Holotoxin A1 Induces Apoptosis by Activating Acid Sphingomyelinase and Neutral Sphingomyelinase in K562 and Human Primary Leukemia Cells


Abstract
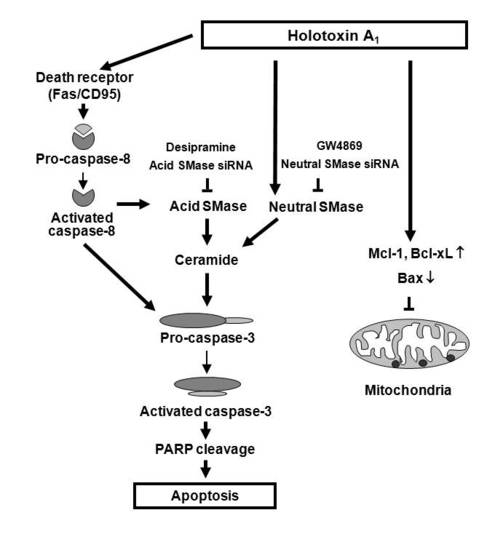
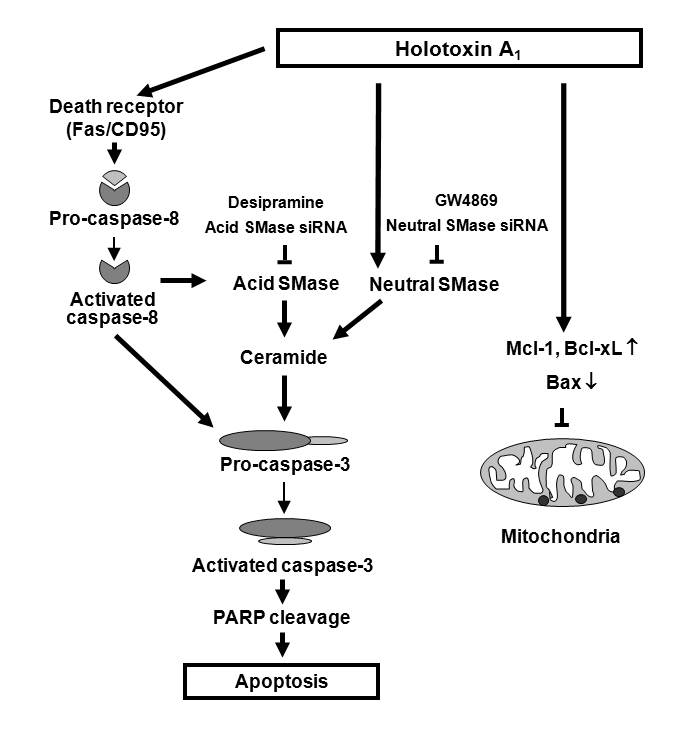
:
1. Introduction
2. Results
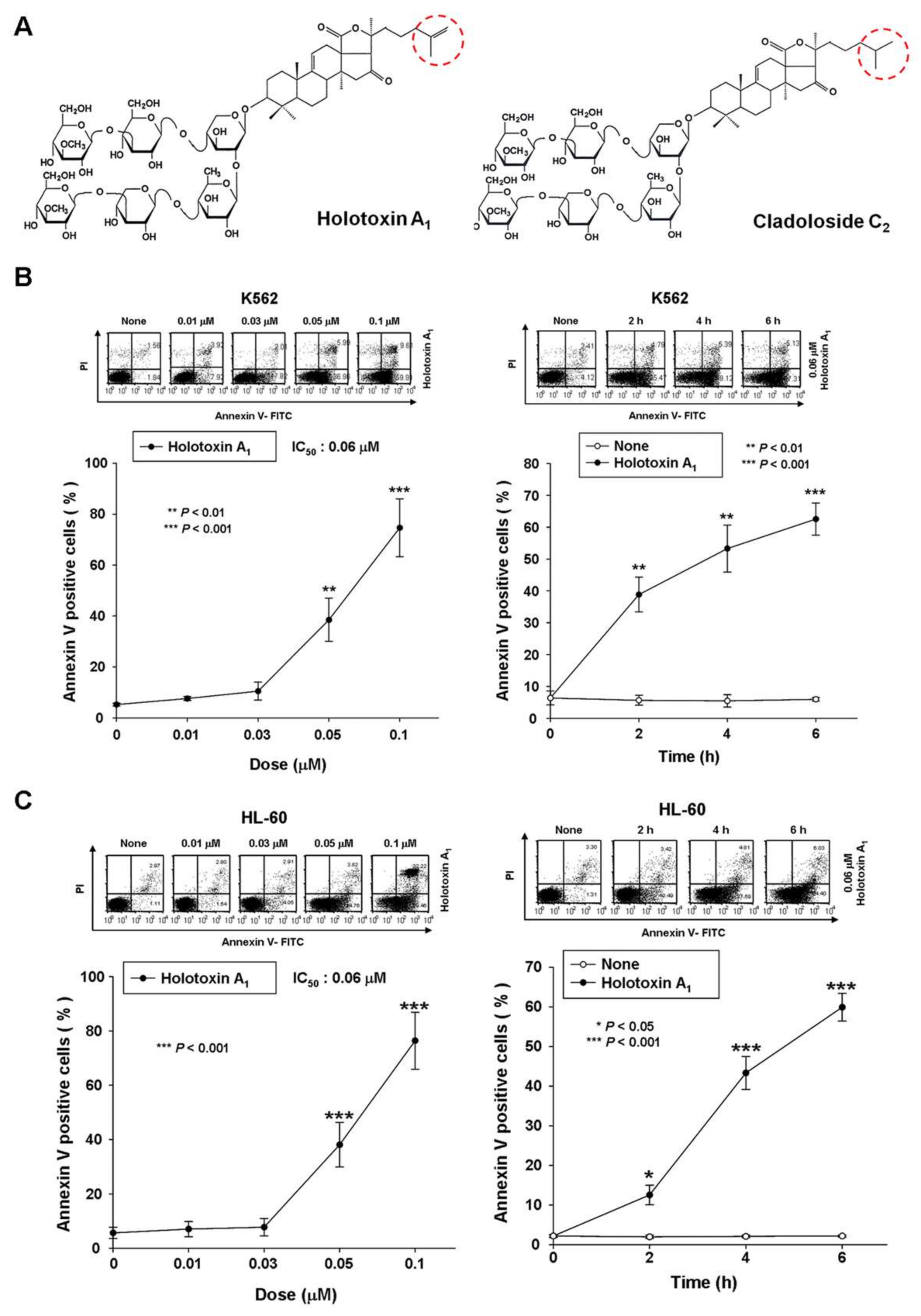
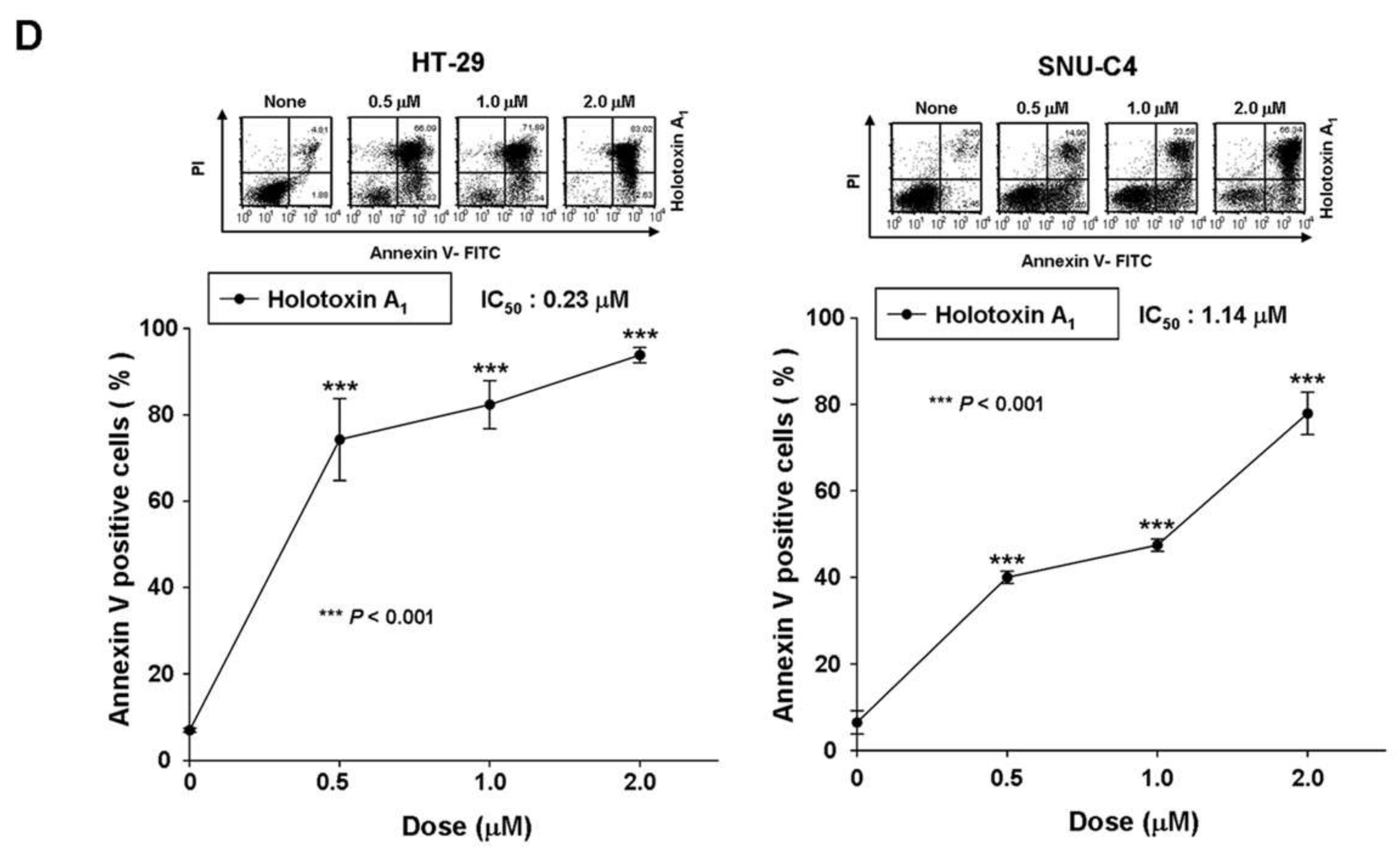
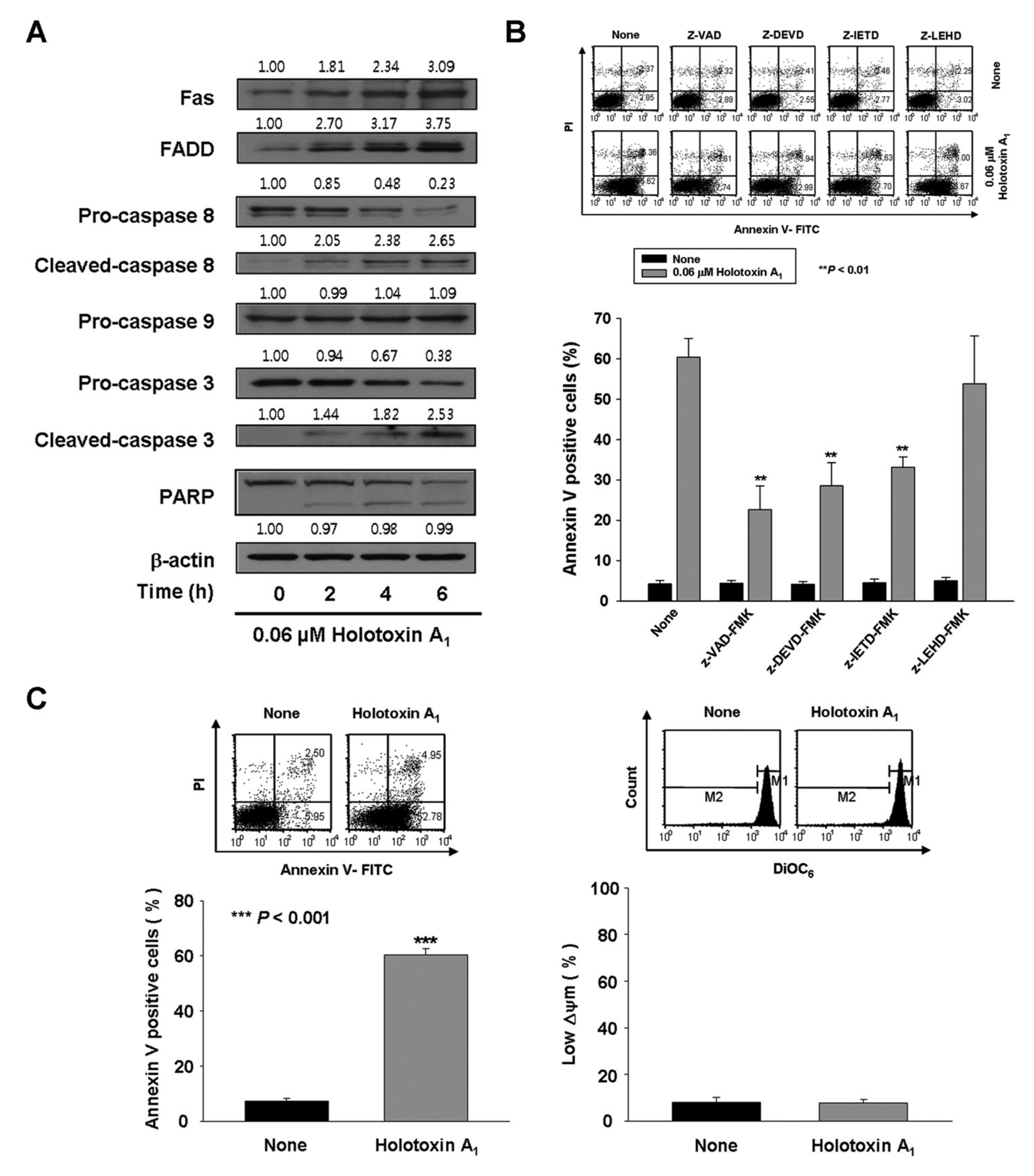
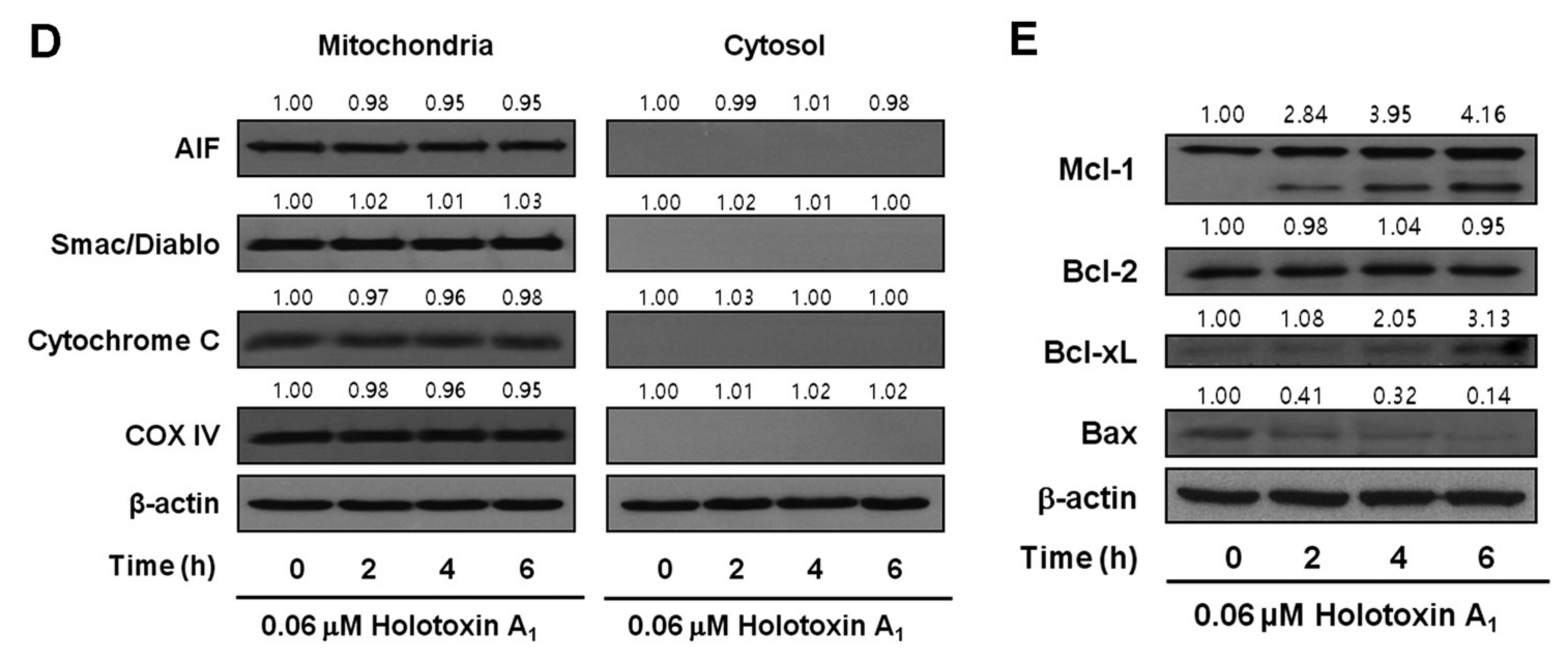
2.1. Holotoxin A1 Induces Apoptosis in K562 Cells by Activating the Extrinsic Pathway
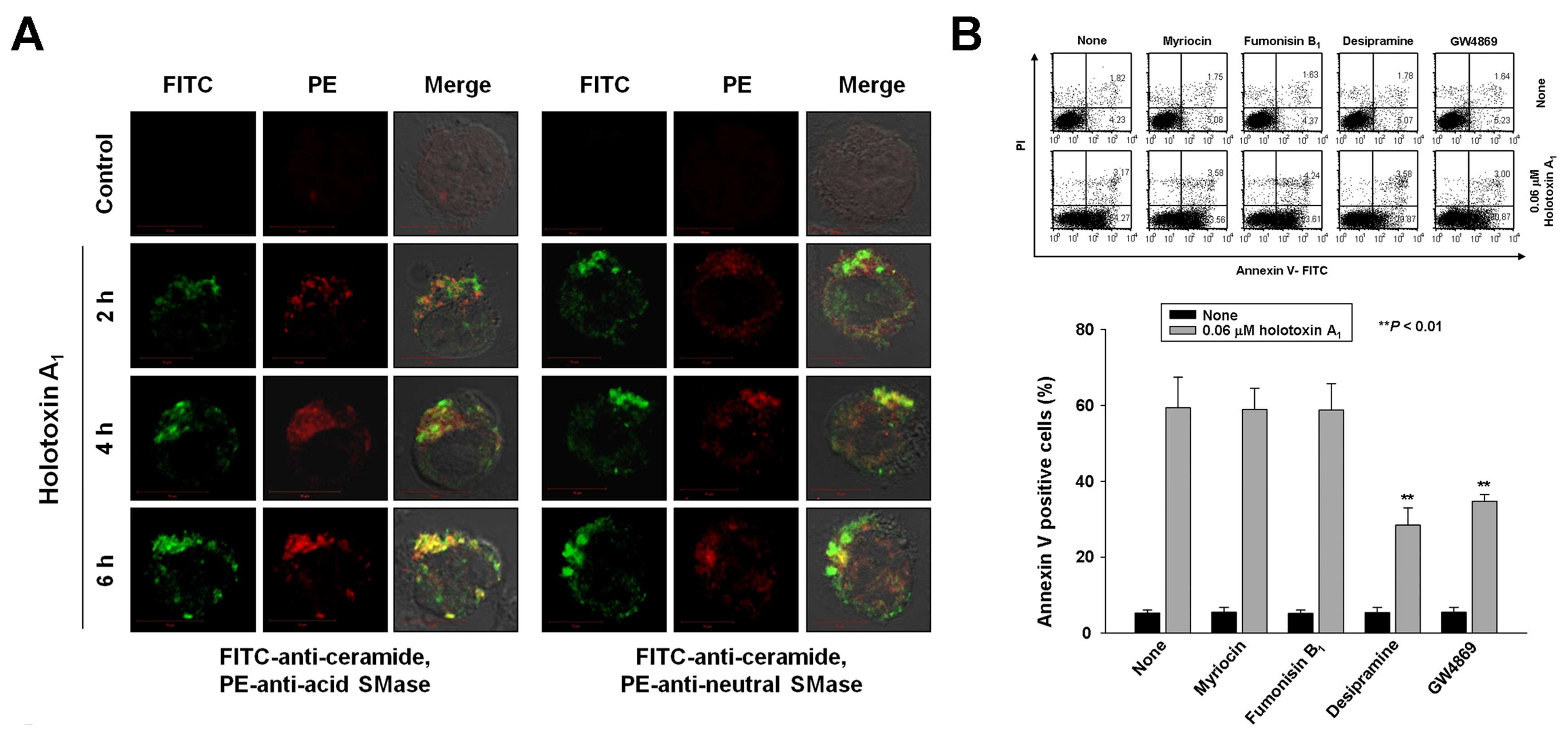
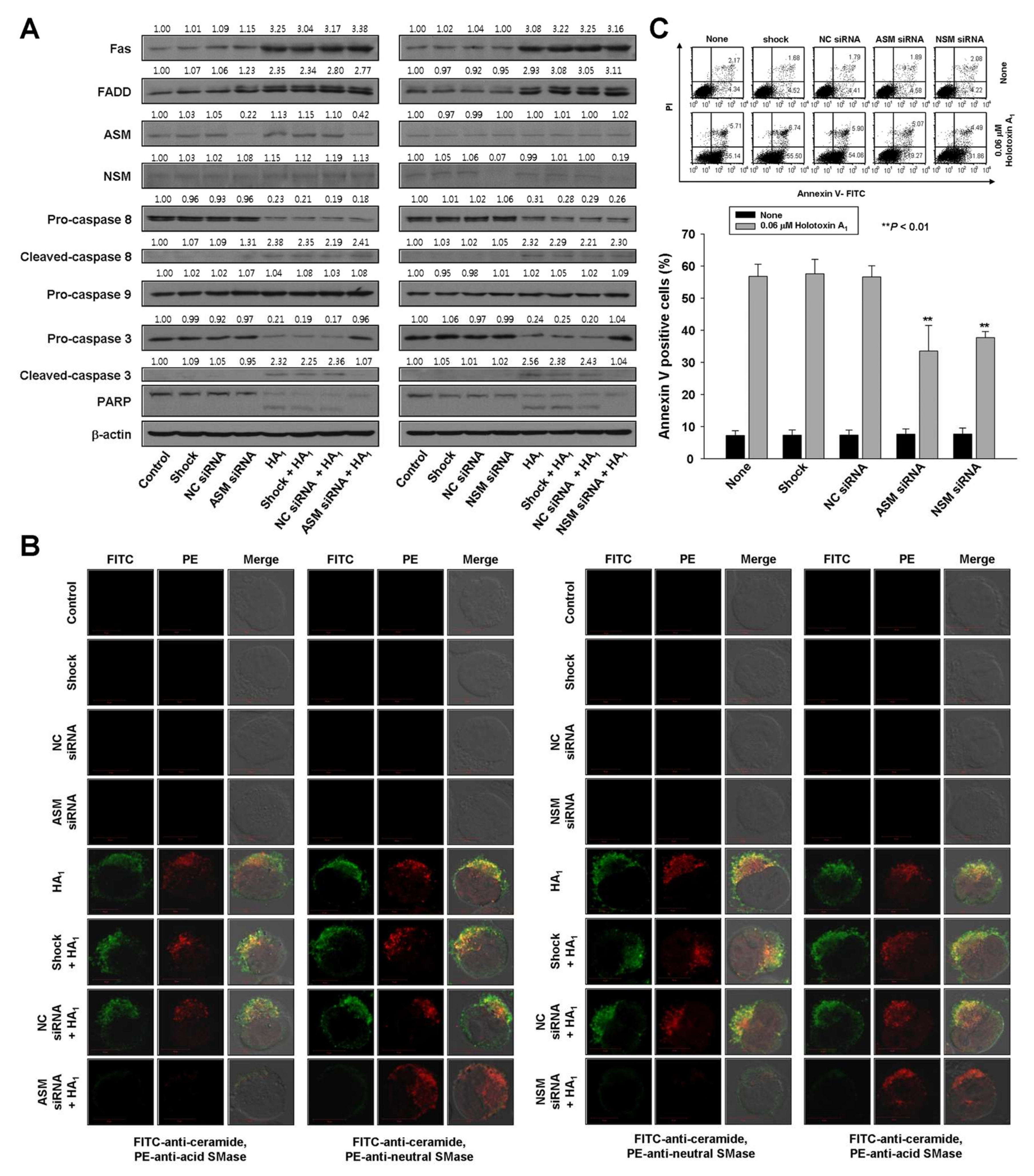
2.2. Holotoxin A1 Induces Apoptosis in K562 Cells by Activating Fas, then Acid SMase and Neutral SMase
2.3. Holotoxin A1 Induces Apoptosis via Activation of Acid and Neutral SMases in Primary Human Leukemia Cells, But not in Normal Human Hematopoietic Progenitor Cells (CD34+ Cells)
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Apoptosis Analysis
4.4. Measurement of MMP
4.5. Separation of the Mitochondrial and Cytosolic Proteins
4.6. Separation of the Cytosolic and Membrane Proteins
4.7. Western Blot Analysis
4.8. Immunofluorescence Staining
4.9. siRNA Transfection
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shipley, J.L.; Butera, J.N. Acute myelogenous leukemia. Exp. Hamatol. 2009, 37, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, B.C.; Gillet, J.P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug resistance: Still a daunting challenge to the successful treatment of AML. Drug Resist. Updates 2012, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Tumor suppressive functions of ceramide: Evidence and mechanisms. Apoptosis 2015, 20, 689–711. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.N.; Goni, F.M.; Alonso, A. Compartmentalization of ceramide signaling: Physical foundations and biological effects. J. Cell. Physiol. 2000, 184, 285–300. [Google Scholar] [CrossRef]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signaling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Strum, J.C.; Ghosh, S.; Bell, R.M. Lipid second messengers—A role in cell growth regulation and cell cycle regulation. Adv. Exp. Mol. Biol. 1997, 407, 421–431. [Google Scholar]
- Yun, S.H.; Shin, S.W.; Stonik, V.A.; Park, J.I. Ceramide as a target of marine triterpene glycosides for treatment of human myeloid leukemia. Mar. Drugs 2016, 14, 205. [Google Scholar] [CrossRef] [PubMed]
- Kitatani, K.; Taniguchi, M.; Okazaki, T. Role of sphingolipids and metabolizing enzymes in hematological malignancies. Mol. Cells 2015, 38, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Beckman, B.S.; Foroozesh, M. A review of ceramide analogs as potential anticancer agents. Future Med. Chem. 2013, 5, 1405–1421. [Google Scholar] [CrossRef] [PubMed]
- Aminin, D.; Pislyagin, E.; Astashev, M.; Es’kov, A.; Kozhemyako, V.; Avilov, S.; Zelepuga, Z.; Yurchenko, E.; Kaluzhskiy, L.; Kozlovskaya, E.; et al. Glycosides from edible sea cucumbers stimulate macrophages via purinergic receptors. Sci. Rep. 2016, 6, 39683. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, V.I.; Aminin, D.L.; Avilov, S.A.; Silchenko, A.S.; Stonik, V.A. Triterpene glycosides from sea cucumbers (Holothurioidae, Echinodermata), biological activities and functions. In Studies in Natural Product Chemistry (Bioactive Natural Products); ur Rahman, A., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2008; Volume 35, pp. 135–196. [Google Scholar]
- Bahrami, Y.; Franco, C.M. Acetylated triterpene glycosides and their biological activity from Holothuroidea reported in the past six decades. Mar. Drugs 2016, 14, 147. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.H.; Park, E.S.; Shin, S.W.; Na, W.Y.; Han, J.Y.; Jeong, J.S.; Shastina, V.V.; Stonik, V.A.; Park, J.I.; Kwak, J.Y. Stichoposide C induces apoptosis through the generation of ceramide in leukemia and colorectal cancer cells and shows in vivo antitumor activity. Clin. Cancer Res. 2012, 18, 5934–5948. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.H.; Park, E.S.; Shin, S.W.; Ju, M.H.; Han, J.Y.; Jeong, J.S.; Kim, S.H.; Stonik, V.A.; Kwak, J.Y.; Park, J.I. By activating Fas/ceramide synthase 6/p38 kinase in lipid rafts, stichoposide D inhibits growth of leukemia xenografts. Oncotarget 2015, 6, 27596–27612. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.H.; Sim, E.H.; Han, S.H.; Kim, T.R.; Ju, M.H.; Han, J.Y.; Jeong, J.S.; Kim, S.H.; Sichenko, A.S.; Stonik, V.A.; et al. In vitro and in vivo anti-leukemic effects of cladoloside C2 are mediated by activation of Fas/ceramide synthase 6/p38 kinase/c-Jun NH2-terminal kinase/caspase-8. Oncotraget 2017, 9, 495–511. [Google Scholar]
- Silchenko, A.S.; Kalinovsky, A.I.; Avilov, S.A.; Andryjaschenko, P.V.; Dmitrenok, P.S.; Yurchenko, E.A.; Dolmatov, I.Y.; Kalinin, V.I.; Stonik, V.A. Structure and biological action of cladolosides B1, B2, C, C1, C2 and D, six new triterpene glycosides from the sea cucumber Cladolabes schmeltzii. Nat. Prod. Commun. 2013, 8, 1527–1534. [Google Scholar] [PubMed]
- Maltsev, I.I.; Stonik, V.A.; Kalinovsky, A.I.; Elyakov, G.B. Triterpene glycosides from sea cucumber Stichopus japonicus Selenka. Comp. Biochem. Physiol. 1984, 78, 421–426. [Google Scholar] [CrossRef]
- Aminin, D.L.; Menchinskaya, E.S.; Pislyagin, E.A.; Silchenko, A.S.; Avilov, S.A.; Kalinin, V.I. Sea cucumber triterpene glycosides as anticancer agents. In Studies in Natural Products Chemistry, 1st ed.; ur Rahman, A., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2016; Volume 49, pp. 55–105. ISBN 978-0-444-63601-0. [Google Scholar]
- Wargasetia, T.L.; Widodo. Mechanisms of cancer cell killing by sea cucumber-derived compounds. Investig. New Drugs 2017, 35, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Grant, S. Mcl-1 as a therapeutic target in acute myelogenous leukemia (AML). Leuk. Res. Rep. 2013, 2, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Wang, Z.; Philip, P.A.; Mohammad, R.M.; Sarkar, F.H. Emerging Bcl-2 inhibitors for the treatment of cancer. Expert Opin. Emerg. Drugs 2011, 16, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.A.; Dash, R.; Azab, B.; Sarkar, S.; Das, S.K.; Kumar, S.; Oyesanya, R.A.; Dasgupta, S.; Dent, P.; Grant, S.; et al. Targeting Mcl-1 for the therapy of cancer. Expert Opin. Investig. Drugs 2011, 20, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Mondol, M.A.M.; Shin, H.J.; Rahman, M.A.; Islam, M.T. Sea cucumber glycosides: Chemical structures, producing species and important biological properties. Mar. Drugs 2017, 15, 317. [Google Scholar] [CrossRef] [PubMed]
- Mimaki, Y.; Yokosuka, A.; Kuroda, M.; Sashida, Y. Cytotoxic activities and structure-cytotoxic relationships of steroidal saponins. Biol. Pharm. Bull. 2001, 24, 1286–1289. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.W.; Seo, C.Y.; Han, H.; Han, J.Y.; Jeong, J.S.; Kwak, J.Y.; Park, J.I. 15d-PGJ2 induces apoptosis by reactive oxygen species-mediated inactivation of Akt in leukemia and colorectal cancer cells and shows in vivo antitumor activity. Clin. Cancer Res. 2009, 15, 5414–5425. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Han, J.S.; Seo, C.Y.; Park, T.H.; Kwon, H.C.; Jeong, J.S.; Kim, I.H.; Yun, J.; Bae, Y.S.; Kwak, J.Y.; et al. Pioglitazone, a synthetic ligand for PPARγ, induces apoptosis in RB-deficient human colorectal cancer cells. Apoptosis 2006, 11, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Shin, S.W.; Seo, C.Y.; Kwon, H.C.; Han, J.Y.; Kim, I.H.; Kwak, J.Y.; Park, J.I. 15-Deoxy-∆12,14-prostaglandin J2 (15d-PGJ2) sensitizes human leukemic HL-60 cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through Akt downregulation. Apoptosis 2007, 12, 2101–2114. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Sex/Age | Diagnosis (FAB) | Conventional Karyotype Analysis | IC50 of Holotoxin A1 (μM) |
|---|---|---|---|---|
| P1 | F/63 | AML M2 | Normal female chromosome | 0.013 |
| P2 | M/51 | AML M3 | Abnormal male chromosome 46,XY,t(15;17)(q24.1;q21.1) [26] | 0.081 |
| P3 | F/37 | AML M2 | Normal female chromosome | 0.068 |
| P4 | F/70 | AML M4E | Abnormal female chromosome 46,XX,inv(16)(p13.1q22) [24]/46,XX [1] | 0.058 |
| P5 | M/86 | AML M4 | Normal male chromosome | 0.061 |
| P6 | F/53 | AML M1 | Normal female chromosome | 0.051 |
| P7 | F/33 | AML M4 | Abnormal female chromosome 46,XX,t(11;19)(p15;p12) [26]/46,XX [1] | 0.059 |
| P8 | M/72 | AML M5b | Abnormal male chromosome 46,XY,t(6;11)(q27;q23) [26] | 0.030 |
| P9 | M/79 | AML M1 | Abnormal male chromosome 47,XY,+4 [12]/46,XY [14] | 0.076 |
| P10 | M/28 | CML | Abnormal male chromosome 46,XY,t(9;22)(q34;q11.2) [25] | 0.096 |
| P11 | F/40 | CML | Abnormal female chromosome 46,XX,t(9;22)(q34;q11.2) [20] | 0.098 |
| P12 | F/53 | CML | Abnormal female chromosome 46,XX,t(9;22)(q34;q11.2) [22] | 0.036 |
| P13 | F/82 | CML | Abnormal female chromosome 46,XX,t(9;22)(q34;q11.2) [22] | 0.057 |
| P14 | M/47 | T-ALL | Normal male chromosome | 0.070 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, S.-H.; Sim, E.-H.; Han, S.-H.; Han, J.-Y.; Kim, S.-H.; Silchenko, A.S.; Stonik, V.A.; Park, J.-I. Holotoxin A1 Induces Apoptosis by Activating Acid Sphingomyelinase and Neutral Sphingomyelinase in K562 and Human Primary Leukemia Cells. Mar. Drugs 2018, 16, 123. https://doi.org/10.3390/md16040123
Yun S-H, Sim E-H, Han S-H, Han J-Y, Kim S-H, Silchenko AS, Stonik VA, Park J-I. Holotoxin A1 Induces Apoptosis by Activating Acid Sphingomyelinase and Neutral Sphingomyelinase in K562 and Human Primary Leukemia Cells. Marine Drugs. 2018; 16(4):123. https://doi.org/10.3390/md16040123
Chicago/Turabian StyleYun, Seong-Hoon, Eun-Hye Sim, Sang-Heum Han, Jin-Yeong Han, Sung-Hyun Kim, Alexandra S. Silchenko, Valentin A. Stonik, and Joo-In Park. 2018. "Holotoxin A1 Induces Apoptosis by Activating Acid Sphingomyelinase and Neutral Sphingomyelinase in K562 and Human Primary Leukemia Cells" Marine Drugs 16, no. 4: 123. https://doi.org/10.3390/md16040123