Lactones from the Sponge-Derived Fungus Talaromyces rugulosus

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
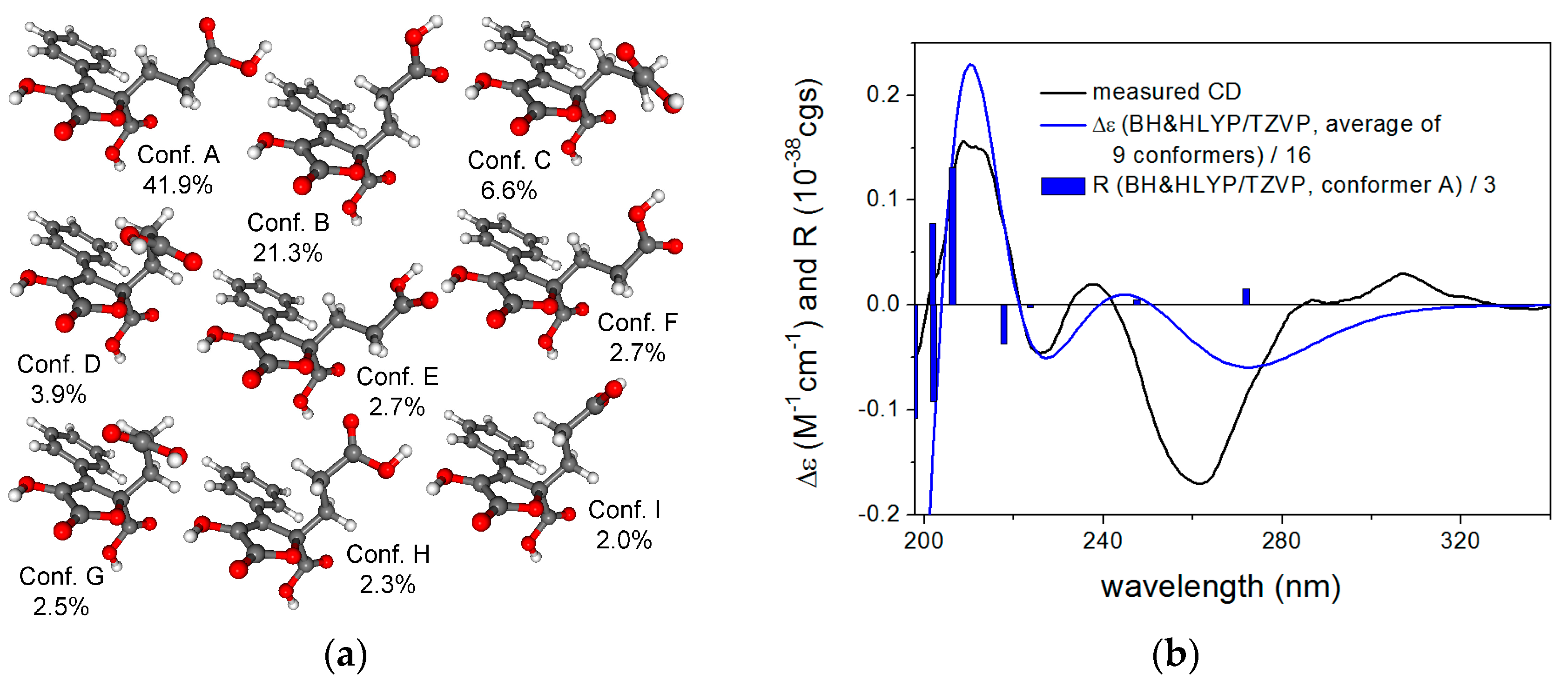
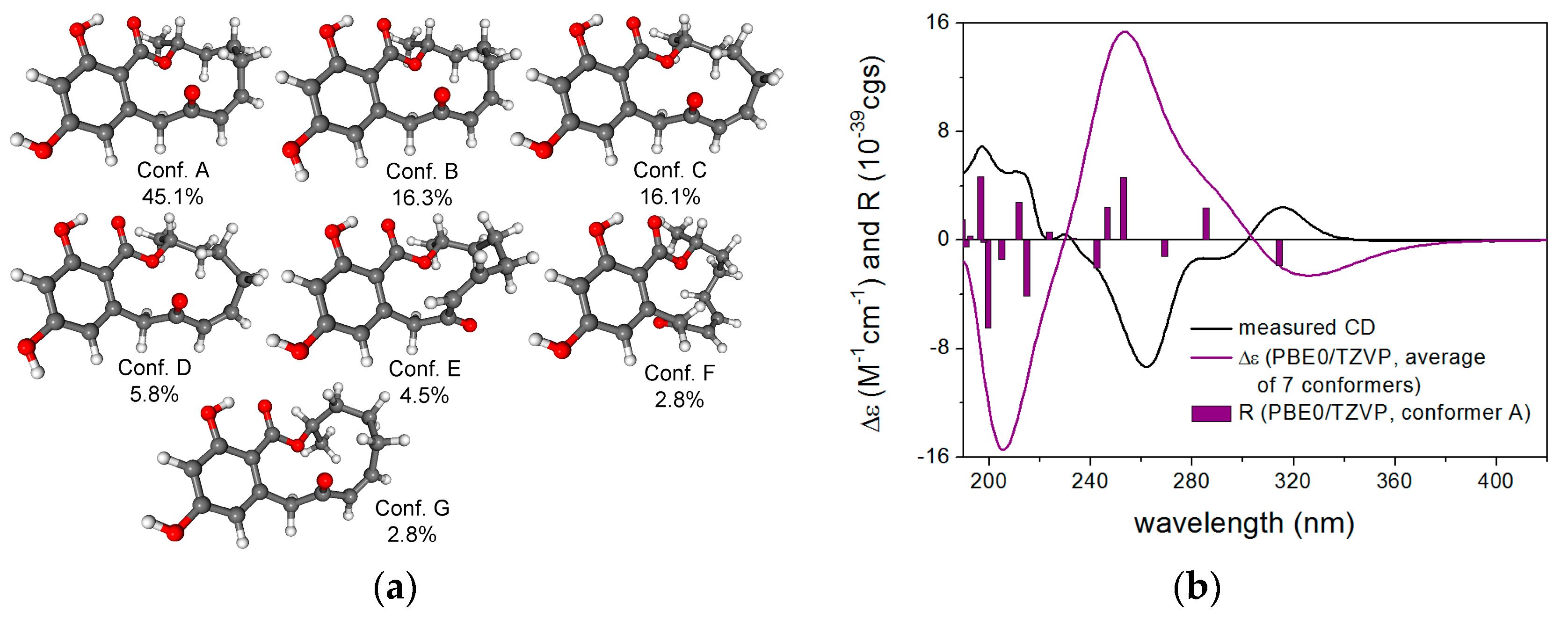
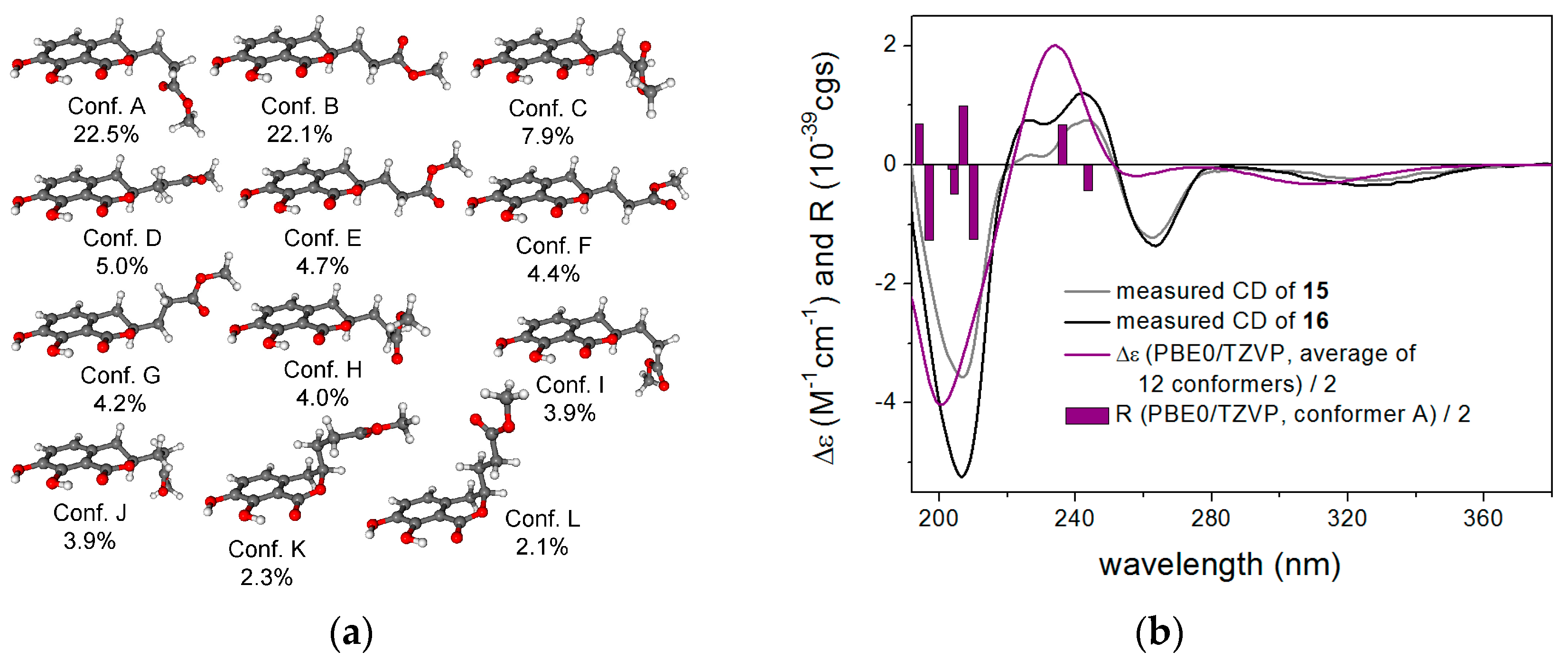
2.1. Structure Elucidation
2.2. Biological Activities
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Fungal Material
4.3. Fermentation, Extraction and Isolation
4.4. Computational Section
4.5. Cytotoxicity Assay
4.6. Antimicrobial Assay
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hong, J.H.; Jang, S.; Heo, Y.M.; Min, M.; Lee, H.; Lee, Y.M.; Lee, H.; Kim, J.J. Investigation of marine-derived fungal diversity and their exploitable biological activities. Mar. Drugs 2015, 13, 4137–4155. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Ebada, S.; Proksch, P. Marine-Derived Fungal Metabolites. In Springer Handbook of Marine Biotechnology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 759–788. [Google Scholar]
- Ebrahim, W.; El-Neketi, M.; Lewald, L.I.; Orfali, R.S.; Lin, W.; Rehberg, N.; Kalscheuer, R.; Daletos, G.; Proksch, P. Metabolites from the fungal endophyte Aspergillus austroafricanus in axenic culture and in fungal-bacterial mixed cultures. J. Nat. Prod. 2016, 79, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dai, H.; Makhloufi, G.; Heering, C.; Janiak, C.; Hartmann, R.; Mándi, A.; Kurtán, T.; Müller, W.E.G.; Kassack, M.U.; et al. Cytotoxic 14-membered macrolides from a mangrove-derived endophytic fungus, Pestalotiopsis microspora. J. Nat. Prod. 2016, 79, 2332–2340. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, M.S.; Ebada, S.S.; Ashour, M.L.; Ebrahim, W.; Müller, W.E.G.; Mándi, A.; Kurtán, T.; Singab, A.; Lin, W.; Liu, Z.; et al. Xanthones and sesquiterpene derivatives from a marine-derived fungus Scopulariopsis sp. Tetrahedron 2016, 72, 2411–2419. [Google Scholar] [CrossRef]
- Elnaggar, M.S.; Ebada, S.S.; Ashour, M.L.; Ebrahim, W.; Singab, A.; Lin, W.; Liu, Z.; Proksch, P. Two new triterpenoids and a new naphthoquinone derivative isolated from a hard coral-derived fungus Scopulariopsis sp. Fitoterapia 2017, 116, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lin, J.; Lu, X.; Zheng, Z.; Ren, X.; Zhang, H.; He, J.; Yang, J. Cathepsin B inhibitory tetraene lactones from the fungus Talaromyces wortmannii. Helv. Chim. Acta 2009, 92, 567–574. [Google Scholar] [CrossRef]
- Liu, F.; Cai, X.L.; Yang, H.; Xia, X.K.; Guo, Z.Y.; Yuan, J.; Li, M.F.; She, Z.G.; Lin, Y.C. The bioactive metabolites of the mangrove endophytic fungus Talaromyces sp. ZH-154 isolated from Kandelia candel (L.) Druce. Planta Med. 2010, 76, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Bara, R.; Aly, A.H.; Pretsch, A.; Wray, V.; Wang, B.; Proksch, P.; Debbab, A. Antibiotically active metabolites from Talaromyces wortmannii, an endophyte of Aloe vera. J. Antibiot. 2013, 66, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Zhai, M.M.; Li, J.; Jiang, C.X.; Shi, Y.P.; Di, D.L.; Crews, P.; Wu, Q.X. The bioactive secondary metabolites from Talaromyces species. Nat. Prod. Bioprospect. 2016, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, N.; Visagie, C.M.; Houbraken, J.; Frisvad, J.C.; Samson, R.A. Polyphasic taxonomy of the genus Talaromyces. Stud. Mycol. 2014, 78, 175–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghisalberti, E.L.; Hargreaves, J.R.; McConville, E. Butenolides from a cultured microfungus, Acremonium sp. Nat. Prod. Res. 2004, 18, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Ancheeva, E.; Küppers, L.; Akone, S.H.; Ebrahim, W.; Liu, Z.; Mándi, A.; Kurtán, T.; Lin, W.; Orfali, R.; Rehberg, N.; et al. Expanding the metabolic profile of the fungus Chaetomium sp. through co-culture with autoclaved Pseudomonas aeruginosa. Eur. J. Org. Chem. 2017, 2017, 3256–3264. [Google Scholar] [CrossRef]
- Pescitelli, G.; Bruhn, T. Good computational practice in the assignment of absolute configurations by TDDFT calculations of ECD spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Mándi, A.; Mudianta, I.W.; Kurtán, T.; Garson, M.J. Absolute configuration and conformational study of psammaplysins A and B from the Balinese marine sponge Aplysinella strongylata. J. Nat. Prod. 2015, 78, 2051–2056. [Google Scholar] [CrossRef] [PubMed]
- Barrow, C.J. New macrocyclic lactones from a Penicillium species. J. Nat. Prod. 1997, 60, 1023–1025. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, T.; Espinosa-Artiles, P.; Tang, Y.; Zhan, J.; Molnár, I. Insights into the biosynthesis of 12-membered resorcylic acid lactones from heterologous production in Saccharomyces cerevisiae. ACS Chem. Biol. 2014, 9, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Meng, L.H.; Mándi, A.; Li, X.M.; Kurtán, T.; Wang, B.G. Structure, absolute configuration, and conformational study of resorcylic acid derivatives and related congeners from the fungus Penicillium brocae. RSC Adv. 2015, 5, 39870–39877. [Google Scholar] [CrossRef]
- An, Y.N.; Zhang, X.; Zhang, T.Y.; Zhang, M.Y.; Zhang, Q.; Deng, X.Y.; Zhao, F.; Zhu, L.J.; Wang, G.; Zhang, J.; et al. Penicimenolides A–F, resorcylic acid lactones from Penicillium sp., isolated from the rhizosphere soil of Panax notoginseng. Sci. Rep. 2016, 6, 27396–27409. [Google Scholar] [CrossRef] [PubMed]
- Kerti, G.; Kurtán, T.; Illyés, T.Z.; Kövér, K.E.; Sólyom, S.; Pescitelli, G.; Fujioka, N.; Berova, N.; Antus, S. Enantioselective synthesis of 3-methylisochromans and determination of their absolute configurations by circular dichroism. Eur. J. Org. Chem. 2007, 296–305. [Google Scholar] [CrossRef]
- Krohn, K.; Kock, I.; Elsässer, B.; Flörke, U.; Schulz, B.; Dräger, S.; Pescitelli, G.; Antus, S.; Kurtán, T. Bioactive natural products from the endophytic fungus Ascochyta sp. from meliotus dentatus—Configurational assignment by solid-state CD and TDDFT calculations. Eur. J. Org. Chem. 2007, 1123–1129. [Google Scholar]
- Hussain, H.; Akhtar, N.; Dräger, S.; Schulz, B.; Pescitelli, G.; Salvadori, P.; Antus, S.; Kurtán, T.; Krohn, K. New bioactive 2,3-epoxycyclohexenes and isocoumarins from the endophytic fungus Phomopsis sp. from Laurus azorica. Eur. J. Org. Chem. 2009, 749–756. [Google Scholar] [CrossRef]
- Li, S.; Wei, M.; Chen, G.; Lin, Y. Two new dihydroisocoumarins from the endophytic fungus Aspergillus sp. collected from the South China Sea. Chem. Nat. Compd. 2012, 48, 371–373. [Google Scholar] [CrossRef]
- Yadav, J.S.; Mishra, A.K.; Dachavaram, S.S.; Kumar, S.G.; Das, S. First enantioselective total synthesis of penicimarin B, aspergillumarins A and B. Tetrahedron Lett. 2014, 55, 2921–2923. [Google Scholar] [CrossRef]
- El Amrani, M.; Debbab, A.; Aly, A.H.; Wray, V.; Dobretsov, S.; Müller, W.E.G.; Lin, W.; Lai, D.; Proksch, P. Farinomalein derivatives from an unidentified endophytic fungus isolated from the mangrove plant Avicennia marina. Tetrahedron Lett. 2012, 53, 6721–6724. [Google Scholar] [CrossRef]
- Ebrahim, W.; Aly, A.H.; Mándi, A.; Totzke, F.; Kubbutat, M.H.G.; Wray, V.; Lin, W.H.; Dai, H.; Proksch, P.; Kurtán, T.; et al. Decalactone derivatives from Corynespora cassiicola, an endophytic fungus of the mangrove plant Laguncularia racemosa. Eur. J. Org. Chem. 2012, 2012, 3476–3484. [Google Scholar] [CrossRef]
- Zhuravleva, O.I.; Sobolevskaya, M.P.; Leshchenko, E.V.; Kirichuk, N.N.; Denisenko, V.A.; Dmitrenok, P.S.; Dyshlovoy, S.A.; Zakharenko, A.M.; Kim, N.Y.; Afiyatullov, S.S. Meroterpenoids from the alga-derived fungi Penicillium thomii Maire and Penicillium lividum Westling. J. Nat. Prod. 2014, 77, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.J.; Yang, L.Q.; Hua, L.; Gao, K. Labdanediterpenoids and shikimic acid derivatives from Araucaria cunninghamii. Planta Med. 2011, 77, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Dangroo, N.A.; Raina, A.; Qayum, A.; Singh, S.; Kumar, A.; Sangwan, P.L. Biologically active xanthones from Codonopsis ovate. Phytochemistry 2016, 132, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Kjer, J.; Debbab, A.; Aly, A.H.; Proksch, P. Methods for isolation of marine-derived endophytic fungi and their bioactive secondary products. Nat. Protoc. 2010, 5, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Xu, D.X.; Mándi, A.; Kurtán, T.; Li, T.J.; Schulz, B.; Zhang, W. Structure, absolute configuration, and conformational study of 12-membered macrolides from the fungus Dendrodochium sp. associated with the sea cucumber Holothuria nobilis Selenka. J. Org. Chem. 2013, 78, 7030–7047. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010.
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- MacroModel. Schrödinger, LLC, 2015. Available online: http://www.schrodinger.com/ (accessed on 31 December 2013).
- Varetto, U. MOLEKEL, version 5.4; Swiss National Supercomputing Centre: Manno, Switzerland, 2009.
- Ashour, M.; Edrada, R.; Ebel, R.; Wray, V.; Wätjen, W.; Padmakumar, K.; Müller, W.E.G.; Lin, W.H.; Proksch, P. Kahalalide derivatives from the Indian sacoglossan mollusk Elysia grandifolia. J. Nat. Prod. 2006, 69, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standard, 9th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 a | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 171.0, C | 168.3, C | 170.0, C | |||
| 2 | 139.5, C | 142.7, C | 141.6, C | |||
| 3 | 130.6, C | 124.6, C | 128.7, C | |||
| 4 | 79.3, CH | 5.52, dd (8.1, 2.2) | 109.8, C | 86.8, C | ||
| 5 | 30.5, CH2 | 2.45, m | 34.6, CH2 | 2.54, ddd (14.5, 7.9, 6.5) | 30.0, CH2 | 2.68, ddd (15.2, 8.7, 6.4) |
| 1.75, m | 2.18, ddd (14.5, 7.9, 6.5) | 2.48, ddd (15.2, 8.7, 6.4) | ||||
| 6 | 30.0, CH2 | 2.44, m | 29.4, CH2 | 2.42, ddd (16.0, 7.9, 6.5) | 29.1, CH2 | 2.25, ddd (15.8, 8.7, 6.4) |
| 2.31, ddd (16.0, 7.9, 6.5) | 2.15, ddd (15.8, 8.7, 6.4) | |||||
| 7 | 174.6, C | 174.5, C | 174.2, C | |||
| 8 | 131.9, C | 131.3, C | 131.3, C | |||
| 9, 13 | 128.6, CH | 7.75, d (7.5) | 128.6, CH | 7.93, d (7.5) | 128.6, CH | 7.76, d (7.4) |
| 10, 12 | 129.7, CH | 7.44, t (7.5) | 129.8, CH | 7.45, t (7.5) | 129.7, CH | 7.42, t (7.4) |
| 11 | 129.7, CH | 7.35, t (7.5) | 129.8, CH | 7.36, t (7.5) | 129.9, CH | 7.35, t (7.4) |
| 14 | 171.9, C | |||||
| 1′ | 65.6, CH2 | 4.08, dt (10.8, 6.6) | 65.6, CH2 | 4.02, dt (10.8, 6.7) | 65.7, CH2 | 4.00, dt (10.8, 6.7) |
| 4.04, dt (10.8, 6.6) | 3.97, dt (10.8, 6.7) | 3.94, dt (10.8, 6.7) | ||||
| 2′ | 31.7, CH2 | 1.60, m | 31.7, CH2 | 1.56, m | 31.6, CH2 | 1.54, m |
| 3′ | 20.2, CH2 | 1.38, m | 20.2, CH2 | 1.36, m | 20.1, CH2 | 1.34, m |
| 4′ | 14.0, CH3 | 0.94, t (7.3) | 14.0, CH3 | 0.93, t (7.3) | 14.0, CH3 | 0.91, t (7.3) |
| OMe-4 | 50.7, CH3 | 3.18, s | ||||
| No. | 6 a | 7 b | 8 b | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type c | δH (J in Hz) | δC, Type c | δH (J in Hz) | |
| 1 | 172.9, C | n.d. d | n.d. d | |||
| 3 | 76.9, CH | 5.03, m | 75.5, CH | 4.95, m | 73.1, CH | 5.19, m |
| 4 | 32.2, CH2 | 1.87, m | 33.1, CH2 | 1.91, m | 32.9, CH2 | 1.68, m |
| 1.65, m | 1.75, m | |||||
| 5 | 26.4, CH2 | 1.68, m | 21.5, CH2 | 1.70, m | 18.9, CH2 | 1.58, m |
| 1.43, m | ||||||
| 6 | 27.6, CH2 | 2.52, m | 36.5, CH2 | 1.58, m | 36.8, CH2 | 1.81, m |
| 2.21, m | 1.50, m | |||||
| 7 | 139.9, CH | 5.81, ddd (11.8, 10.0, 5.7) | 66.2, CH | 4.39, m | 66.9, CH | 4.36, m |
| 8 | 132.8, CH | 6.51, br d (11.8) | 53.5, CH2 | 3.03, dd (13.0, 3.1) | 51.3, CH2 | 2.94, dd (15.4, 10.1) |
| 2.42, dd (13.0, 10.3) | 2.61, dd (15.4, 1.6) | |||||
| 9 | 204.7, C | 206.3, C | 205.2, C | |||
| 10 | 51.5, CH2 | 4.58, d (18.4) | 50.2, CH2 | 4.84, d (18.4) | 52.3, CH2 | 4.59, d (18.5) |
| 3.65, d (18.4) | 3.75, d (18.4) | 3.90, d (18.5) | ||||
| 11 | 140.1, C | 140.0, C | 140.2, C | |||
| 12 | 113.9, CH | 6.16, d (2.5) | 113.5, CH | 6.23, d (2.5) | 113.2, CH | 6.23, d (2.5) |
| 13 | 164.1, C | 162.7, C | 162.9, C | |||
| 14 | 103.0, CH | 6.26, d (2.5) | 102.2, CH | 6.31, d (2.5) | 102.3, CH | 6.32, d (2.5) |
| 15 | 167.1, C | 165.5, C | 166.4, C | |||
| 16 | 106.2, C | 106.3, C | 105.6, C | |||
| 17 | 21.5, CH3 | 1.28, d (6.2) | 20.9, CH3 | 1.31, d (6.1) | 18.2, CH3 | 1.29, d (6.4) |
| 7-OMe | ||||||
| No. | 9 a | 10 a | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 172.5, C | 172.3, C | ||
| 3 | 76.1, CH | 4.93, m | 73.8, CH | 5.19, m |
| 4 | 33.8, CH2 | 1.82, m | 33.5, CH2 | 1.63, m |
| 1.75, m | ||||
| 5 | 21.7, CH2 | 1.68, m | 19.0, CH2 | 1.58, m |
| 1.40, m | ||||
| 6 | 34.4, CH2 | 1.69, m | 34.0, CH2 | 1.87, m |
| 1.50, m | 1.49, m | |||
| 7 | 77.1, CH | 3.95, m | 78.2, CH | 3.90, m |
| 8 | 49.6, CH2 | 3.10, dd (13.3, 2.8) | 47.4, CH2 | 2.89, dd (15.7, 9.7) |
| 2.40, dd (13.3, 10.2) | 2.71, dd (15.7, 1.0) | |||
| 9 | 208.8, C | 208.1, C | ||
| 10 | 51.0, CH2 | 4.68, d (18.7) | 53.2, CH2 | 4.63, d (18.8) |
| 3.80, d (18.7) | 3.84, d (18.8) | |||
| 11 | 139.6, C | 139.9, C | ||
| 12 | 113.9, CH | 6.14, d (2.5) | 113.7, CH | 6.13, d (2.5) |
| 13 | 163.7, C | 163.9, C | ||
| 14 | 102.9, CH | 6.25, d (2.5) | 103.0, CH | 6.25, d (2.5) |
| 15 | 166.0, C | 166.9, C | ||
| 16 | 107.0, C | 106.4, C | ||
| 17 | 21.1, CH3 | 1.32, d (6.1) | 18.5, CH3 | 1.29, d (6.4) |
| 7-OMe | 56.7, CH3 | 3.42, s | 56.5, CH3 | 3.34, s |
| No. | 11 a | 12 b | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 172.5, C | 172.3, C | ||
| 3 | 76.2, CH | 4.94, m | 73.9, CH | 5.19, m |
| 4 | 33.8, CH2 | 1.80, m | 33.6, CH2 | 1.64, m |
| 1.75, m | ||||
| 5 | 21.9, CH2 | 1.69, m | 19.1, CH2 | 1.58, m |
| 1.40, m | ||||
| 6 | 34.6, CH2 | 1.66, m | 34.3, CH2 | 1.84, m |
| 1.50, m | 1.51, m | |||
| 7 | 75.4, CH | 4.04, m | 76.5, CH | 3.98, m |
| 8 | 50.3, CH2 | 3.09, dd (13.2, 3.0) | 48.2, CH2 | 2.90, dd (15.6, 9.8) |
| 2.40, dd (13.2, 10.3) | 2.69, dd (15.6, 1.0) | |||
| 9 | 208.9, C | 208.3, C | ||
| 10 | 50.9, CH2 | 4.69, d (18.7) | 53.1, CH2 | 4.61, d (18.8) |
| 3.78, d (18.7) | 3.84, d (18.8) | |||
| 11 | 139.6, C | 139.9, C | ||
| 12 | 114.0, CH | 6.13, d (2.5) | 113.8, CH | 6.12, d (2.5) |
| 13 | 163.7, C | 163.9, C | ||
| 14 | 102.9, CH | 6.25, d (2.5) | 103.1, CH | 6.25, d (2.5) |
| 15 | 166.0, C | 166.8, C | ||
| 16 | 107.0, C | 106.5, C | ||
| 17 | 21.1, CH3 | 1.31, d (6.1) | 18.6, CH3 | 1.29, d (6.4) |
| 1′ | 69.7, CH2 | 3.63, dt (9.2, 6.4) | 69.5, CH2 | 3.51, dt (9.3, 6.5) |
| 3.51, dt (9.2, 6.4) | 3.45, dt (9.3, 6.5) | |||
| 2′ | 33.2, CH2 | 1.56, m | 33.2, CH2 | 1.53, m |
| 3′ | 20.4, CH2 | 1.42, m | 20.4, CH2 | 1.38, m |
| 4′ | 14.3, CH3 | 0.95, t (7.3) | 14.2, CH3 | 0.93, t (7.3) |
| Compounds | 7 | 9 | 11 | 13 | 8 | 10 | 12 | 14 |
|---|---|---|---|---|---|---|---|---|
| 3J7,8a (Hz) | 3.1 | 2.8 | 3.0 | 3.1 | 10.1 | 9.6 | 9.8 | 10.4 |
| 3J7,8b (Hz) | 10.3 | 10.2 | 10.3 | 10.4 | 1.6 | 1.0 | 1.0 | - a |
| C-3 | 75.5 | 76.1 | 76.2 | 76.1 | 73.1 | 73.8 | 73.9 | 74.0 |
| H-3 | 4.95 | 4.93 | 4.94 | 4.93 | 5.19 | 5.19 | 5.19 | 5.13 |
| C-17 | 20.9 | 21.1 | 21.1 | 20.9 | 18.2 | 18.5 | 18.6 | 18.9 |
| No. | 13 a | 14 a | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 172.4, C | 172.2, C | ||
| 3 | 76.1, CH | 4.93, m | 74.0, CH | 5.13, m |
| 4 | 33.4, CH2 | 1.75, m | 33.8, CH2 | 1.66, m |
| 1.61, m | ||||
| 5 | 21.2, CH2 | 1.39, m | 19.0, CH2 | 1.52, m |
| 1.31, m | ||||
| 6 | 33.2, CH2 | 1.57, m | 32.8, CH2 | 1.84, m |
| 1.56, m | ||||
| 7 | 72.9, CH | 5.54, m | 74.2, CH | 5.53, m |
| 8 | 49.2, CH2 | 2.94, dd (13.3, 3.1) | 46.9, CH2 | 3.07, dd (15.8, 10.4) |
| 2.58, dd (13.3, 10.4) | 2.49, d (15.8) | |||
| 9 | 206.6, C | 206.1, C | ||
| 10 | 50.8, CH2 | 4.67, d (18.8) | 52.8, CH2 | 4.52, d (18.8) |
| 3.78, d (18.8) | 3.73, d (18.8) | |||
| 11 | 139.3, C | 139.6, C | ||
| 12 | 114.1, CH | 6.14, d (2.4) | 113.8, CH | 6.07, d (2.5) |
| 13 | 163.8, C | 163.9, C | ||
| 14 | 103.0, CH | 6.26, d (2.4) | 103.0, CH | 6.24, d (2.5) |
| 15 | 166.2, C | 166.8, C | ||
| 16 | 106.7, C | 106.2, C | ||
| 17 | 20.9, CH3 | 1.28, d (6.2) | 18.9, CH3 | 1.27, d (6.4) |
| 1′ | 169.8, C | 169.7, C | ||
| 2′ | 141.9, C | 141.8, C | ||
| 3′ | 128.1, C | 128.6, C | ||
| 4′ | 86.6, C | 86.6, CH | ||
| 5′ | 30.1, CH2 | 2.77, ddd (15.2, 8.6, 6.4) | 30.0, CH2 | 2.72, ddd (15.3, 8.7, 6.5) |
| 2.59, ddd (15.2, 8.6, 6.4) | 2.49, ddd (15.3, 8.7, 6.5) | |||
| 6′ | 28.7, CH2 | 2.29, ddd (16.0, 8.6, 6.4) | 28.7, CH2 | 2.23, ddd (15.9, 8.7, 6.5) |
| 2.22, ddd (16.0, 8.6, 6.4) | 2.14, ddd (15.9, 8.7, 6.5) | |||
| 7′ | 174.4, C | 175.9, C | ||
| 8′ | 131.0, C | 131.0, C | ||
| 9′, 13′ | 128.6, CH | 7.73, d (7.7) | 128.5, CH | 7.70, d (7.4) |
| 10′, 12′ | 129.9, CH | 7.44, t (7.7) | 129.9, CH | 7.44, t (7.4) |
| 11′ | 130.1, CH | 7.37, t (7.7) | 130.1, CH | 7.37, t (7.4) |
| 14′ | 169.9, C | 169.5, C | ||
| OMe | 52.3, C | 3.58, s | ||
| No. | 15 a | 16 b | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 169.6, C | 169.5, C | ||
| 3 | 79.3, CH | 4.63, m | 80.8, CH | 4.63, m |
| 4 | 31.2, CH2 | 2.94, dd (16.4, 3.6) | 32.7, CH2 | 2.96, dd (16.1, 4.1) |
| 2.83, dd (16.4, 10.9) | 2.87, dd (16.1, 10.4) | |||
| 5 | 117.5, CH | 6.63, d (8.0) | 118.6, CH | 6.65, d (8.0) |
| 6 | 121.6, CH | 7.01, d (8.0) | 122.4, CH | 7.01, d (8.0) |
| 7 | 144.4, C | 145.7, C | ||
| 8 | 149.9, C | 151.0, C | ||
| 9 | 108.4, C | 109.4, C | ||
| 10 | 129.1, C | 130.7, C | ||
| 11 | 29.3, CH2 | 1.96, m | 30.7, CH2 | 2.09, m |
| 12 | 29.1, CH2 | 2.42, m | 30.3, CH2 | 2.58, m |
| 13 | 173.8, C | 174.4, C | ||
| 1′ | 65.3, CH2 | 4.11, t (6.6) | ||
| 2′ | 31.6, CH2 | 1.63, m | ||
| 3′ | 19.9, CH2 | 1.40, m | ||
| 4′ | 13.7, CH3 | 0.95, t (7.4) | ||
| OH-7 | 9.33, s | |||
| OH-8 | 10.82, s | |||
| OH-13 | 12.21, s | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Küppers, L.; Ebrahim, W.; El-Neketi, M.; Özkaya, F.C.; Mándi, A.; Kurtán, T.; Orfali, R.S.; Müller, W.E.G.; Hartmann, R.; Lin, W.; et al. Lactones from the Sponge-Derived Fungus Talaromyces rugulosus. Mar. Drugs 2017, 15, 359. https://doi.org/10.3390/md15110359
Küppers L, Ebrahim W, El-Neketi M, Özkaya FC, Mándi A, Kurtán T, Orfali RS, Müller WEG, Hartmann R, Lin W, et al. Lactones from the Sponge-Derived Fungus Talaromyces rugulosus. Marine Drugs. 2017; 15(11):359. https://doi.org/10.3390/md15110359
Chicago/Turabian StyleKüppers, Lisa, Weaam Ebrahim, Mona El-Neketi, Ferhat C. Özkaya, Attila Mándi, Tibor Kurtán, Raha S. Orfali, Werner E. G. Müller, Rudolf Hartmann, Wenhan Lin, and et al. 2017. "Lactones from the Sponge-Derived Fungus Talaromyces rugulosus" Marine Drugs 15, no. 11: 359. https://doi.org/10.3390/md15110359