
Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, ADMET, and Molecular Dynamics (MD) Simulation of Potential Inhibitors of PD-L1 from the Library of Marine Natural Products


Abstract
:1. Introduction
2. Results
2.1. Structure-Based Pharmacophore Modeling and Virtual Screening
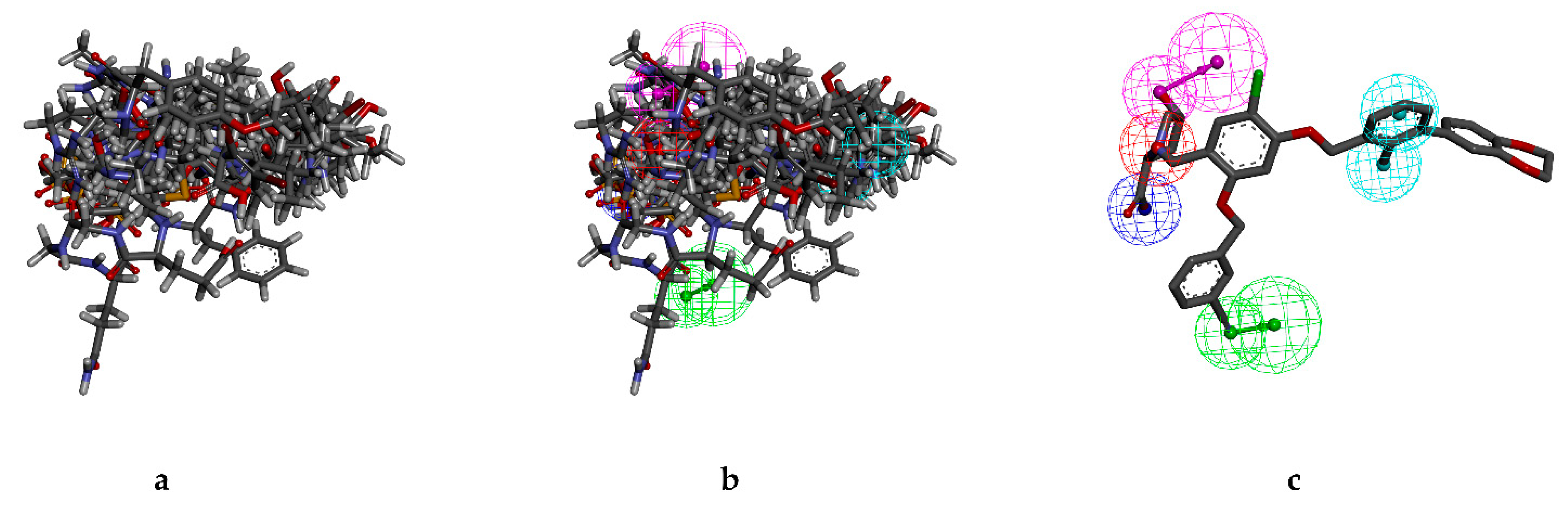
2.1.1. Pharmacophore Model Establishment
2.1.2. Pharmacophore Model Validation
2.1.3. Virtual Screening Based on Pharmacophore
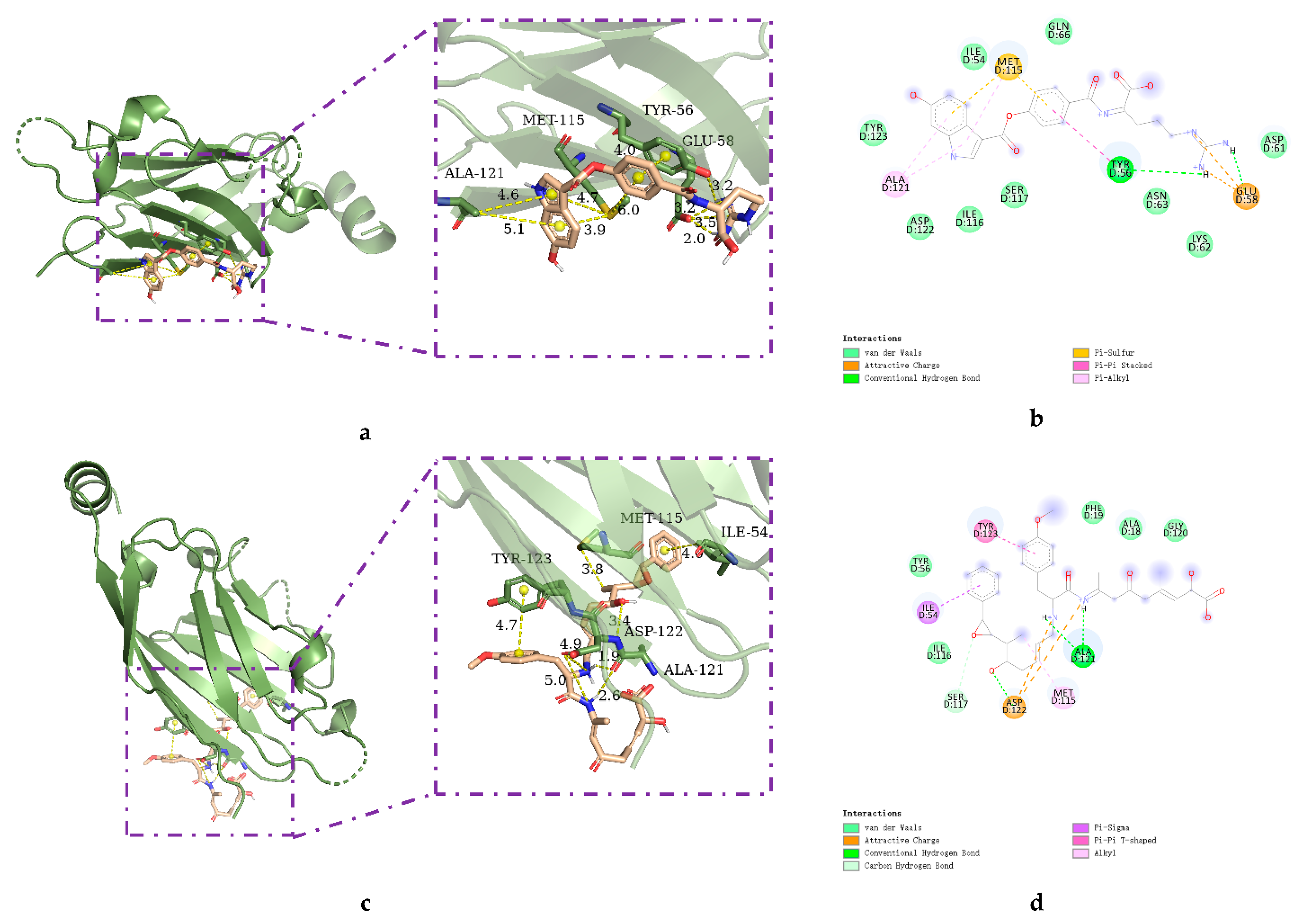
2.2. Molecular Docking
2.3. Analysis of Pharmacophore Characteristics
2.4. ADME and Toxicity Test ADME Properties Analysis
2.5. Toxicity Analysis
2.6. Structure-Based Pharmacophore Modeling and Virtual Screening
2.6.1. RMSD Analysis
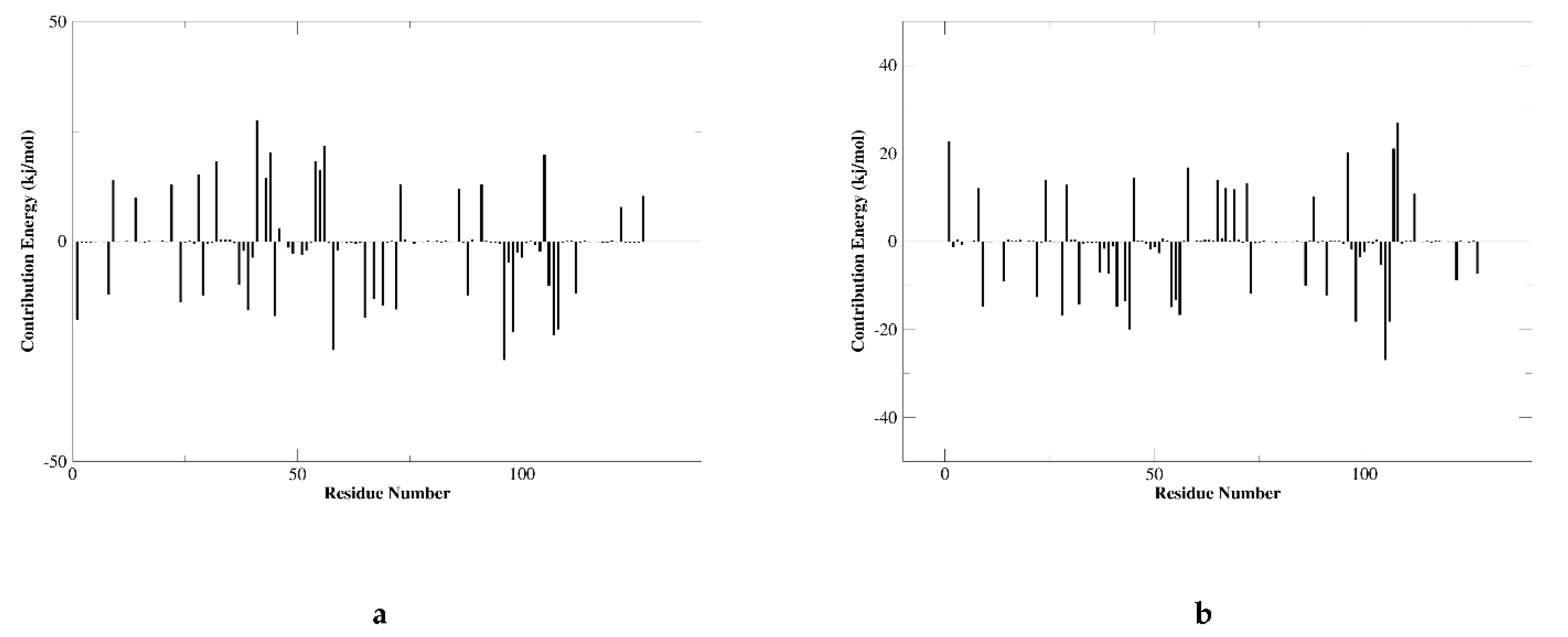
2.6.2. RMSF Analysis
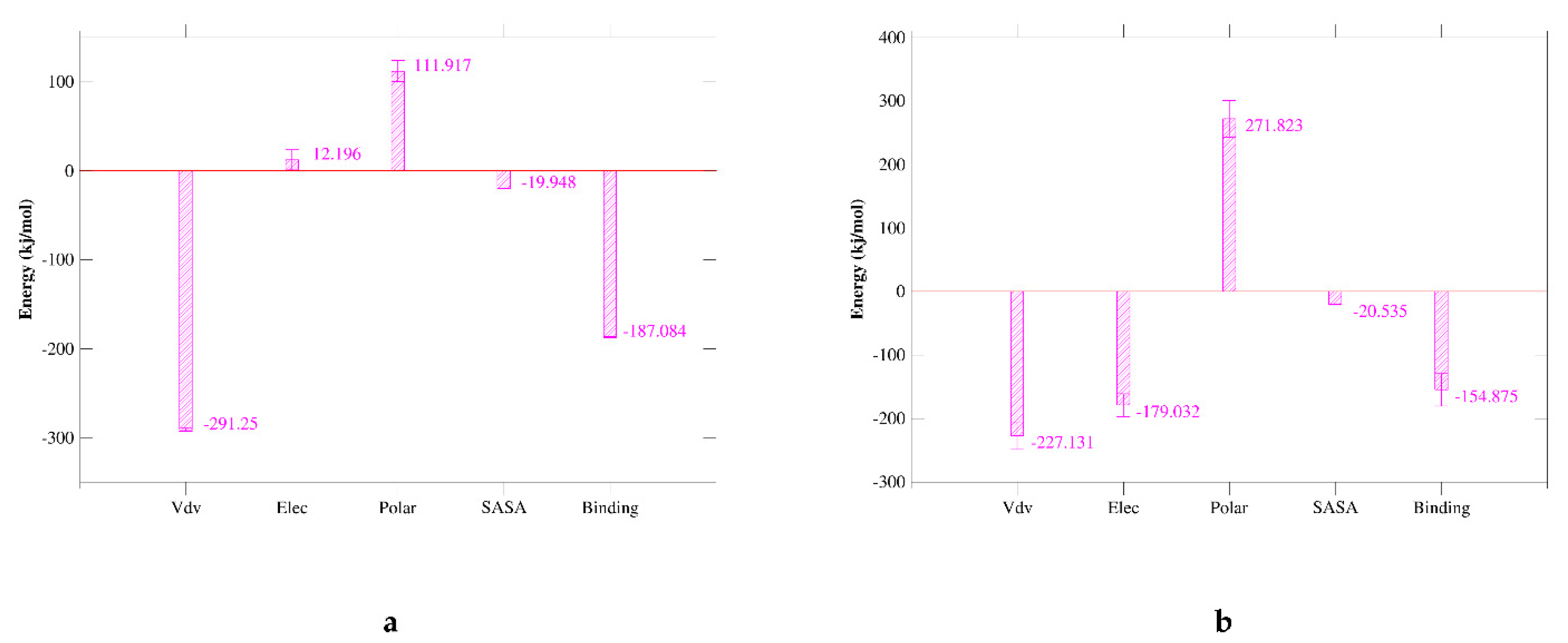
2.7. MM/GBSA Analysis
3. Discussion
4. Materials and Methods
4.1. Structure-Based Pharmacophore Modeling and Virtual Screening
4.1.1. Complex-Based Pharmacophore Modeling
4.1.2. Pharmacophore Model Validation
4.1.3. Virtual Screening Based on Pharmacophore
4.2. ADME and Toxicity Test
4.2.1. ADME
4.2.2. Toxicity Test
4.3. MD Simulation
4.4. MM/GBSA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef]
- Webb, E.S.; Peng, L.; Baleeiro, R.; Lemoine, N.R.; Ming, Y.; Wang, Y.J. Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2018, 32, 317–326. [Google Scholar] [PubMed]
- Ferrara, R.; Mezquita, L.; Texier, M.; Lahmar, J.; Audigier-Valette, C.; Tessonnier, L.; Mazieres, J.; Zalcman, G.; Brosseau, S.; Le Moulec, S.; et al. Hyperprogressive Disease in Patients with Advanced Non–Small Cell Lung Cancer Treated with PD-1/PD-L1 Inhibitors or with Single-Agent Chemotherapy. JAMA Oncol. 2018, 4, 1543–1552. [Google Scholar] [CrossRef]
- Nishijima, T.F.; Shachar, S.S.; Nyrop, K.A.; Muss, H.B. Safety and Tolerability of PD-1/PD-L1 Inhibitors Compared with Chemotherapy in Patients with Advanced Cancer: A Meta-Analysis. Oncologist 2017, 22, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Mühlbauer, M.; Fleck, M.; Schütz, C.; Weiss, T.; Froh, M.; Blank, C.; Schölmerich, J.; Hellerbrand, C. PD-L1 is induced in hepatocytes by viral infection and by interferon-α and -γ and mediates T cell apoptosis. J. Hepatol. 2006, 45, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Bari, S.; Muzaffar, J.; Chan, A.; Jain, S.R.; Haider, A.M.; Adams Curry, M.; Hostler, C.J. Outcomes of Programmed Cell Death Protein 1 (PD-1) and Programmed Death-Ligand 1(PD-L1) Inhibitor Therapy in HIV Patients with Advanced Cancer. J. Oncol. 2019, 2019, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Cimadamore, A.; Massari, F.; Santoni, M.; Lopez-Beltran, A.; Cheng, L.; Scarpelli, M.; Montironi, R.; Moch, H. PD1 and PD-L1 Inhibitors for the Treatment of Kidney Cancer: The Role of PD-L1 Assay. Curr. Drug Targets 2020, 21, 1664–1671. [Google Scholar] [CrossRef]
- Sun, J.; Zheng, Y.; Mamun, M.A.A.; Li, X.; Chen, X.; Gao, Y. Research progress of PD-1/PD-L1 immunotherapy in gastrointestinal tumors. Biomed. Pharmacother. 2020, 129, 110504. [Google Scholar] [CrossRef]
- Seetharamu, N.; Preeshagul, I.; Sullivan, K. New PD-L1 inhibitors in non-small cell lung cancer—Impact of atezolizumab. Lung Cancer Targets Ther. 2017, 8, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Collins, D.; Dolly, S.; McDonald, F.; O’Brien, M.E.R.; Yap, T.A. Targeting the PD-1/PD-L1 axis in non–small cell lung cancer. Curr. Probl. Cancer 2017, 41, 111–124. [Google Scholar] [CrossRef]
- Cha, J.H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Kim, S.E.; Keam, B.; Park, H.R.; Kim, S.; Kim, M.; Kim, T.M.; Doh, J.; Kim, D.W.; Heo, D.S. Anti-tumor effects of NK cells and anti-PD-L1 antibody with antibody-dependent cellular cytotoxicity in PD-L1-positive cancer cell lines. J. Immunother. Cancer 2020, 8, e000873. [Google Scholar] [CrossRef]
- Delanoy, N.; Michot, J.-M.; Comont, T.; Kramkimel, N.; Lazarovici, J.; Dupont, R.; Champiat, S.; Chahine, C.; Robert, C.; Herbaux, C.; et al. Haematological immune-related adverse events induced by anti-PD-1 or anti-PD-L1 immunotherapy: A descriptive observational study. Lancet Haematol. 2019, 6, e48–e57. [Google Scholar] [CrossRef]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Zhang, J.; Hong, S.; Zhan, J.; Zhang, L.J.O. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget 2014, 5, 12189. [Google Scholar] [CrossRef]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Cha, J.-H.; Chan, L.-C.; Wu, Y.; Chang, S.-S.; Lin, W.-C.; Hsu, J.-M.; Hsu, Y.-H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [Green Version]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2021, 38, 362–413. [Google Scholar] [CrossRef]
- Villa, F.A.; Gerwick, L. Marine natural product drug discovery: Leads for treatment of inflammation, cancer, infections, and neurological disorders. Immunopharmacol. Immunotoxicol. 2010, 32, 228–237. [Google Scholar] [CrossRef]
- Abdelmohsen, U.R.; Balasubramanian, S.; Oelschlaeger, T.A.; Grkovic, T.; Pham, N.B.; Quinn, R.J.; Hentschel, U. Potential of marine natural products against drug-resistant fungal, viral, and parasitic infections. Lancet Infect. Dis. 2017, 17, e30–e41. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, C. Marine Natural Products in Medicinal Chemistry. ACS Med. Chem. Lett. 2018, 9, 959–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunt, J.W.; Copp, B.R.; Hu, W.P.; Munro, M.H.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2008, 25, 35–94. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.D.J.; Vasanthi, A.H.R. Seaweed metabolite database (SWMD): A database of natural compounds from marine algae. Bioinformation 2011, 5, 361. [Google Scholar] [CrossRef] [Green Version]
- Lyu, C.; Chen, T.; Qiang, B.; Liu, N.; Wang, H.; Zhang, L.; Liu, Z. CMNPD: A comprehensive marine natural products database towards facilitating drug discovery from the ocean. Nucleic Acids Res. 2020, 49, D509–D515. [Google Scholar] [CrossRef]
- Muneer, I.; ul Qamar, M.T.; Tusleem, K.; Abdul Rauf, S.; Hussain, H.M.J.; Siddiqi, A.R. Discovery of selective inhibitors for cyclic AMP response element-binding protein: A combined ligand and structure-based resources pipeline. Anti-Cancer Drugs 2019, 30, 363–373. [Google Scholar] [CrossRef]
- Ul Qamar, M.T.; Kiran, S.; Ashfaq, U.A.; Javed, M.R.; Anwar, F.; Ali, M.A.; Gilani, A.u.H. Discovery of novel dengue NS2B/NS3 protease inhibitors using pharmacophore modeling and molecular docking based virtual screening of the zinc database. Int. J. Pharmacol. 2016, 12, 621–632. [Google Scholar]
- Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D.J.M. Pharmacophore models and pharmacophore-based virtual screening: Concepts and applications exemplified on hydroxysteroid dehydrogenases. Molecules 2015, 20, 22799–22832. [Google Scholar] [CrossRef] [Green Version]
- Muszak, D.; Surmiak, E.; Plewska, J.; Magiera-Mularz, K.; Holak, T. Terphenyl-based Small-Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein—Protein Interaction. J. Med. Chem. 2021, 64, 15. [Google Scholar] [CrossRef] [PubMed]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167–72181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Törner, R.; Skalniak, L.; Dömling, A.; Dubin, G.; Holak, T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Perry, E.; Mills, J.J.; Zhao, B.; Wang, F.; Sun, Q.; Christov, P.P.; Tarr, J.C.; Rietz, T.A.; Olejniczak, E.T.; Lee, T.; et al. Fragment-based screening of programmed death ligand 1 (PD-L1). Bioorganic Med. Chem. Lett. 2019, 29, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.; Kokh, D.B.; Bomke, J.; Wegener, A.; Buchstaller, H.P.; Eggenweiler, H.M.; Matias, P.; Sirrenberg, C.; Wade, R.C.; Frech, M.J.N.C. Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nat. Commun. 2017, 8, 2276. [Google Scholar] [CrossRef] [Green Version]
- Temml, V.; Garscha, U.; Romp, E.; Schubert, G.; Gerstmeier, J.; Kutil, Z.; Matuszczak, B.; Waltenberger, B.; Stuppner, H.; Werz, O.J.S.R. Discovery of the first dual inhibitor of the 5-lipoxygenase-activating protein and soluble epoxide hydrolase using pharmacophore-based virtual screening. Sci. Rep. 2017, 7, 42751. [Google Scholar] [CrossRef] [Green Version]
- Tai, W.; Tao, L.; Yuan, H.; Wang, F.; Liu, H.; Lu, S.; Leng, Y.; Zhang, W.; Jiang, Y.; Chen, Y. Pharmacophore modeling and virtual screening studies to identify new c-Met inhibitors. J. Mol. Model. 2012, 18, 3087–3100. [Google Scholar] [CrossRef]
- Antoine Daina, O.M.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- El-Hasab, E.M.; El-Bastawissy, E.E.; El-Moselhy, T.F. Identification of potential inhibitors for HCV NS3 genotype 4a by combining protein–ligand interaction fingerprint, 3D pharmacophore, docking, and dynamic simulation. J. Biomol. Struct. Dyn. 2018, 36, 1713–1727. [Google Scholar] [CrossRef]
- Alamri, M.A.; Qamar, M.T.U.; Mirza, M.U.; Bhadane, R.; Alqahtania, S.M.; Muneerf, I.; Froeyen, M.; Salo-Ahen, O.M.H. Pharmacoinformatics and molecular dynamics simulation studies reveal potential covalent and FDA-approved inhibitors of SARS-CoV-2 main protease 3CLpro. J. Biomol. Struct. Dyn. 2020, 39, 4936–4948. [Google Scholar] [CrossRef] [PubMed]
- Ryde, U. QM/MM Calculations on Proteins. Methods Enzym. 2016, 577, 119–158. [Google Scholar] [CrossRef]
- Kuca, K.; Musilek, K.; Jun, D.; Zdarova-Karasova, J.; Nepovimova, E.; Soukup, O.; Hrabinova, M.; Mikler, J.; Franca, T.C.C.; Da Cunha, E.F.F.; et al. A newly developed oxime K203 is the most effective reactivator of tabun-inhibited acetylcholinesterase. BMC Pharm. Toxicol. 2018, 19, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalho, T.C.; Cunha, E.F.F.D.; Castro, A.A.D.; Pereira, A.F.; de Lima, W.E.A. Flexibility in the Molecular Design of Acetylcholinesterase Reactivators: Probing Representative Conformations by Chemometric Techniques and Docking/QM Calculations. Lett. Drug Des. Discov. 2016, 13, 360–371. [Google Scholar]
- Kirchmair, J.; Markt, P.; Distinto, S.; Wolber, G.; Langer, T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes? J. Comput.-Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Pin, J.P.; Bertrand, O.; Triballeau, N.; Brabet, I.; Acher, F. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar]
- Drwal, M.N.; Priyanka, B.; Mathias, D.; Wettig, M.R.; Robert, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A.; Open Source Drug Discovery Consortium. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Number of Features | Feature Set | Selectivity Score | References |
|---|---|---|---|---|
| 6R3K | 6 | DHHHNP | 16.25 | [31] |
| 5NIU | 6 | DDHHHP | 15.635 | [32] |
| 5N2F | 6 | AAHHNP | 12.936 | [33] |
| 5N2D | 6 | AHHHPR | 12.848 | [33] |
| 5J89 | 5 | HHHHP | 11.196 | [17] |
| 6NM8 | 6 | HHHHHP | 10.996 | [34] |
| 5J8O | 6 | HHHHHR | 9.2594 | [35] |
| Molecules | 2D Structure | Binding Affinity (kcal/mol) | Formula |
|---|---|---|---|
| 37080 |  | −6.5 | C22H23N5O6 |
| 51320 |  | −6.3 | C35H44N2O8 |
| 37113 |  | −6.2 | C24H34N2O9 |
| 38010 |  | −5.7 | C22H25Br4N10O8S |
| 41160 |  | −5.2 | C18H29NO3 |
| 32979 |  | −5.1 | C22H35NO6S |
| 35432 |  | −4.7 | C16H24Br2N6O5S |
| 21793 |  | −4.6 | C23H34N2O4S2 |
| 23671 |  | −4.6 | C13H16Br2N6O5S |
| 41159 |  | −4.5 | C16H31NO3 |
| 35433 |  | −4.4 | C16H24Br2N6O5S |
| 50094 |  | −3.9 | C47H63N11O16 |
| JQT |  | −6.2 | C36H33ClN2O7 |
| Molecule | MW | Rotatable Bonds | H-bond Acceptors | H-bond Donors | ESOL Log S | TPSA | WLOGP | GI Absorption | log Kp (cm/s) |
|---|---|---|---|---|---|---|---|---|---|
| 37080 | 453.45 | 12 | 7 | 7 | −3.18 | 190.62 | 1.54 | 0.78 | Low |
| 51320 | 620.73 | 19 | 9 | 5 | −3.82 | 157.72 | 3.33 | 0.99 | Low |
| 37113 | 494.53 | 9 | 10 | 6 | −1.91 | 174.65 | −0.09 | 0.04 | Low |
| 38010 | 909.18 | 10 | 8 | 11 | −5.32 | 271.15 | −2.38 | 0.43 | Low |
| 41160 | 307.43 | 12 | 4 | 3 | −1.6 | 69.56 | 3.22 | 0.35 | High |
| 32979 | 441.58 | 11 | 7 | 4 | −2.04 | 132.31 | 3.13 | 1.28 | High |
| 35432 | 572.27 | 11 | 7 | 5 | −2 | 165 | 0.94 | 0.31 | Low |
| 21793 | 466.66 | 9 | 6 | 3 | −3.74 | 143.96 | 3.07 | 2.18 | Low |
| 23671 | 528.18 | 9 | 6 | 5 | −3.99 | 166.6 | 1.06 | −0.33 | Low |
| 41159 | 285.42 | 13 | 4 | 3 | −2.01 | 69.56 | 3.11 | 0.14 | High |
| 35433 | 572.27 | 11 | 7 | 5 | −2 | 165 | 0.94 | 0.31 | Low |
| 50094 | 1038.07 | 40 | 17 | 15 | −0.68 | 449.83 | −3.85 | −4.93 | Low |
| JQT | 641.11 | 10 | 9 | 2 | −5.81 | 121.48 | 5.30 | 2.64 | Low |
| Endpoint | Target | JQT | 51320 |
|---|---|---|---|
| Organ toxicity | Hepatotoxicity | Inactive | Inactive |
| Toxicity end points | Carcinogenicity | Inactive | Inactive |
| Immunotoxicity | Inactive | Inactive | |
| Mutagenicity | Inactive | Inactive | |
| Cytotoxicity | Active | Inactive | |
| LD50 (mg/kg) | 800 | 765 | |
| Toxicity class | 4 | 4 | |
| Tox21-nuclear receptor signaling pathways | Aryl hydrocarbon receptor (AhR) | Inactive | Inactive |
| Androgen receptor (AR) | Inactive | Inactive | |
| Tox21-stress response pathways | Heat shock factor response element (HSE) | Inactive | Inactive |
| Criteria | JQT | 51320 |
|---|---|---|
| Van der Waal energy (kJ/mol) | −291.250 ± 1.797 | −227.131 ± 21.896 |
| Electrostatic energy (kJ/mol) | 12.196 ± 11.229 | −179.032 ± 18.056 |
| Polar solvation energy (kJ/mol) | 111.917 ± 12.103 | 271.823 ± 28.866 |
| SASA energy (kJ/mol) | −19.948 ± 0.176 | −20.535 ± 0.727 |
| Binding energy (kJ/mol) | −187.084 ± 0.748 | −154.875 ± 25.470 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, L.; Zhong, A.; Wang, Q.; Zheng, T. Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, ADMET, and Molecular Dynamics (MD) Simulation of Potential Inhibitors of PD-L1 from the Library of Marine Natural Products. Mar. Drugs 2022, 20, 29. https://doi.org/10.3390/md20010029
Luo L, Zhong A, Wang Q, Zheng T. Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, ADMET, and Molecular Dynamics (MD) Simulation of Potential Inhibitors of PD-L1 from the Library of Marine Natural Products. Marine Drugs. 2022; 20(1):29. https://doi.org/10.3390/md20010029
Chicago/Turabian StyleLuo, Lianxiang, Ai Zhong, Qu Wang, and Tongyu Zheng. 2022. "Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, ADMET, and Molecular Dynamics (MD) Simulation of Potential Inhibitors of PD-L1 from the Library of Marine Natural Products" Marine Drugs 20, no. 1: 29. https://doi.org/10.3390/md20010029