1. Introduction
Designing multitarget drugs (agents that can simultaneously interact with multiple biological entities) for the treatment of multifactorial pathologies is a promising approach and a new challenge in medicinal chemistry [
1]. Single-target drugs, indeed, have been clearly demonstrated to be inadequate to achieve a therapeutic effect in the case of some complex multifactorial diseases [
2,
3]. On the other hand, it is now clear that many drugs may derive their therapeutic effect (as well as their undesirable off-target activities) from the interactions with multiple targets and, thus, it looks as if many clinically useful drugs are multitarget agents. Therefore, we are moving beyond the traditional one-target-one-drug model in drug discovery to a more effective strategy, in which compounds with a well-defined multitarget profile are addressed and rationally designed. These are the so-called designed multiple ligands (DMLs), well-distinguished from nonselective agents with activity against targets not involved in the selected disease, being designed and developed to achieve highly specific modulation of two or more selected biological targets [
4,
5,
6,
7].
A multitarget approach could be particularly beneficial for treatment of type 2 diabetes mellitus (T2DM), which is increasing at an alarming rate, owing to incorrect lifestyle and dietary habits, in addition to population ageing [
8,
9,
10]. T2DM is a multifactorial chronic disease characterised by hyperglycaemia, originating from reduced sensitivity of target tissues to insulin (insulin resistance), and other metabolic dysfunctions. It is hyperglycaemia, indeed, that triggers abnormal cellular signalling and induces cellular dysfunctions, which are, in turn, responsible for physical damages, failure of different organs, and serious chronic complications, including nephropathy and cardiovascular diseases (CVD). Moreover, a relationship between T2DM and obesity has been recognised, and the major basis for this link is the ability of obesity to engender insulin resistance [
11]. Nowadays, effective therapeutics are currently available to control hyperglycaemia and prevent chronic complications in diabetic patients; however, in most cases, combinations of various drugs are required, leading to drug–drug interactions and inadequate patient adherence. The identification of common pathways as drug targets could surely be useful to determine molecules with a multitarget profile able to interact with them to contrast the disorder.
Recently, the challenge of developing small-molecule inhibitors of protein tyrosine phosphatase 1B (PTP1B) has resulted in intense research; it can be considered a novel target for T2DM and its inhibition as a point of intervention could provide a new therapeutic option to patients with at-risk obesity or T2DM. PTP1B is crucially implicated in the development of insulin resistance; it is a negative regulator of insulin action, by dephosphorylating specific residues of phosphotyrosine (pTyr) of the activated insulin receptor (IR) and, thus, interrupting the signalling pathways mediated by the hormone. Among the insulin target organs, adipose tissue reacts to endocrine and metabolic signals by either increasing or decreasing a variety of adipokines and cytokines that can act at both the local and systemic level [
12]. PTP1B also downregulates the signal of leptin, an adipokine that controls food intake and increases energy expenditure and, thus, has a central importance in the global obesity and cardiovascular disease problem [
13]. PTP1B overexpression is strictly related to insulin resistance, whereas PTP1B inhibition or genetic ablation improve glucose homeostasis, cellular sensitivity to both insulin and leptin, and resistance to diet-induced obesity, without inducing hypoglycaemia or toxic effects [
14]. The efforts made in recent decades have allowed the identification of new inhibitors, which prove to be well tolerated and effective, both as insulin-sensitising agents and as antidiabetic drugs in animal models of T2DM [
15]. Together, this evidence confirms that development of potent and safe PTP1B inhibitors could represent one of the most interesting challenges for the discovery of new therapeutic interventions in T2DM and its complications in the coming years.
Aldose reductase (AR) has become another potential drug target for developing new therapeutics for treatment of T2DM and its complications. It is a cytosolic enzyme involved in the polyol pathway, in which glucose is converted to sorbitol that is, in turn, transformed in fructose through the NAD
+-dependent action of sorbitol dehydrogenase. In hyperglycaemic conditions, due to saturation of the glycolytic enzyme hexokinase, an increased flux of glucose is channelled through the polyol pathway, leading to several metabolic alterations. In particular, the accumulation of the osmolyte sorbitol increases cellular osmolarity, inducing a deleterious cell swelling. At the same time, the advanced glycation end product formation, linked to increased fructose levels, promotes ROS generation and NF-κB expression, leading to oxidative stress. In addition, the NADPH depletion linked to AR activity impairs the glutathione-reductase-dependent recovery of reduced glutathione. On the other hand, the increase in NADH/NAD
+ ratio might lead to NADH oxidase upregulation, thus further promoting oxidative stress. Under these conditions, lipid peroxidation occurs, with the consequent formation of cytotoxic aldehydes, including 4-hydroxy-
trans-2-nonenal and its derivative conjugated with glutathione (GS-HNE). AR reduces GS-HNE in the corresponding alcohol (3-glutathionyl-1,4-dihydroxynonane), which promotes the expression of NF-κB. In conclusion, the enhanced activity of AR promotes not only the onset of oxidative stress linked to insulin resistance, but also the long-term complications linked to T2DM [
16,
17]. Based on the above, the potential of facing the complex and multifactorial T2DM disease by combining two selective inhibitors into one molecule with dual PTP1B and AR modulator activity is evident. A dual inhibitor of the PTP1B and AR enzymes could be useful to treat both the condition of insulin resistance and chronic complications associated with T2DM [
18,
19,
20].
T2DM is spreading rapidly, not only in industrialised countries, but also in developing ones, where the economic resources available to tackle this pandemic are very limited [
21]. Most people living in these countries rely on remedies of traditional medicine to counteract metabolic alterations induced by diabetes [
22]. Such extracts obtained using different parts of plants, algae, and terrestrial or aquatic organisms contain a plethora of natural molecules able to act as multiple target ligands [
23]. Therefore, natural products (NPs), used either unaltered or developed through chemical manipulation and synthesis, represent one of the most important sources of scaffold molecules showing innate multitarget activity that could be used for the development of new DMLs [
24,
25]. The marine environment, in particular, offers a chemical arsenal of unique structures with different structural motifs compared to NPs of terrestrial origin because of the different conditions of salinity, pressure, and temperature that influence biosynthetic pathways [
26,
27,
28]. Several NPs of marine origin have been reported as inhibitors of both PTP1B and AR enzymes; they include terpenes, polyketides, steroids, phenols, and alkaloids, mainly isolated from marine invertebrates [
29,
30,
31,
32].
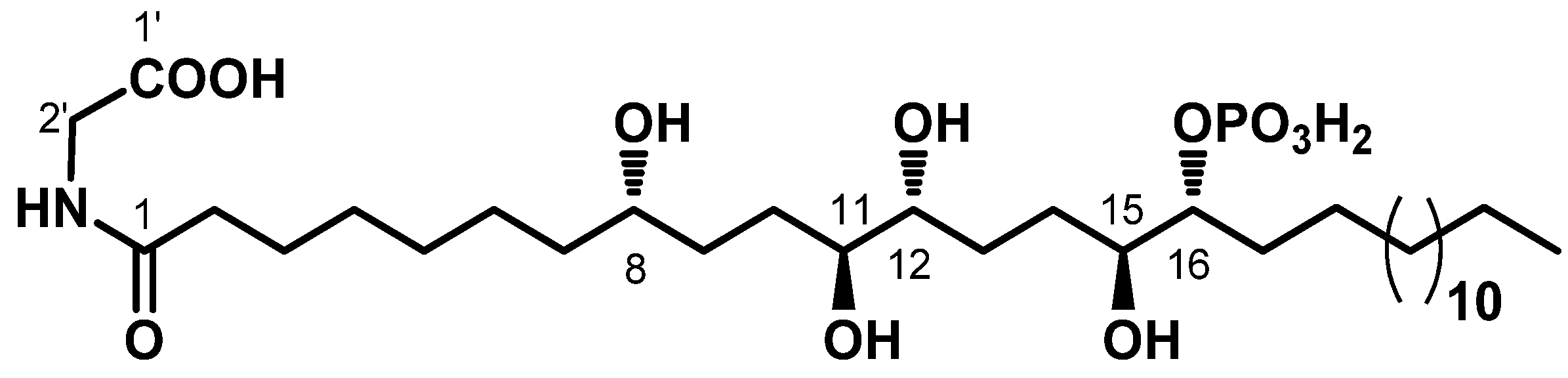
Recently, we have isolated from the Mediterranean ascidian
Sidnyum elegans a novel phosphorylated compound and potent PTP1B inhibitor, phosphoeleganin (PE,
Figure 1) [
33,
34].
In order to explore the potential of PE to be developed as a multitarget drug to treat insulin resistance and chronic complications associated with T2DM, we have carried out an in-depth study on its inhibitory mechanism, including analysis of the insulin signalling pathway; in parallel, we have evaluated the AR inhibitory activity of PE. The results reported here show that PE inhibits both enzymes, acting, respectively, as a mixed-type inhibitor of AR and a pure non-competitive inhibitor of PTP1B. In addition, in silico docking analyses were performed to describe how PE interacts with both enzymes. Finally, tests carried out using HepG2 cells confirmed that PE possesses an insulin-sensitising activity.
2. Results
2.1. Chemistry
PE was obtained in pure form (
Figures S1 and S2) by extraction of a sample of the Mediterranean ascidian
S. elegans from the existing collection of natural sources available at the Department of Pharmacy of University of Naples Federico II. PE was isolated and purified according to the previously described procedure [
33] and unequivocally identified by comparison of its HRMS and NMR data with those reported in the literature [
33].
2.2. Preliminary Screening on PTP1B and AR In Vitro
To assess the effect of PE on PTP1B and AR catalytic activities, a preliminary evaluation of IC
50 was performed. The obtained results confirm the ability of PE to inhibit PTP1B and highlight that PE can also act as an AR inhibitor, despite a minor efficiency (IC
50 = 28.7 ± 1.1 μM). The IC
50 value determined for PTP1B (1.3 ± 0.04 μM) is almost eight times lower than that previously determined [
33]. We think that such differences may be attributable to the different experimental conditions used to perform the activity assays, such as a different pH value and the use of a GST–PTP1 fusion protein instead of purified PTP1B alone (1-302 aa).
2.3. Definition of PTP1B and AR Inhibition Mechanism by PE
The mechanism of the enzyme inhibition by PE was investigated and it was shown that it behaves as a reversible inhibitor of both AR and PTP1B. As far as AR is concerned, indeed, we found that 100% of enzyme activity was recovered after the inhibitor had been removed by extensive dialysis (see Methods for details). Analogously, dilution assay test showed that PTP1B recovered its activity almost completely after incubation with a saturating concentration of PE, thereby confirming that PE behaves as a reversible inhibitor of PTP1B (
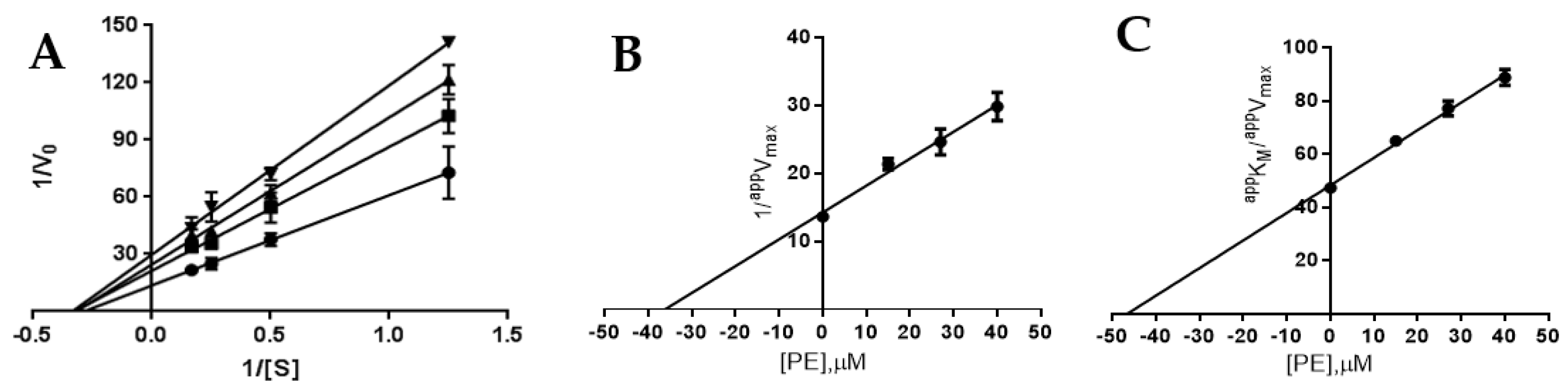
Figures S3 and S4). Kinetic characterisation of PE as an AR and PTP1B inhibitor was carried out. Kinetic analysis revealed that PE acts as a mixed inhibitor of AR (
Figure 2A), being able to affect to a different extent both
Vmax and
KM. The analysis of the dependence of
appVmax and
appKM/
appVmax on the inhibitor concentration allowed the measurement of
K’i (36 ± 1 μM,
Figure 2B) and
Ki (47 ± 1 μM,
Figure 2C).
These values indicated a slight, but significant (
p < 0.001), preference of PE toward the ES complex with respect to the free enzyme. On the other hand, PE acts as a pure non-competitive inhibitor of PTP1B; the analysis of the Lineweaver–Burk plot, indeed, indicated that only
Vmax is affected by the presence of the inhibitor (
Figure 3). In fact, the analysis of the dependence of
appVmax and
appKM/
appVmax on the inhibitor concentration allowed the measurement of an identical value of 0.7 ± 0.1 µM for
Ki and
K’i. 2.4. Dissecting the Interaction of PE with PTP1B and AR by In Silico Docking Analyses
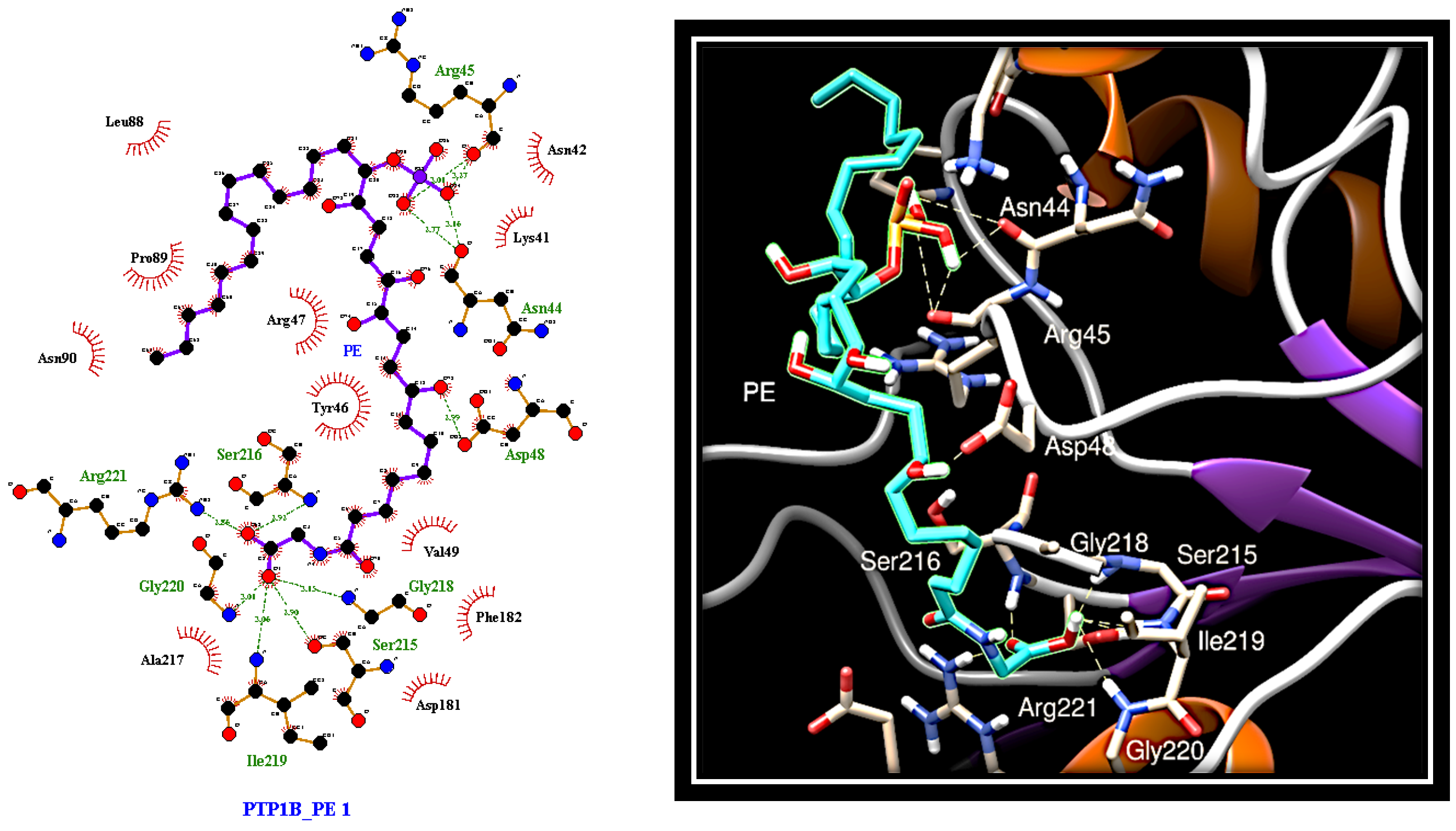
To evaluate the interaction mode of PE with PTP1B, in silico docking analysis was carried out using AutoDock software. The compound was docked to the whole protein surface of PTP1B. We found that PE was able to fit to different sites that can be categorised depending on the docking energy. The most favourable position places PE between the active site and the region that defines the “C” site [
35], which differs from the secondary aryl phosphate site (identified by Arg24 and Arg254) previously described as the docking site for aryl difluorophosphonate groups (
Figure 4) [
36].
Interestingly, the phosphate group of PE creates hydrogen bonds with the carbonyl groups of Asn44 and Arg45 that lie beyond the residues forming the "C" site. This suggests an alternative bonding mode of PE on the surface of PTP1B compared to fluorine-bisphosphonates. Further hydrogen bonds involve the Asp48 residue belonging to the “C” site and a hydroxyl group present on the aliphatic chain (C12) of PE. Finally, the carbon chain of PE extends to the active site where the carboxyl group of PE forms several hydrogen bonds with the side chain of Arg221 and the nitrogen atoms of the main chains of Ser215, Ser216, Gly218, Ile219, and Gly220.
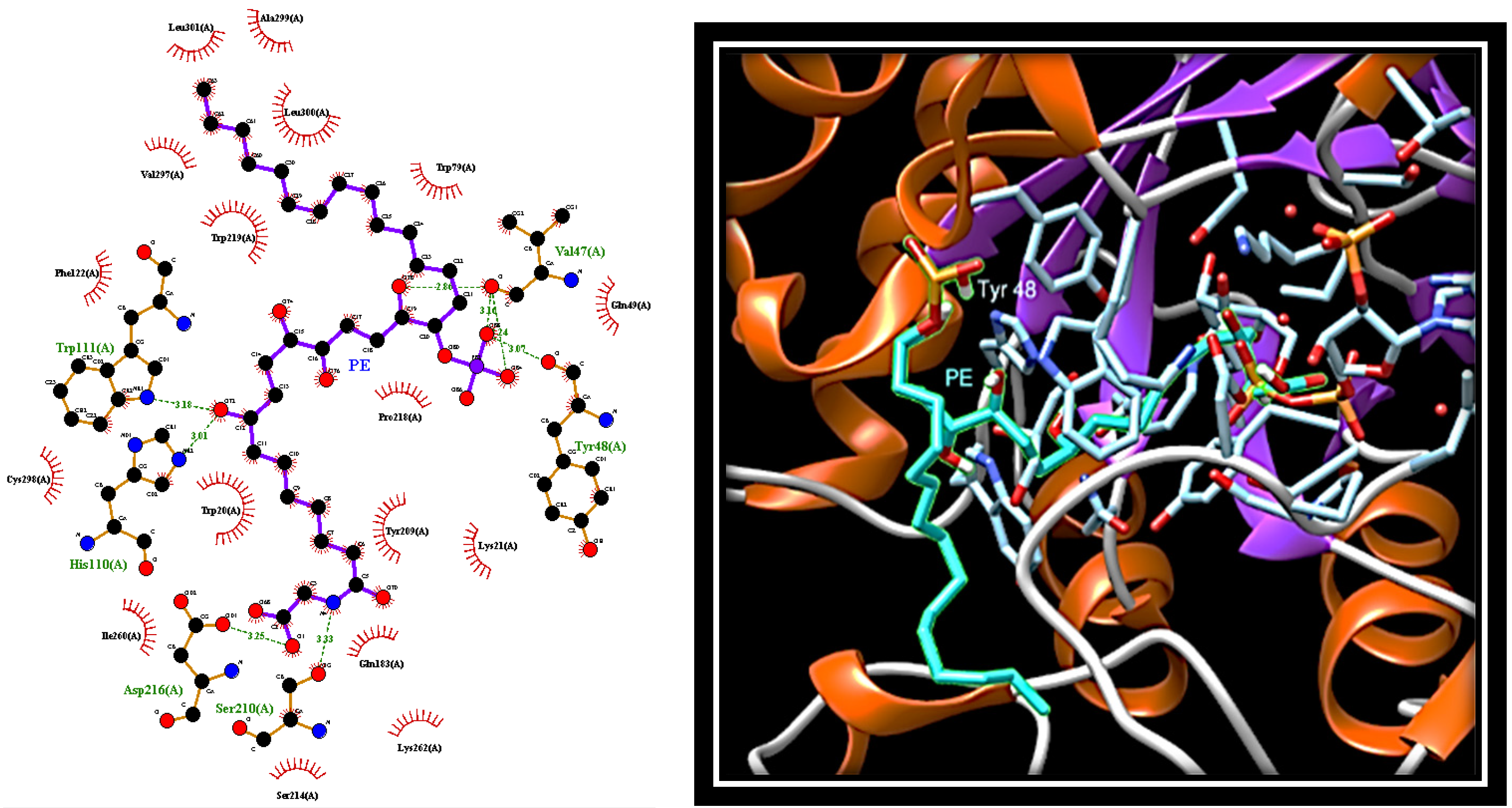
The docking analysis carried out in the absence of substrate localises PE in the active site of AR (
Figure 5).
Here, the OH group at C8 forms hydrogen bonds with nitrogen atoms of His110 and Trp111, whereas the OH at C15, as well as the phosphate group, form hydrogen bonds with carbonyl groups of the main chain of Val47 and Tyr48. In addition, the aliphatic portion of PE (C17-30) is firmly anchored to the selectivity pocket, where it makes several hydrophobic interactions with Trp79, Trp219, Val297, Ala299, Leu300, and Leu301. Finally, the portion from the carboxyl group at C7 interacts with Trp20 and Tyr209, and extends up to a more hydrophilic region where the carboxyl group forms hydrogen bonds with side chains of Asp216 and Ser210.
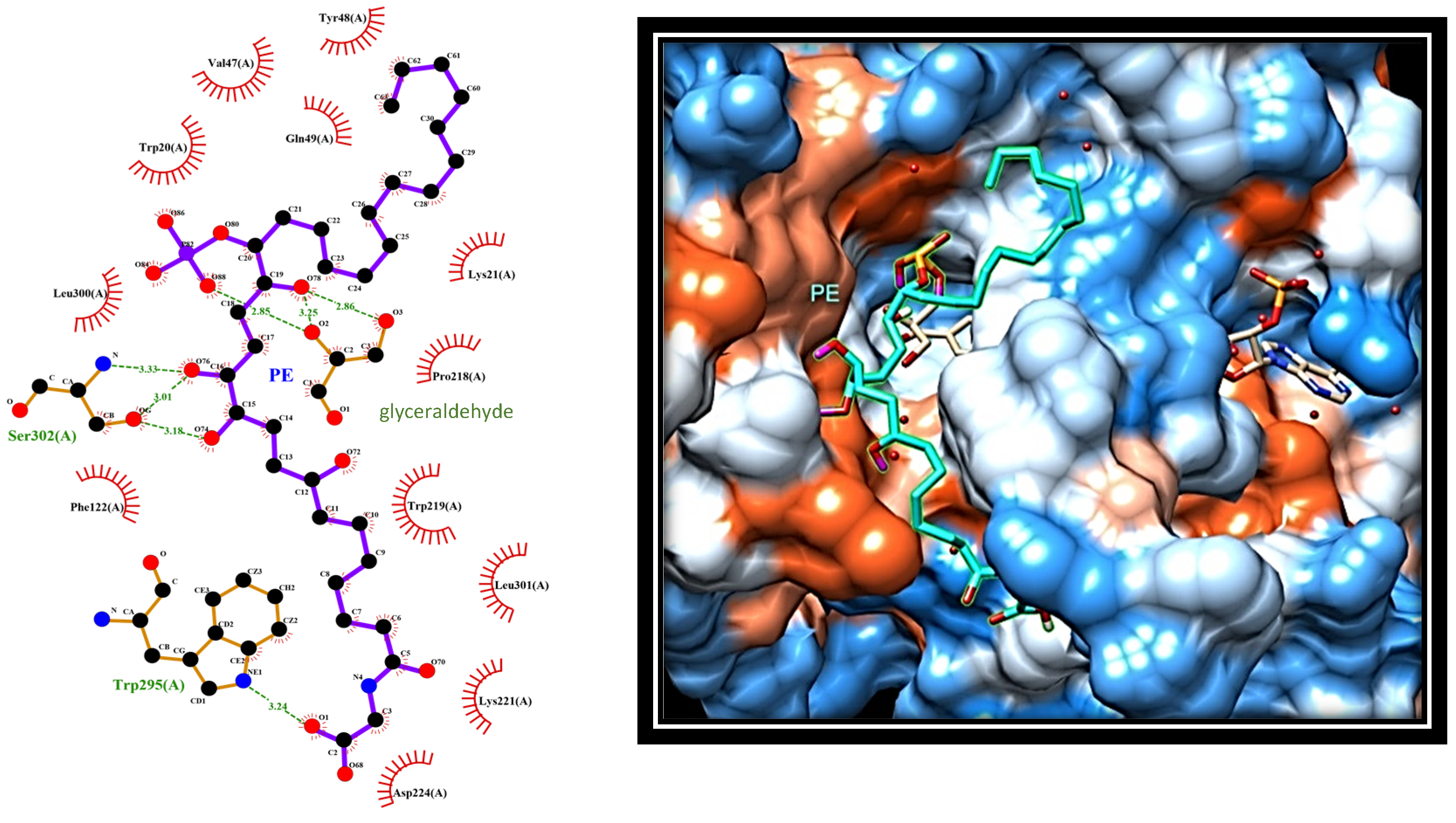
When PE was docked in the AR–substrate complex, the interaction with the enzyme appeared different, since it involved amino acids located in the external portion of the active site (
Figure 6).
In this arrangement, the terminal carboxyl portion of PE (C7–C1’) extends out of the active site, making a hydrophobic interaction with Trp219, Leu301, and Lys221, while the carboxyl group of PE form a hydrogen bond with the nitrogen atom of Trp295. On the other side, the hydroxyl groups at both C15 and C16 face the mouth of the active site, where they form hydrogen bonds with the oxygen atom of Ser302. In addition, phosphate moiety moves into the active site, where it forms hydrogen bonds with glyceraldehyde. Finally, the C17–C30 portion of PE remains excluded from the active site, interacting with amino acids surrounding the entrance to the active site, including the aliphatic chain of Lys21, Tyr48, and Gln49. These data concur with kinetic analysis in confirming the ability of PE to efficiently interact with AR.
2.5. Analysis of the Insulin Signalling Pathway in Liver Cells Treated with PE
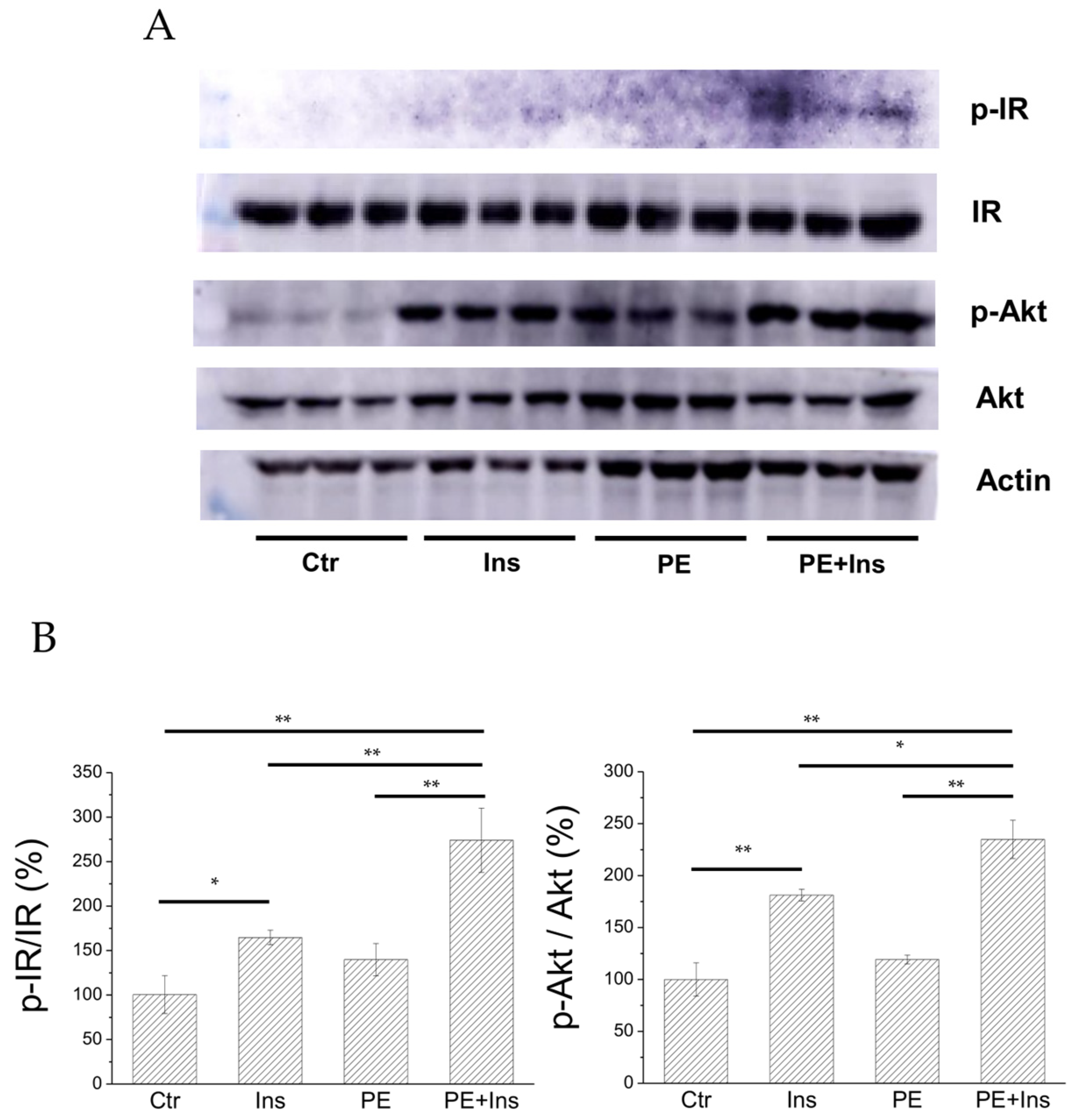
To evaluate whether PE is able to improve the insulin signalling pathway in vitro, we analysed the effect of the compound on HepG2 cell line. For this purpose, we treated HepG2 cells with PE (25 µM final concentration) for 30 minutes in the presence or absence of insulin (10 nM). After treatment, cells were lysed and cellular extracts were analysed to evaluate the phosphorylation status of insulin receptor (IR) and Akt, a kinase downstream IR (
Figure 7).
We found that, in liver cells treated with PE alone, the phosphorylation levels of both IR and Akt are similar to those detected in control cells, suggesting that PE did not behave as an insulin-mimetic agent. Nevertheless, a significant increase in IR and Akt phosphorylation levels was observed, combining insulin stimulation with PE treatment. Taken together, these results demonstrated that PE acts as an insulin-sensitising agent.
3. Discussion
Diabetic patients may rely on different types of oral hypoglycaemic drugs to keep blood glucose below physiological levels; however, most of these drugs cause patients to develop various and serious diabetes-related diseases in the medium- to long-term that contribute to deterioration of the quality and duration of their life [
37]. This is probably because actual antihyperglycemic drugs are designed to compensate for a single metabolic defect and, therefore, are not able to reproduce the physiological effects of insulin. Although the synthesis of DMLs is a very exciting challenge, the identification of the appropriate scaffolds represents the real greatest challenge in this approach because of the difficulty to obtain multifunctional molecules showing a balanced activity toward the selected targets [
5]. Therefore, many researchers are concretely exploring the possibility of exploiting natural molecules, even of marine origin, to discover new DMLs to be used for the treatment of T2DM [
29,
30,
31,
32,
33].
Here, we demonstrated that PE, a phosphorylated polyketide isolated from the Mediterranean ascidian
S. elegans, behaves as a multitarget ligand, targeting both PTP1B and AR. The ability of PE to inhibit PTP1B was previously described [
33]; however, its mechanism of action and its in vitro activity had not yet been studied. Being endowed with a phosphate group, we expected that PE could bind to the active site of PTP1B, acting as a classical competitive inhibitor. However, unexpectedly, our results showed that PE acts as a pure non-competitive inhibitor of PTP1B, showing a Ki value of 0.7 µM. Interestingly, in silico docking analyses suggested that the nonfunctionalised aliphatic portion of PE structure interacts with various hydrophobic residues present on the surface of the enzyme, whereas the phosphate group binds firmly to some amino acids close to the "C" site, a region previously described to act as a binding site for negatively charged difluorophenylmethylene phosphonates [
35]. The other functionalised part of the molecule forms hydrogen or hydrophobic bonds with numerous other amino acids, extending to the area of the active site, where the carboxyl group of the PE forms further bonds with the residues of the P-loop. Therefore, for the first time, we investigated the way in which PE interacts with PTP1B, also clarifying the role of the phosphate group in stabilising the enzyme–inhibitor complex. Concerning AR, it is worth noting the effort in the last decades in synthetising AR inhibitors (ARI), with the aim to treat secondary complications related to diabetes [
38]. Despite the proved clinical efficacy of some of these, the main limitation to their use in clinic is the severe side effects they induce [
20]. Even if the identification of new and safe ARI remains one of the priority objectives in the pharmaceutical industry, the interest for ARIs with a well-defined multitarget profile has grown in the last years, based on the belief that they could have greater efficacy and less toxic effects than traditional ARI. The ability of PE to inhibit AR had not been reported until now. We showed, for the first time, that PE acts as a mixed-type inhibitor of AR (IC
50 of 28.7 µM), being able, in the absence of substrate, to move inside the active site of enzyme or, in the presence of substrate, to interact on the upper part of the AR active site, positioning the phosphate group at the entry of the active site. In this position, the phosphate can interact with substrate, thereby interfering with the catalytic process.
Tests performed on liver cells showed that, after short-term PE treatment, the phosphorylation levels of IR and Akt kinase increased in hormone-stimulated, but not in non-stimulated, cells. Akt kinase is an essential effector of the insulin signalling pathway and its enhanced phosphorylation is an expected/essential event to propagate the signalling wave trigger by insulin [
39]. Similarly, a plethora of studies demonstrated that the enhancement of Akt phosphorylation is a common landmark observed in both muscle and liver cells treated with PTP1B inhibitors [
40,
41,
42,
43]. In analogy with this evidence, the data reported in this study show that treatment of liver cells with PE enhances insulin activity, suggesting that it acts as an insulin-sensitising agent. Moreover, our data indicate that PE, despite the presence of a charged phosphate group, can cross the plasma membrane and move within the cytoplasm of cells where it inhibits PTP1B, thus enhancing the insulin signalling pathway. This finding is quite surprising, considering that most low molecular weight inhibitors with a phosphate group show poor permeability across the cell membrane [
44]. Although no information is currently available on the mechanisms promoting PE internalisation in liver cells, it can be hypothesised that its entry into cells may be mediated by fatty acid transporters present on the cell surface or, alternatively, by the process of endocytosis. Further tests will be needed in the future to confirm this hypothesis.
4. Material and Methods
4.1. Materials
Solvents and commercial reagents: Sigma-Aldrich (Saint Louis, MO, USA). TLC: Silica Gel 60 F254, plates 5 × 20, 0.25 mm, Merck (Kenilworth, NJ, USA). Deuterated solvents: Sigma-Aldrich-Merck. High-resolution MS (negative mode) was performed on a Thermo LTQ Orbitrap XL mass spectrometer (Thermo-Fisher, San Josè, CA, USA). The MS spectrum of PE was recorded by infusion into the ESI source using MeOH as solvent. 1H (700 MHz) and 13C (175 MHz) NMR were carried out on a Bruker Avance Neo spectrometer (Bruker BioSpin Corporation, Billerica, MA, USA); chemical shifts were referenced to the residual solvent signal (CD3OD: δH = 3.31, δC = 49.0). A Jasco P-2000 polarimeter (Jasco Europe s.r.l., Cremella, Italy) at the sodium D line was used to measure optical rotations. High-performance liquid chromatography (HPLC) separation was achieved on a Knauer K-501 apparatus equipped with a Knauer K-2301 RI detector (LabService Analytica s.r.l., Anzola dell’Emilia, Italy).
L-idose and NADPH were from Carbosynth (Compton, England); p-nitrophenylphosphate (p-NPP) was from Chemcruz, Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other chemicals were obtained from Merck Life Science s.r.l. (St. Louis, MO, USA), unless otherwise specified. Cell culture media and foetal bovine serum (FBS) were from Euroclone (Pero, Italy). The following antibodies were used: pIR β subunit, Y1162/1163 (sc-25103-R) and β-actin, clone C-4 (sc-47778) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); IR β subunit, clone CT-3 (MABS65) was from Merck-Millipore (Burlington, MA, USA). Akt (9272S) and p-Akt (9271S) antibodies were from Cell Signaling Technology (Danvers, MA, USA). Secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
The antibodies used were purchased by Merck (anti-insulin receptor, MABS65), Santa Cruz Biotechnology (p-insulin Rβ, sc-81500; β-actin, sc-47778), and Cell Signaling (Akt, 9272S; p-Akt, 9271S). Primary antibodies were diluted 1:1000 in PBS solution containing 0.05% Tween 20 and 5% BSA; secondary antibodies were diluted 1: 2500 in the same solution. Primary antibodies were incubated with PVDF membrane overnight at 4 °C; secondary antibodies were incubated for 1 hour at room temperature. Detection was performed using Clarity Western ECL substrates (Bio-Rad Laboratories S.r.l., Segrate, Milan, Italy).
4.2. Collection, Extraction, and Isolation of PE from Specimens of S. elegans
Specimens of
S. elegans were collected at Pozzuoli (Naples, Italy, April 2019). They were frozen immediately after collection and kept frozen until extraction. Fresh thawed animals (21.3 g of dry weight after extraction) were extracted according to a previously reported procedure [
33]. The most polar organic layer (n-BuOH material) was chromatographed by MPLC over a C-18 column following a gradient elution H
2O → MeOH → CHCl
3. The fraction eluted with MeOH/H
2O 7:3 (
v/
v) was further purified by HPLC on reversed phase (column: Synergi RP-MAX 4 μm, eluent: MeOH/H
2O 8:2 + 0.1% of TFA) and afforded compound PE (t
R = 20.5 min, 15.8 mg) in pure form.
Phosphoeleganin (PE): colourless oil; all data agree with those reported in the literature [
33,
34].
4.3. Expression and Purification of AR and PTP1B
The human recombinant AR was expressed in BL21(DE3) pLysS
E. coli cells and purified to electrophoretic homogeneity, as previously described [
45]. Purified AR (5.0 U/mg) was stored at −80 °C in 10 mM sodium phosphate buffer pH 7.0 containing 2 mM dithiothreitol and 30 % (
w/v) glycerol; the enzyme was extensively dialysed against 10 mM sodium phosphate buffer pH 7.0 before use.
The human recombinant PTP1B (1-302 aa) and LMW-PTP (full-length) were expressed as fusion proteins containing N-terminus polyhistidine tags in BL21(DE3) pLysS E. coli and purified by using affinity chromatography. After purification, enzymatic assays were carried out to identify the fraction containing the enzyme. Such fractions were collected and concentrated using centrifugal filters. Finally, concentrate solution was purified using an FPLC system equipped with a Superdex 200 pg 16/600 semi-preparative column equilibrate in 50 mM Tri-HCl buffer pH 8.0 containing 150 mM NaCl, 1 mM DTT. Fractions containing the enzyme were collected, concentrated, and batched in cryovials that were stored at −80 °C. The degree of purity of the enzymes was evaluated by SDS PAGE.
4.4. Determination of Enzymatic Activities
AR activity was determined at 37 °C, as previously described [
46], evaluating the decrease in absorbance at 340 nm linked to NADPH oxidation using a Biochrom Libra S60 spectrophotometer. The standard assay mixture contained 0.25 M sodium phosphate buffer pH 6.8, 0.18 mM NADPH, 2.4 M ammonium sulphate, 0.5 mM EDTA, and 4.7 mM
d,l-glyceraldehyde. One unit of enzyme activity is the amount that catalyses the conversion of 1 μmol of substrate/min in the above assay conditions. For AR inhibition studies, L-idose (at the indicated concentrations) was used as substrate in the conditions described above.
PTP1B activity was determined as previously described [
47]. Briefly, each assay was started by adding an aliquot of human recombinant PTP1B (1-302) in the assay buffer (0.075 M β,β-dimethylglutarate pH 7.0, containing 1 mM EDTA, 0.1 mM DTT, and
p-nitrophenylphosphate (
p-NPP)). All reactions were performed at 37 °C in a final volume of 1 mL. After the incubation time, the reactions were stopped by adding 2 mL of 0.1 M KOH to the samples. The concentration of the released
p-nitrophenol was determined by measuring the absorbance at 400 nm in a 1 cm pathlength cuvette. One unit of enzyme activity is the amount that catalyses the conversion of 1 μmol of substrate/min in the above assay conditions.
4.5. Inhibition Studies
PE was dissolved in DMSO at a final concentration of 10 mM and added to the assay mixture containing 10 mU of purified AR (84 nM on the basis of a 34 KDa molecular weight) or 8 mU purified PTP1B (52 nM on the basis of a 36 KDa molecular weight). The reaction was started by the addition of the substrate. The DMSO concentration in the assays for AR determination was kept constant at 0.7 % (
v/
v) in order to avoid effects on AR activity [
48]. The inhibitory effect of PE was determined by measuring the reaction rates in the presence of a fixed substrate concentration (2.5 mM
p-NPP and 0.8 mM L-idose for PTP1B and AR, respectively) and increasing inhibitor concentrations (at least 14-16 different concentrations). The IC
50 (the inhibitor concentration able to decrease the enzymatic activity up to 50%) values were determined by nonlinear regression analysis using Prism GraphPad 6.0 (GraphPad Software, San Diego, CA, USA), fitting experimental data to the following equation:
where
Vi/
V0 represents the relative activity calculated in presence of each inhibitor concentration; “
Max” and “
Min” represent the maximum and minimum value of the activity, respectively; “
x” is the concentration of the inhibitor; and “slope” represents the slope of the curve in the transition zone.
In order to define the mechanism of action of PE, reaction rates were measured in assay mixtures containing different L-idose concentrations (between 0.8 and 6 mM) in the case of AR, and different p-NPP concentrations (between 0.5 and 25 mM) in the case of PTP1B, in the absence and in the presence of different PE concentrations. Data were analysed by Lineweaver–Burk plots. The apparent dissociation constants Ki’ (for the ESI complex) and Ki (for the EI complex) were determined from secondary plots of 1/appVmax and appKM/appVmax as a function of the inhibitor concentration, respectively.
In order to evaluate the reversibility of the inhibitory action, 10 mU of purified AR was incubated for 90 min at 0 °C in the presence of PE (3 mM) in order to completely inhibit AR activity. The mixture was then extensively dialysed on Amicon ultrafiltration membrane (cut-off 3 KDa) against 10 mM sodium phosphate buffer, pH 7.0. After dialysis, the enzyme activity was again measured using 0.8 mM L-idose as substrate and compared to that of a mixture treated in the same conditions but in the absence of inhibitor. In order to evaluate the reversibility of the inhibitory action on PTP1B, 8 mU of PTP1B was incubated in the presence of a saturating concentration of PE (3 µM) and then incubated at 37 °C for 1 h. After this time, an aliquot of solution was withdrawn and extensively diluted in the assay buffer containing 2.5 mM of p-NPP. Residual activity of PTP1B was determined by calculating the amount of pNP released by measuring the absorbance of solution at 400 nm.
4.6. Cell Cultures
HepG2 liver cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). HepG2 were routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM)-high glucose (4500 mg/L) supplemented with 10% foetal bovine serum (FBS, Euroclone), 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were incubated at 37 °C in a humidified atmosphere with 5% CO2.
4.7. Analysis of the Insulin Signalling Pathway
In order to evaluate the effect of PE on the insulin signalling pathway, HepG2 cells were seeded in 35-mm culture dishes and, after 24 hours, were incubated for 30 minutes with starvation medium (DMEM supplemented with 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin) containing 25 μM of PE and/or 10 nM insulin (Humalog, lispro insulin). The medium was then removed, cells were washed with PBS, and lysed with sample buffer (2% SDS, 10% glycerol, 5% 2-mercaptoethanol, 0.002% bromophenol blue, and 0.0625 M Tris HCl, pH 6.8) for cellular protein extraction. Cellular extracts were boiled for 5 minutes, and then an aliquot (20 µg of protein) was analysed by SDS-PAGE and Western blot to evaluate both total and phosphorylated expression levels of Akt and IR. β-actin was used as a reference protein to normalise protein loading.
4.8. Molecular Docking
Docking simulations were carried out with AutoDock (version 4.2.5.1, The Scripps Reasearch Institute, La Jolla, CA, USA) and AutoDock Tools (version 1.5.6rc3, The Scripps Reasearch Institute, La Jolla, CA, USA) [
49] using the crystal structures PDB 1PTY and 3V36 structures for PTP1B and AR, respectively [
50,
51]. The grid map of the interaction energies for various atoms of the ligand with the macromolecule was calculated using AutoGrid software (version N 1.5.6., The Scripps Reasearch Institute, La Jolla, CA, USA). Specifically, for 1PTY, the grid search size was 126 × 100 × 90 points, with a spacing of 0.400 Å. The Lamarckian genetic algorithm (LGA) was used to identify potential binding pockets using the following parameters: The number of GA runs was 100, the number of individuals in the population was 150, the maximum number of energy evaluation was 250,000, and the maximum number of generations was 27,000. The LGA resulted in 100 ligand poses. These poses were clustered according to root-mean-square deviation (RMSD), with a similarity of 10 rms. Among all, five poses were selected from the obtained clusters. Graphical representations of the ligand, the protein, and the complexes were performed using UCSF Chimera [
52].
The potential intermolecular interactions between the ligand and the protein were calculated automatically by LigPlot
+ software (version 2.2, EMBL-EBI, Hinxton, Cambridgeshire, UK), which generated 2D-schematic representations of protein–ligand complexes displaying hydrogen bonds and hydrophobic interactions within the binding pocket [
53,
54].
4.9. Statistical Analysis of Data
Data obtained were analysed using OriginPro 2021 software (OriginLab Corporation, Northampton, MA, USA). Statistical significance between data was evaluated using ANOVA, followed by Tukey HSD. Data were checked for appropriate normality and homoscedasticity using the Shapiro–Wilk normality test and the Levene’s test, respectively. All tests were carried out in triplicate.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






