole: From discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012, 29, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Al-Awadhi, F.H.; Salvador-Reyes, L.A.; Elsadek, L.A.; Ratnayake, R.; Chen, Q.-Y.; Luesch, H. Largazole is a brain-penetrant class I HDAC inhibitor with extended applicability to glioblastoma and CNS diseases. ACS Chem. Neurosci. 2020, 11, 1937–1943. [Google Scholar] [CrossRef] [PubMed]
- Dey, B.; Lerner, D.L.; Lusso, P.; Boyd, M.R.; Elder, J.H.; Berger, E.A. Multiple antiviral activities of cyanovirin-N: Blocking of human immunodeficiency virus type 1 gp120 interaction with CD4 and coreceptor and inhibition of diverse enveloped viruses. J. Virol. 2000, 74, 4562–4569. [Google Scholar] [CrossRef]
- Huskens, D.; Férir, G.; Vermeire, K.; Kehr, J.-C.; Balzarini, J.; Dittmann, E.; Schols, D. Microvirin, a novel α(1,2)-mannose-specific lectin isolated from Microcystis aeruginosa, has anti-HIV-1 activity comparable with that of cyanovirin-N but a much higher safety profile. J. Biol. Chem. 2010, 285, 24845–24854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A. Early probe and drug discovery in academia: A minireview. High-Throughput 2018, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Leikoski, N.; Liu, L.; Jokela, J.; Wahlsten, M.; Gugger, M.; Calteau, A.; Permi, P.; Kerfeld, C.A.; Sivonen, K.; Fewer, D.P. Genome mining expands the chemical diversity of the cyanobactin family to include highly modified linear peptides. Chem. Biol. 2013, 20, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Dong, S.-H.; Sarkar, S.; Nair, S.K.; Schmidt, E.W. The biochemistry and structural biology of cyanobactin pathways: Enabling combinatorial biosynthesis. Methods Enzymol. 2018, 604, 113–163. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Donia, M.S.; Schmidt, E.W. Insights into heterocyclization from two highly similar enzymes. J. Am. Chem. Soc. 2010, 132, 4089–4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Czekster, C.M.; Miller, O.K.; Botting, C.H.; Schwarz-Linek, U.; Naismith, J.H. Insights into the mechanism of the cyanobactin heterocyclase enzyme. Biochemistry 2019, 58, 2125–2132. [Google Scholar] [CrossRef]
- McIntosh, J.A.; Donia, M.S.; Nair, S.K.; Schmid, E.W. Enzymatic basis of ribosomal peptide prenylation in cyanobacteria. J. Am. Chem. Soc. 2011, 133, 13698–13705. [Google Scholar] [CrossRef] [Green Version]
- Donia, M.D.; Schmidt, E.W. Linking chemistry and genetics in the growing cyanobactin natural products family. Chem. Biol. 2011, 18, 508–519. [Google Scholar] [CrossRef] [Green Version]
- Phan, C.-S.; Matsuda, K.; Balloo, N.; Fujita, K.; Wakimoto, T.; Okino, T. Argicyclamides A-C unveil enzymatic basis for guanidine bis-prenylation. J. Am. Chem. Soc. 2021, 143, 10083–10087. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, M.; Sarkar, S.; Morita, M.; Gugger, M.; Schmidt, E.W.; Morinaka, B.I. Genome-mining-based discovery of the cyclic peptide tolypamide and TolF, a Ser/Thr forward O-prenyltransferase. Angew. Chem. Int. Ed. Engl. 2021, 60, 8460–8465. [Google Scholar] [CrossRef]
- Martins, J.; Vasconcelos, V. Cyanobactins from cyanobacteria: Current genetic and chemical state of knowledge. Mar. Drugs 2015, 13, 6910–6946. [Google Scholar] [CrossRef] [Green Version]
- Lawton, L.A.; Morris, L.A.; Jaspars, M. A bioactive modified peptide, aeruginosamide, isolated from the cyanobacterium Microcystis aeruginosa. J. Org. Chem. 1999, 64, 5329–5332. [Google Scholar] [CrossRef]
- Cegłowska, M.; Szubert, K.; Wieczerzak, E.; Kosakowska, A.; Mazur-Marzec, H. Eighteen new aeruginosamide variants produced by the Baltic cyanobacterium Limnoraphis CCNP1324. Mar. Drugs 2020, 18, 446. [Google Scholar] [CrossRef]
- Chena, Z.; Ye, T. The first total synthesis of aeruginosamide. New J. Chem. 2006, 30, 518–520. [Google Scholar] [CrossRef]
- Huang, I.-S.; Zimba, P.V. Cyanobacterial bioactive metabolites-a review of their chemistry and biology. Harmful Algae 2019, 83, 42–94. [Google Scholar] [CrossRef]
- Jones, M.R.; Pinto, E.; Torres, M.A.; Dörr, F.; Mazur-Marzec, H.; Szubert, K.; Tartaglione, L.; Dell’Aversano, C.; Miles, C.O.; Beach, D.G.; et al. CyanoMetDB, a comprehensive public database of secondary metabolites from cyanobacteria. Water Res. 2021, 196, 117017. [Google Scholar] [CrossRef] [PubMed]
- Wätjen, W.; Weber, N.; Lou, Y.-J.; Wang, Z.-Q.; Chovolou, Y.; Kampkötter, A.; Kahl, R.; Proksch, P. Prenylation enhances cytotoxicity of apigenin and liquiritigenin in rat H4IIE hepatoma and C6 glioma cells. Food Chem. Toxicol. 2007, 45, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mukwaya, E.; Wong, M.-S.; Zhang, Y. A systematic review on biological activities of prenylated flavonoids. Pharm. Biol. 2014, 52, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Tianero, Ma.D.; Pierce, E.; Raghuraman, S.; Sardar, D.; McIntosh, J.A.; Heemstra, J.R.; Schonrock, Z.; Covington, B.C.; Maschek, J.A.; Cox, J.E.; et al. Metabolic model for diversity-generating biosynthesis. Proc. Natl. Acad. Sci. USA 2016, 113, 1772–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offerman, S.C.; Kadirvel, M.; Abusara, O.H.; Bryant, J.L.; Telfer, B.A.; Brown, G.; Freeman, S.; White, A.; Williams, K.J.; Aojula, H.S. N-tert-prenylation of the indole ring improves the cytotoxicity of a short antagonist G analogue against small cell lung cancer. Medchemcomm 2017, 8, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Magee, A.I.; Seabra, M.C. Are prenyl groups on proteins sticky fingers or greasy handles? Biochem. J. 2003, 376, e3–e4. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.; Costa-Rodrigues, J.; Fernandes, M.H.; Barros, P.; Vasconcelos, V.; Martins, R. Marine cyanobacteria compounds with anticancer properties: A review on the implication of apoptosis. Mar. Drugs 2012, 10, 2181–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador-Reyes, L.A.; Luesch, H. Biological targets and mechanisms of action of natural products from marine cyanobacteria. Nat. Prod. Rep. 2015, 32, 478–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacFarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, function and role in cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.F. Drug target miRNAs: Chances and challenges. Trends Biotechnol. 2014, 32, 578–585. [Google Scholar] [CrossRef]
- Annese, T.; Tamma, R.; De Giorgis, M.; Ribatti, D. microRNAs biogenesis, functions and role in tumor angiogenesis. Front. Oncol. 2020, 10, 581007. [Google Scholar] [CrossRef]
- Brzuzan, P.; Mazur-Marzec, H.; Florczyk, M.; Stefaniak, F.; Fidor, A.; Konkel, R.; Woźny, M. Luciferase reporter assay for small-molecule inhibitors of MIR92b-3p function: Screening cyanopeptolins produced by Nostoc from the Baltic Sea. Toxicol. Vitr. 2020, 68, 104951. [Google Scholar] [CrossRef]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-induced oxidative stress and toxicity. J. Toxicol. 2012, 2012, 645460. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, C.; Sueyoshi, Y.; Ema, M.; Mori, Y.; Takaishi, K.; Hisatomi, H. Induction of oxidative stress by anticancer drugs in the presence and absence of cells. Oncol. Lett. 2017, 14, 6066–6070. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-inducing strategy in anticancer therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Alharbi, Y.; Kapur, A.; Felder, M.; Barroilhet, L.; Pattnaik, B.R.; Patankar, M.S. Oxidative stress induced by the anti-cancer agents, plumbagin, and atovaquone, inhibits ion transport through Na+/K+-ATPase. Sci. Rep. 2020, 10, 19585. [Google Scholar] [CrossRef]
- Pritchard, J.F.; Jurima-Romet, M.; Reimer, M.L.J.; Mortimer, E.; Rolfe, B.; Cayen, M.N. Making better drugs: Decision gates in non-clinical drug development. Nat. Rev. Drug Discov. 2003, 2, 542–553. [Google Scholar] [CrossRef]
- Richardson, S.J.; Bai, A.; Kulkarni, A.A.; Moghaddam, M.F. Efficiency in drug discovery: Liver S9 fraction assay as a screen for metabolic stability. Drug Metab. Lett. 2016, 10, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Markert, Y.; Köditz, J.; Ulbrich-Hofmann, R.; Arnold, U. Proline versus charge concept for protein stabilization against proteolytic attack. Protein Eng. 2003, 16, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Trubetskoy, O.V.; Gibson, J.R.; Marks, B.D. Highly miniaturized formats for in vitro drug metabolism assays using vivid fluorescent substrates and recombinant human cytochrome P450 enzymes. J. Biomol. Screen. 2005, 10, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Esteves, F.; Rueff, J.; Kranendonk, M. The central role of cytochrome P450 in xenobiotic metabolism-a brief review on a fascinating enzyme family. J. Xenobiot. 2021, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. A history of the roles of cytochrome P450 enzymes in the toxicity of drugs. Toxicol. Res. 2020, 37, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Khojasteh, S.C.; Prabhu, S.; Kenny, J.R.; Halladay, J.S.; Lu, A.Y.H. Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: A re-evaluation of P450 isoform selectivity. Eur. J. Drug Metab. Pharmacokinet. 2011, 36, 1–16. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M.S. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Wang, B.; Zhou, S.-F. Synthetic and natural compounds that interact with human cytochrome P450 1A2 and implications in drug development. Curr. Med. Chem. 2009, 16, 4066–4218. [Google Scholar] [CrossRef]
- Li, M.; Chen, P.-Z.; Yue, Q.-X.; Li, J.-Q.; Chu, R.-A.; Zhang, W.; Wang, H. Pungent ginger components modulates human cytochrome P450 enzymes in vitro. Acta Pharmacol. Sin. 2013, 34, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Liu, D.; Yang, Y. Anti-cancer peptides: Classification, mechanism of action, reconstruction and modification. Open Biol. 2020, 10, 200004. [Google Scholar] [CrossRef] [PubMed]
- Chiangjong, W.; Chutipongtanate, S.; Hongeng, S. Anticancer peptide: Physicochemical property, functional aspect and trend in clinical application. Int. J. Oncol. 2020, 57, 678–696. [Google Scholar] [CrossRef] [PubMed]
- Roxin, Á.; Zheng, G. Flexible or fixed: A comparative review of linear and cyclic cancer-targeting peptides. Future Med. Chem. 2012, 4, 1601–1618. [Google Scholar] [CrossRef]
- Shnyder, S.D.; Cooper, P.A.; Millington, N.J.; Pettit, G.R.; Bibby, M.C. Auristatin PYE, a novel synthetic derivative of dolastatin 10, is highly effective in human colon tumour models. Int. J. Oncol. 2007, 31, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Maderna, A.; Leverett, C.A. Recent advances in the development of new auristatins: Structural modifications and application in antibody drug conjugates. Mol. Pharm. 2015, 12, 1798–1812. [Google Scholar] [CrossRef]
- Schrezenmeier, E.; Zollmann, F.S.; Seidel, K.; Böhm, C.; Schmerbach, K.; Kroh, M.; Kirsch, S.; Klare, S.; Bernhard, S.; Kappert, K.; et al. Moderate correlations of in vitro versus in vivo pharmacokinetics questioning the need of early microsomal stability testing. Pharmacology 2012, 90, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Felczykowska, A.; Pawlik, A.; Mazur-Marzec, H.; Toruńska-Sitarz, A.; Narajczyk, M.; Richert, M.; Węgrzyn, G.; Herman-Antosiewicz, A. Selective inhibition of cancer cells’ proliferation by compounds included in extracts from Baltic Sea cyanobacteria. Toxicon 2015, 108, 1–10. [Google Scholar] [CrossRef]
- Wen, X.; Walle, T. Methylation protects dietary flavonoids from rapid hepatic metabolism. Xenobiotica 2006, 36, 387–397. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cegłowska, M.; Kwiecień, P.; Szubert, K.; Brzuzan, P.; Florczyk, M.; Edwards, C.; Kosakowska, A.; Mazur-Marzec, H. Biological Activity and Stability of Aeruginosamides from Cyanobacteria. Mar. Drugs 2022, 20, 93. https://doi.org/10.3390/md20020093
Cegłowska M, Kwiecień P, Szubert K, Brzuzan P, Florczyk M, Edwards C, Kosakowska A, Mazur-Marzec H. Biological Activity and Stability of Aeruginosamides from Cyanobacteria. Marine Drugs. 2022; 20(2):93. https://doi.org/10.3390/md20020093
Chicago/Turabian StyleCegłowska, Marta, Patrycja Kwiecień, Karolina Szubert, Paweł Brzuzan, Maciej Florczyk, Christine Edwards, Alicja Kosakowska, and Hanna Mazur-Marzec. 2022. "Biological Activity and Stability of Aeruginosamides from Cyanobacteria" Marine Drugs 20, no. 2: 93. https://doi.org/10.3390/md20020093