
Grafting (S)-2-Phenylpropionic Acid on Coordinatively Unsaturated Metal Centers of MIL−101(Al) Metal–Organic Frameworks for Improved Enantioseparation


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
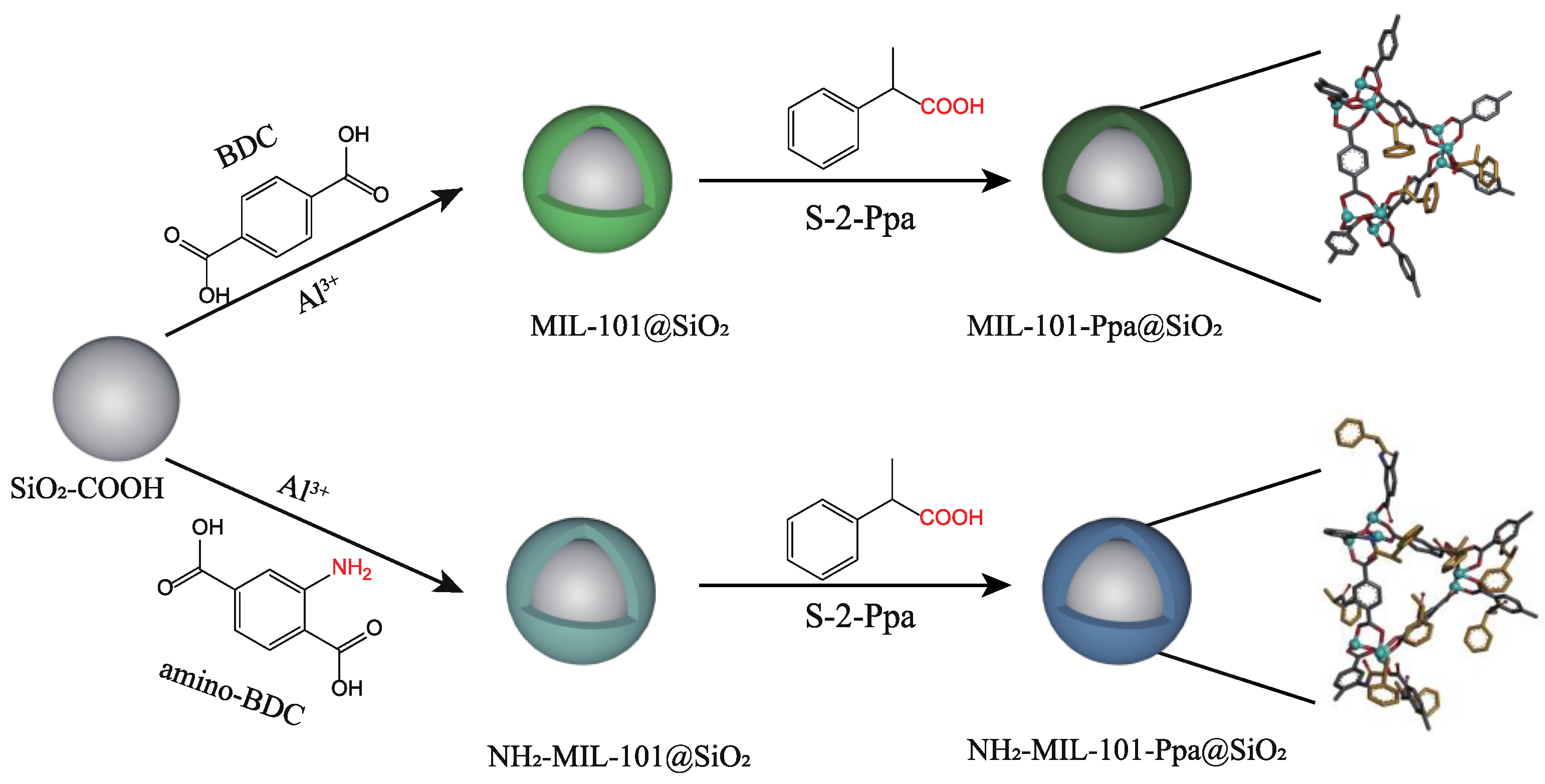
2.2. Preparation of Chiral Stationary Phases
2.2.1. Synthesis of SiO2-NH2
2.2.2. Synthesis of MIL−101@SiO2
2.2.3. Synthesis of NH2−MIL−101@SiO2
2.2.4. Synthesis of MIL−101−Ppa@SiO2
2.2.5. Synthesis of NH2−MIL−101−Ppa@SiO2
2.3. Characterization
2.4. Column Packing
2.5. HPLC
2.6. Molecular Docking
3. Results and Discussion
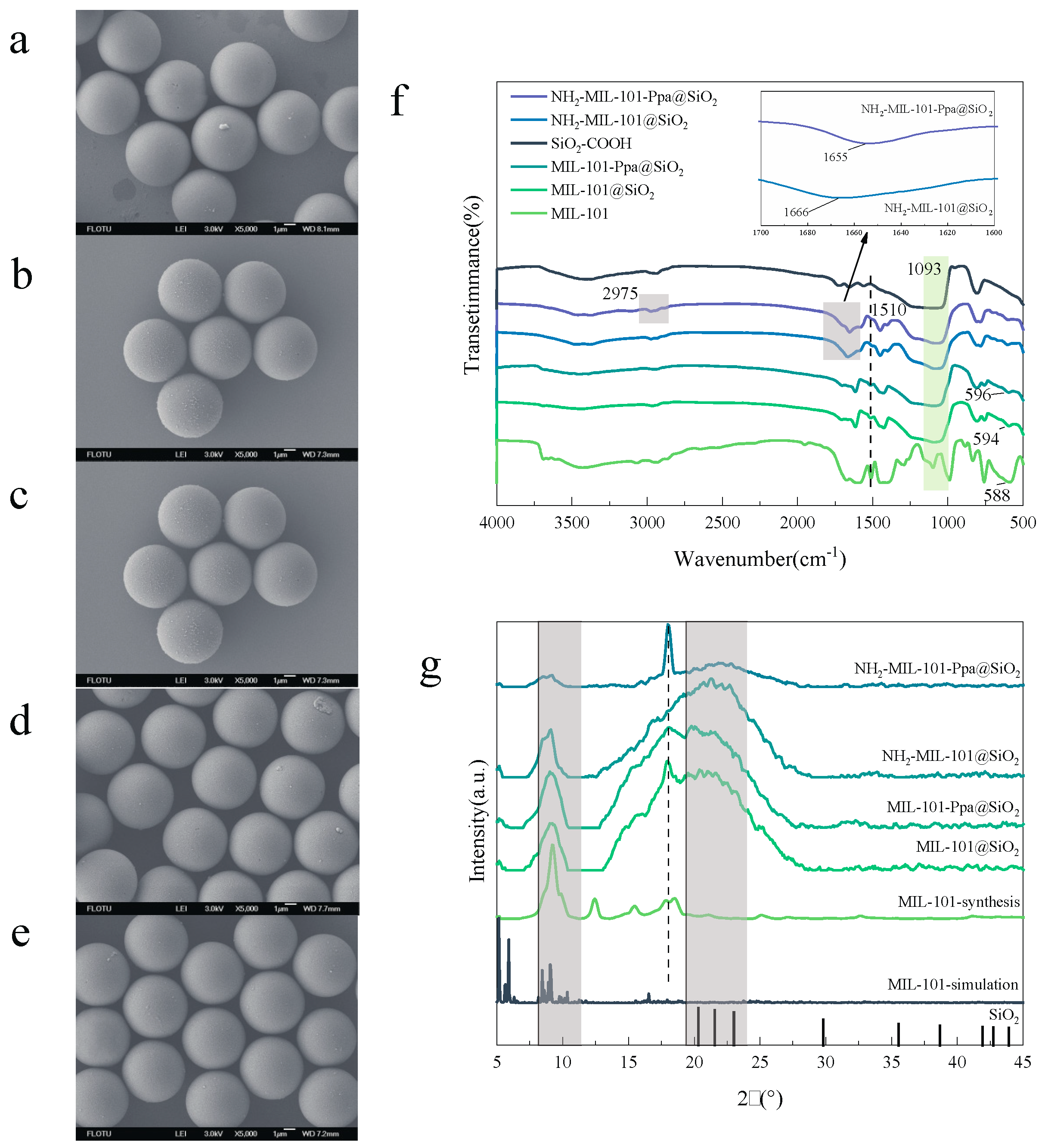
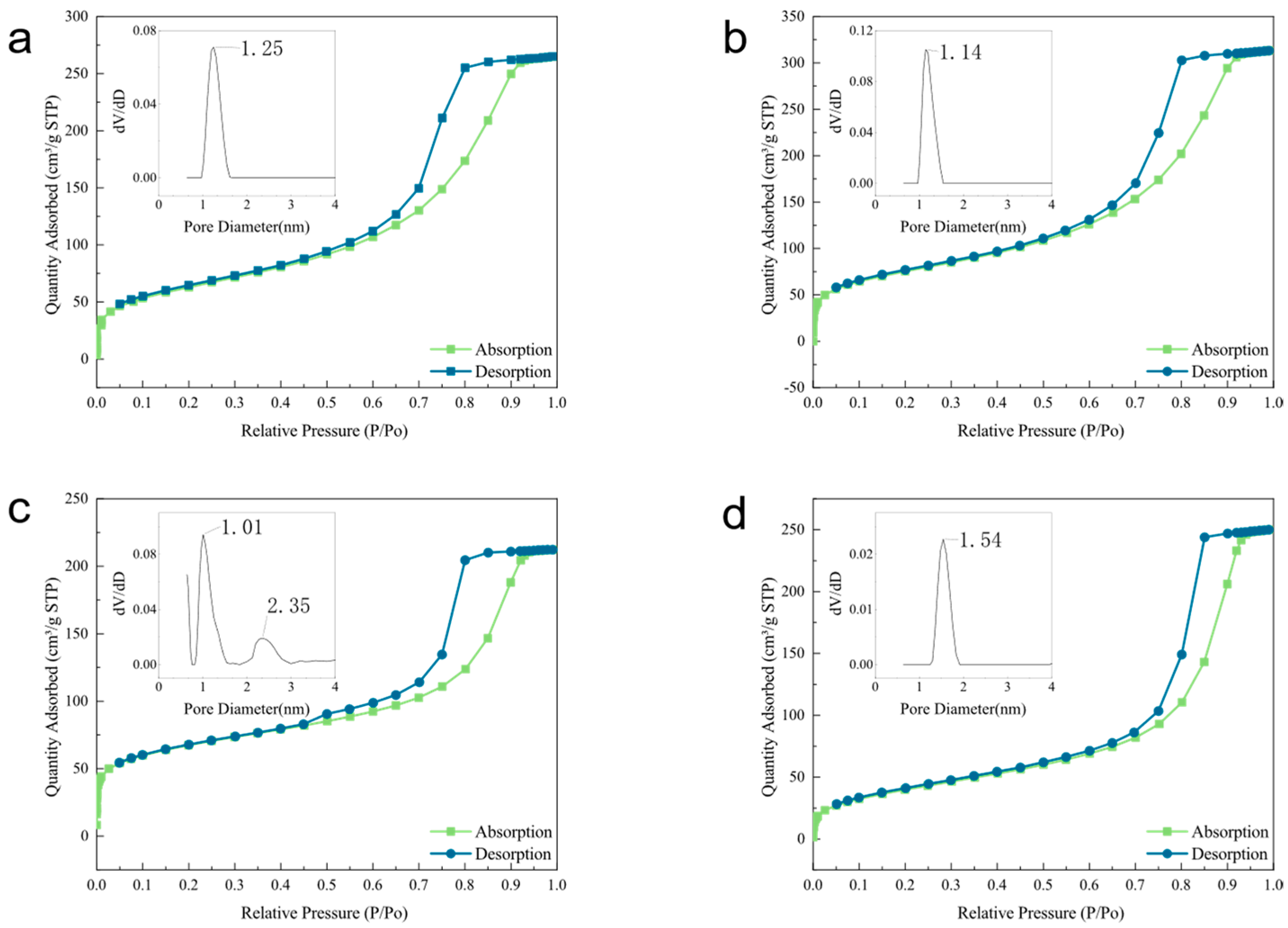
3.1. Characterization
3.2. HPLC Separation of Racemic Compounds
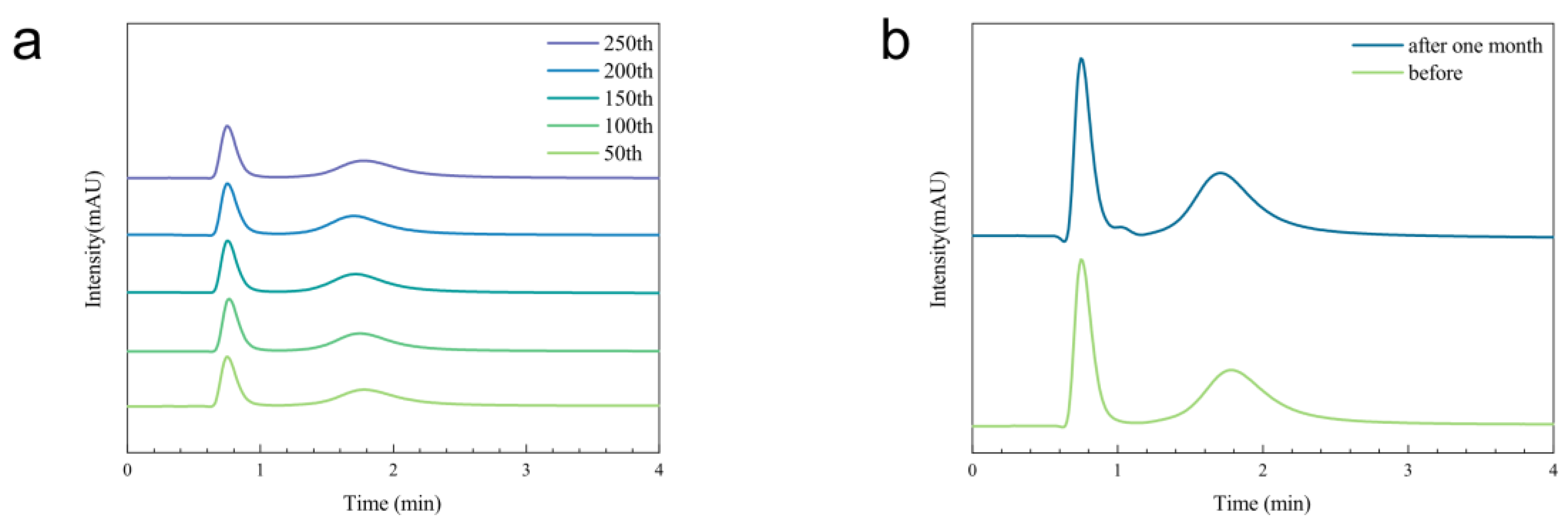
3.3. Evaluation of Separation Performance
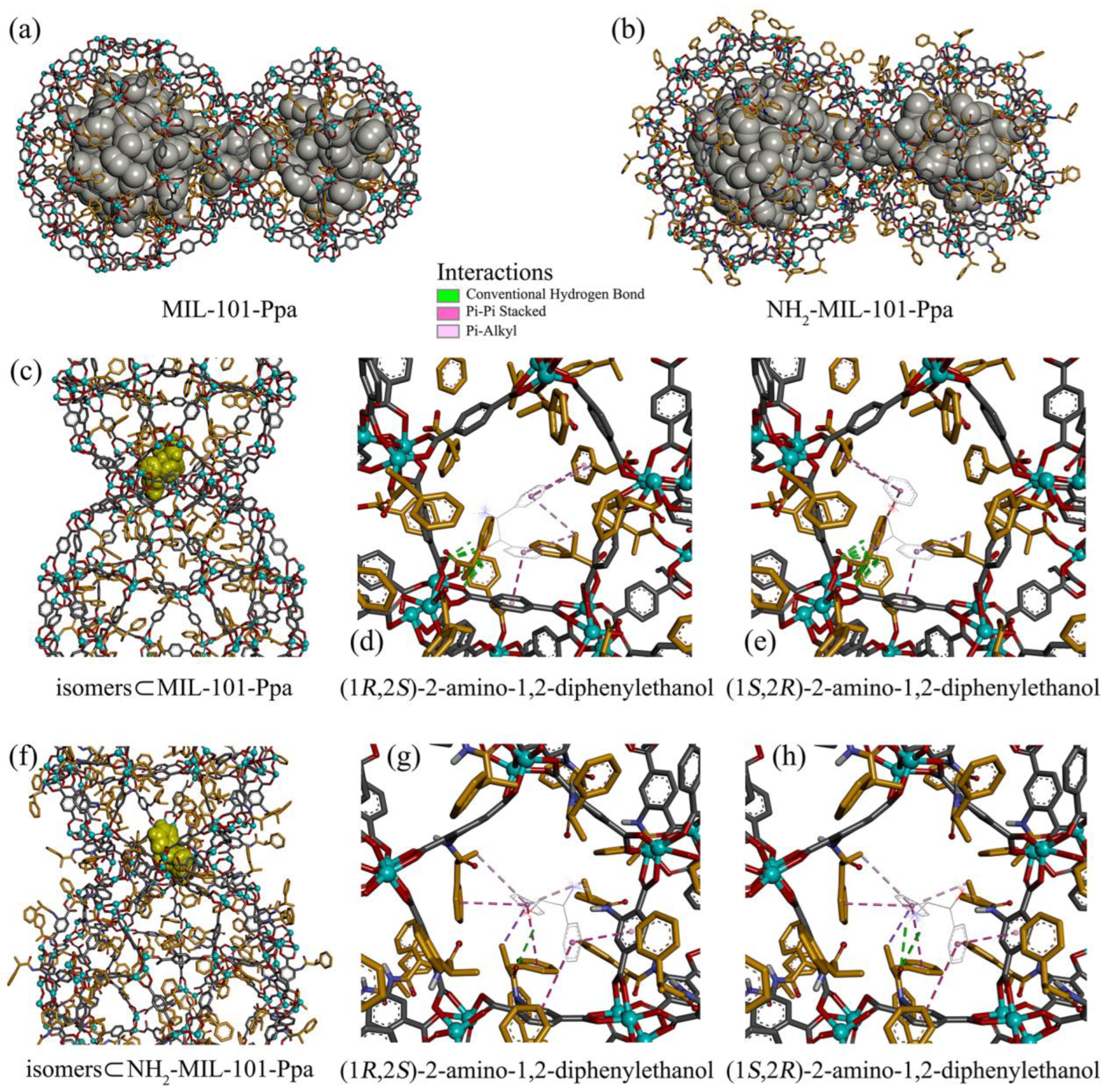
3.4. Docking Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Toyo’oka, T. Resolution of Chiral Drugs by Liquid Chromatography Based upon Diastereomer Formation with Chiral Derivatization Reagents. J. Biochem. Bioph. Meth. 2002, 54, 25–56. [Google Scholar] [CrossRef]
- Karadurmus, L.; Gumustas, M.; Bakirhan, N.K.; Ozkan, S.A. Chiral Sensing as a Future Challenge in Electroanalytical Chemistry: Cyclodextrin-Based Chiral Sensors. Crit. Rev. Anal. Chem. 2021, 1–22. [Google Scholar]
- Grybinik, S.; Bosakova, Z. An Overview of Chiral Separations of Pharmaceutically Active Substances by HPLC (2018–2020). Monatsh. Chem. 2021, 152, 1033–1043. [Google Scholar] [CrossRef]
- Niu, X.H.; Yang, X.; Li, H.X.; Liu, J.; Liu, Z.Y.; Wang, K.J. Application of Chiral Materials in Electrochemical Sensors. Microchim. Acta 2020, 187, 676. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.; Gedanken, A.; Mastai, Y. Enantioselective Separation Using Chiral Mesoporous Spherical Silica Prepared by Templating of Chiral Block Copolymers. ACS Appl. Mater. Inter. 2009, 1, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.; Wen, Y.H.; Hu, S.M.; Sheng, T.L.; Fu, R.B.; Xue, Z.Z.; Zhang, H.; Li, H.R.; Yuan, J.G.; Chen, X.; et al. Homochiral Metal-Organic Frameworks with Tunable Nanoscale Channel Array and Their Enantioseparation Performance against Chiral Diols. Inorg. Chem. 2017, 56, 6275–6280. [Google Scholar] [CrossRef]
- Zhang, Q.; Curran, D.P. Quasienantiomers and Quasiracemates: New Tools for Identification, Analysis, Separation, and Synthesis of Enantiomers. Chem.-Eur. J. 2005, 11, 4866–4880. [Google Scholar] [CrossRef]
- Cheng, Q.S.; Ma, Q.; Pei, H.B.; Mo, Z.L. Chiral Membranes for Enantiomer Separation: A Comprehensive Review. Sep. Purif. Technol. 2022, 292, 121034. [Google Scholar] [CrossRef]
- Elfassy, E.; Basel, Y.; Mastai, Y. Crystallization of Amino Acids at the Chiral Ionic Liquid/Water Interface. Crystengcomm 2016, 18, 8769–8775. [Google Scholar] [CrossRef]
- Higuchi, A.; Tamai, M.; Ko, Y.A.; Tagawa, Y.I.; Wu, Y.H.; Freeman, B.D.; Bing, J.T.; Chang, Y.; Ling, Q.D. Polymeric Membranes for Chiral Separation of Pharmaceuticals and Chemicals. Polym. Rev. 2010, 50, 113–143. [Google Scholar] [CrossRef]
- Wang, C.; Park, M.J.; Seo, D.H.; Drioli, E.; Matsuyama, H.; Shon, H. Recent Advances in Nanomaterial-Incorporated Nanocomposite Membranes for Organic Solvent Nanofiltration. Sep. Purif. Technol. 2021, 268, 118657. [Google Scholar] [CrossRef]
- Jose, C.; Toledo, M.V.; Briand, L.E. Enzymatic Kinetic Resolution of Racemic Ibuprofen: Past, Present and Future. Crit. Rev. Biotechnol. 2016, 36, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Suhail, M.; Lone, M.N.; Alothman, Z.A.; Alwarthan, A. Chiral Resolution of Multichiral Center Racemates by Different Modalities of Chromatography. J. Liq. Chromatogr. R. T. 2016, 39, 435–444. [Google Scholar] [CrossRef]
- Liu, Y.; Lantz, A.W.; Armstrong, D.W. High Efficiency Liquid and Super-/Subcritical Fluid-Based Enantiomeric Separations: An Overview. J. Liq. Chromatogr. R. T. 2004, 27, 1121–1178. [Google Scholar] [CrossRef]
- Alajmi, M.F.; Hussain, A.; Suhail, M.; Mukhtar, S.D.; Sahoo, D.R.; Asnin, L.; Ali, I. Chiral HPLC Separation and Modeling of Four Stereomers of DL-Leucine-DL-Tryptophan Dipeptide on Amylose Chiral Column. Chirality 2016, 28, 642–648. [Google Scholar] [CrossRef]
- LIU, J.; SU, P.; ZHU, J.; AI, P.; YUAN, L. Chiral Stationary Phases of Teicoplanin and Vancomycin in HPLC. Chem. Res. 2022, 33, 136–142. [Google Scholar]
- Da, S.; Wei, X.; Dong, Y. Chiral High Performance Liquid Chromatography Stationary Phase. Chemistry 1997, 2, 36–47. [Google Scholar]
- Nazareth, C.; Pereira, S. A Review on Chiral Stationary Phases for Separation of Chiral Drugs. Int. J. Pharm. Phytopharm. Res. 2020, 10, 77–91. [Google Scholar]
- Yin, C.; Chen, W.; Zhang, J.; Zhang, M.; Zhang, J. A Facile and Efficient Method to Fabricate High-Resolution Immobilized Cellulose-Based Chiral Stationary Phases via Thiol-Ene Click Chemistry. Sep. Purif. Technol. 2019, 210, 175–181. [Google Scholar] [CrossRef]
- Chen, Z.H.; Han, Z.S.; Shi, W.; Cheng, P. Design, Synthesis and Applications of Chiral Metal-Organic Frameworks. Acta Chim. Sin. 2020, 78, 1336–1348. [Google Scholar] [CrossRef]
- Zhuo, S.Q.; Zhang, X.Y.; Luo, H.; Wang, X.H.; Ji, Y.B. The Application of Covalent Organic Frameworks for Chiral Chemistry. Macromol. Rapid Comm. 2020, 41, 200404. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Li, T.L.; Ding, Y.; Hu, A.G. Synthesis of Chiral Porous Organic Polymers Through Nucleophilic Substitution for Chiral Separation. ACS Appl. Polym. Mater. 2020, 2, 5414–5422. [Google Scholar] [CrossRef]
- Gu, Z.-Y.; Yang, C.-X.; Chang, N.; Yan, X.-P. Metal–Organic Frameworks for Analytical Chemistry: From Sample Collection to Chromatographic Separation. Acc. Chem. Res. 2012, 45, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xu, N.; Zhang, J.; Wang, B.; Xie, S.; Yuan, L. Chiral Metal-Organic Framework D-His-ZIF-8@SiO2 Core-Shell Microspheres Used for HPLC Enantioseparations. ACS Appl. Mater. Inter. 2020, 12, 16903–16911. [Google Scholar] [CrossRef] [PubMed]
- Jun, B.M.; Al-Hamadani, Y.A.J.; Son, A.; Park, C.M.; Jang, M.; Jang, A.; Kim, N.C.; Yoon, Y. Applications of Metal-Organic Framework Based Membranes in Water Purification: A Review. Sep. Purif. Technol. 2020, 247, 116947. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Khan, M.; Li, X.; Zhu, Q.-L.; Wu, X.-T. Recent Progress in Asymmetric Catalysis and Chromatographic Separation by Chiral Metal–Organic Frameworks. Catalysts 2018, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Ye, N.S.; Ma, J.C.; An, J.X.; Li, J.; Cai, Z.M.; Zong, H. Separation of Amino Acid Enantiomers by a Capillary Modified with a Metal-Organic Framework. RSC Adv. 2016, 6, 41587–41593. [Google Scholar] [CrossRef]
- Wang, Z.-M.; Yang, C.-X.; Yan, X.-P. Polysiloxane Assisted Fabrication of Chiral Crystal Sponge Coated Capillary Column for Chiral Gas Chromatographic Separation. J. Chromatogr. A 2019, 1608, 460420. [Google Scholar] [CrossRef]
- Yang, C.-X.; Zheng, Y.-Z.; Yan, X.-P. γ-Cyclodextrin Metal–Organic Framework for Efficient Separation of Chiral Aromatic Alcohols. RSC Adv. 2017, 7, 36297–36301. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.M.; Zhang, J.H.; Fu, N.; Wang, B.J.; Chen, L.; Yuan, L.M. A Chiral Porous Organic Cage for Molecular Recognition Using Gas Chromatography. Anal. Chim. Acta 2016, 903, 156–163. [Google Scholar] [CrossRef]
- Ameloot, R.; Liekens, A.; Alaerts, L.; Maes, M.; Galarneau, A.; Coq, B.; Desmet, G.; Sels, B.F.; Denayer, J.F.M.; De Vos, D.E. Silica–MOF Composites as a Stationary Phase in Liquid Chromatography. Eur. J. Inorg. Chem. 2010, 2010, 3735–3738. [Google Scholar] [CrossRef]
- Yuan, B.; Li, L.; Yu, Y.; Xu, N.; Fu, N.; Zhang, J.; Zhang, M.; Wang, B.; Xie, S.; Yuan, L. Chiral Metal-Organic Framework [Co2(d-cam)2(TMDPy)]@SiO2 Core-Shell Microspheres for HPLC Separation. Microchem. J. 2021, 161, 105815. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Dong, S.; Zhang, X.; Wu, Q.; Zhao, L.; Shi, Y. Nanocellulose Derivative/Silica Hybrid Core-Shell Chiral Stationary Phase: Preparation and Enantioseparation Performance. Molecules 2016, 21, 561. [Google Scholar] [CrossRef] [Green Version]
- Gadzikwa, T.; Farha, O.K.; Mulfort, K.L.; Hupp, J.T.; Nguyen, S.T. A Zn-based, Pillared Paddlewheel MOF Containing Free Carboxylic Acids via Covalent Post-Synthesis Elaboration. Chem. Commun. 2009, 45, 3720–3722. [Google Scholar] [CrossRef] [PubMed]
- Canivet, J.; Aguado, S.; Bergeret, G.; Farrusseng, D. Amino Acid Functionalized Metal–Organic Frameworks by a Soft Coupling–Deprotection Sequence. Chem. Commun. 2011, 47, 11650–11652. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Li, D.; Guo, D.; Zhang, H.; Shi, W.; Cheng, P.; Wojtas, L.; Zaworotko, M.J. Synthesis of a Chiral Crystal Form of MOF-5, CMOF-5, by Chiral Induction. J. Am. Chem. Soc. 2015, 137, 15406–15409. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, H.; Zhu, Y.; Marriott, P.J.; Wang, H. Emerging Homochiral Porous Materials for Enantiomer Separation. Adv. Funct. Mater. 2021, 31, 2101335. [Google Scholar] [CrossRef]
- Qian, Q.; Chi, W.S.; Han, G.; Smith, Z.P. Impact of Post-Synthetic Modification Routes on Filler Structure and Performance in Metal–Organic Framework-Based Mixed-Matrix Membranes. Ind. Eng. Chem. Res. 2020, 59, 5432–5438. [Google Scholar] [CrossRef]
- Yin, Z.; Wan, S.; Yang, J.; Kurmoo, M.; Zeng, M.-H. Recent Advances in Post-Synthetic Modification of Metal–Organic Frameworks: New Types and Tandem Reactions. Coordin. Chem. Rev. 2019, 378, 500–512. [Google Scholar] [CrossRef]
- LI, T. Summary of Optimization Methods for Metal Organic Framework Materials. Guangzhou Chem. Ind. 2020, 48, 56–60, 90. [Google Scholar]
- Wang, Z.; Cohen, S.M. Tandem Modification of Metal–Organic Frameworks by a Postsynthetic Approach. Angew. Chem. Int. Edit. 2008, 47, 4699–4702. [Google Scholar] [CrossRef]
- Hwang, Y.K.; Hong, D.Y.; Chang, J.S.; Jhung, S.H.; Seo, Y.K.; Kim, J.; Vimont, A.; Daturi, M.; Serre, C.; Ferey, G. Amine grafting on coordinatively unsaturated metal centers of MOFs: Consequences for catalysis and metal encapsulation. Angew. Chem. Int. Edit. 2008, 47, 4144–4148. [Google Scholar] [CrossRef] [PubMed]
- Arnanz, A.; Pintado-Sierra, M.; Corma, A.; Iglesias, M.; Sánchez, F. Bifunctional Metal Organic Framework Catalysts for Multistep Reactions: MOF-Cu(BTC)-[Pd] Catalyst for One-Pot Heteroannulation of Acetylenic Compounds. Adv. Synth. Catal. 2012, 354, 1347–1355. [Google Scholar] [CrossRef]
- Zou, Y.; Park, M.; Hong, S.; Lah, M.S. A Designed Metal–Organic Framework Based on a Metal–Organic Polyhedron. Chem. Commun. 2008, 44, 2340–2342. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-J.; Huang, G.; Zhang, S.; Fang, Z.-B.; Liu, T.-F.; Cao, R. An Easy and Low-Cost Method of Embedding Chiral Molecules in Metal–Organic Frameworks for Enantioseparation. Chem. Commun. 2020, 56, 7459–7462. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Xu, N.Y.; Guo, P.; Wang, B.J.; Zhang, J.H.; Xie, S.M.; Yuan, L.M. A Chiral Metal-Organic Framework Core-Shell Microspheres Composite for High-Performance Liquid Chromatography Enantioseparation. J. Sep. Sci. 2021, 44, 3976–3985. [Google Scholar] [CrossRef]
- An, Y.; Chen, M.; Xue, Q.; Liu, W. Preparation and Self-assembly of Carboxylic Acid-Functionalized Silica. J. Colloid Interf. Sci. 2007, 311, 507–513. [Google Scholar] [CrossRef]
- Bromberg, L.; Klichko, Y.; Chang, E.P.; Speakman, S.; Straut, C.M.; Wilusz, E.; Hatton, T.A. Alkylaminopyridine-Modified Aluminum Aminoterephthalate Metal-Organic Frameworks As Components of Reactive Self-Detoxifying Materials. ACS Appl. Mater. Inter. 2012, 4, 4595–4602. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, J.; Legrand, A.; Quadrelli, E.A.; Canivet, J.; Farrusseng, D. Enantiopure Peptide-Functionalized Metal-Organic Frameworks. J. Am. Chem. Soc. 2015, 137, 9409–9416. [Google Scholar] [CrossRef]
- Jacobsen, J.; Achenbach, B.; Reinsch, H.; Smolders, S.; Lange, F.-D.; Friedrichs, G.; De Vos, D.; Stock, N. The First Water-Based Synthesis of Ce(iv)-MOFs with Saturated Chiral and Achiral C4-Dicarboxylate Linkers. Dalton T. 2019, 48, 8433–8441. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, L.; Li, X.; Yu, A.; Zhang, S. Controlled Fabrication of Core-Shell Silica@Chiral Metal-Organic Framework for Significant Improvement Chromatographic Separation of Enantiomers. Talanta 2020, 218, 121155. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed] [Green Version]
- Lebedev, O.I.; Millange, F.; Serre, C.; Van Tendeloo, G.; Férey, G. First Direct Imaging of Giant Pores of the Metal−Organic Framework MIL-101. Chem. Mater. 2005, 17, 6525–6527. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Garberoglio, G. OBGMX: A Web-Based Generator of GROMACS Topologies for Molecular and Periodic Systems using the Universal Force Field. J. Comput. Chem. 2012, 33, 2204–2208. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ge, C.; van der Spoel, D.; Feng, W.; Tan, T. Insight into the Structural Deformations of Beta-Cyclodextrin Caused by Alcohol Cosolvents and Guest Molecules. J. Phys. Chem. B 2012, 116, 3880–3889. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sompornpailin, D.; Ratanatawanate, C.; Sattayanon, C.; Namuangruk, S.; Punyapalakul, P. Selective Adsorption Mechanisms of Pharmaceuticals on Benzene-1,4-dicarboxylic Acid-Based MOFs: Effects of a Flexible Framework, Adsorptive Interactions and the DFT Study. Sci. Total Environ. 2020, 720, 137449. [Google Scholar] [CrossRef]
- Hu, X.; Ma, D.; Yang, T.; Deng, Y. Preparation of Solid Base K2CO3 /Al2O3 and Catalytic Conversion of Waste Cooking Oil to Biodiesel. J. Fuel Chem. Technol. 2014, 42, 683–689. [Google Scholar]
- Tang, J.; Dong, W.; Wang, G.; Yao, Y.; Cai, L.; Liu, Y.; Zhao, X.; Xu, J.; Tan, L. Efficient Molybdenum(vi) Modified Zr-MOF Catalysts for Epoxidation of Olefins. RSC Adv. 2014, 4, 42977–42982. [Google Scholar] [CrossRef]
- Chan, J.Y.; Zhang, H.; Nolvachai, Y.; Hu, Y.; Zhu, H.; Forsyth, M.; Gu, Q.; Hoke, D.E.; Zhang, X.; Marriot, P.J.; et al. Incorporation of Homochirality into a Zeolitic Imidazolate Framework Membrane for Efficient Chiral Separation. Angew. Chem. Int. Edit. 2018, 57, 17130–17134. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; He, W.; Zhang, Q.; Shapour, H.; Bakhtari, M.F. Preparation of a GO/MIL-101(Fe) Composite for the Removal of Methyl Orange from Aqueous Solution. ACS Omega 2021, 6, 4597–4608. [Google Scholar] [CrossRef]
- Wu, M.; Ma, Z.; Fan, Y.; Wu, Y.; An, Z.; Zhao, H.; Liu, Y.; Xu, J. Materials Design, Sensing Performance and Mechanism of Anhydrous Hydrogen Fluoride Gas Sensor Based on Amino-Functionalized MIL-101(Cr) for New Energy Vehicles. Coatings 2022, 12, 260. [Google Scholar] [CrossRef]
- Zhao, R.; Liao, H.; Wu, X.; Cao, X. Selective Adsorption Behaviours of MOFs@SiO2 with Different Pore Sizes and Shell Thicknesses. J. Solid State Chem. 2020, 292, 121693. [Google Scholar] [CrossRef]
- Ma, X.; Guo, Y.; Zhang, L.; Wang, K.; Yu, A.; Zhang, S.; Ouyang, G. Crystal Morphology Tuning and Green Post-Synthetic Modification of Metal Organic Framework for HPLC Enantioseparation. Talanta 2022, 239, 123143. [Google Scholar] [CrossRef] [PubMed]
- Kou, W.T.; Yang, C.X.; Yan, X.P. Post-Synthetic Modification of Metal-Organic Frameworks for Chiral Gas Chromatography. J. Mater. Chem. A 2018, 6, 17861–17866. [Google Scholar] [CrossRef]
- Sun, S.L.; Tan, Z.Y.; Zhang, M.Y.; Yang, H.D.; Zhang, H.X. Influence of the Degree of Grafting on the Morphology and Mechanical Properties of Blends of Poly(butylene terephthalate) and Glycidyl Methacrylate Grafted Poly(ethylene-co-propylene) (EPR). Polym. Int. 2006, 55, 834–842. [Google Scholar] [CrossRef]
- Liang, J.Y.; Zhang, Y.; Sun, A.J.; Deng, D.; Hu, Y.F. Enantioselective Resolution of (+/-)-1-Phenylethanol and (+/-)-1-Phenylethyl Acetate by a Novel Esterase from Bacillus sp SCSIO 15121. Appl. Biochem. Biotech. 2016, 178, 558–575. [Google Scholar] [CrossRef]
- Vosu, J.; Britton, P.; Howard-Jones, A.; Isaacs, D.; Kesson, A.; Khatami, A.; Marais, B.; Nayda, C.; Outhred, A. Is the Risk of Ibuprofen or Other Non-steroidal Anti-inflammatory Drugs Increased in COVID-19? J. Paediatr. Child Health 2020, 56, 1645–1646. [Google Scholar] [CrossRef]
- Hong, J. Natural Product Synthesis at the Interface of Chemistry and Biology. Chem.-Eur. J. 2014, 20, 10204–10212. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Li, Z.; Liu, Y.; Jia, L. Preparation of Chitosan-Based Molecularly Imprinted Material for Enantioseparation of Racemic Mandelic Acid in Aqueous Medium by Solid Phase Extraction. J. Sep. Sci. 2019, 42, 3544–3552. [Google Scholar] [CrossRef]
- Tanaka, K.; Muraoka, T.; Otubo, Y.; Takahashi, H.; Ohnishi, A. HPLC Enantioseparation on a Homochiral MOF-Silica Composite as a Novel Chiral Stationary Phase. RSC Adv. 2016, 6, 21293–21301. [Google Scholar] [CrossRef]
- Dutta, S.; Samanta, P.; Joarder, B.; Let, S.; Mahato, D.; Babarao, R.; Ghosh, S.K. A Water-Stable Cationic Metal–Organic Framework with Hydrophobic Pore Surfaces as an Efficient Scavenger of Oxo-Anion Pollutants from Water. ACS Appl. Mater. Inter. 2020, 12, 41810–41818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xi, W.H.; Zi, M.; Peng, Y.; Xie, S.M.; Yuan, L.M. Enantioseparation of 38 Racemates on Four Chiral Columns in High Performance Liquid Chromatography. Chin. J. Anal. Chem. 2010, 38, 181–186. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
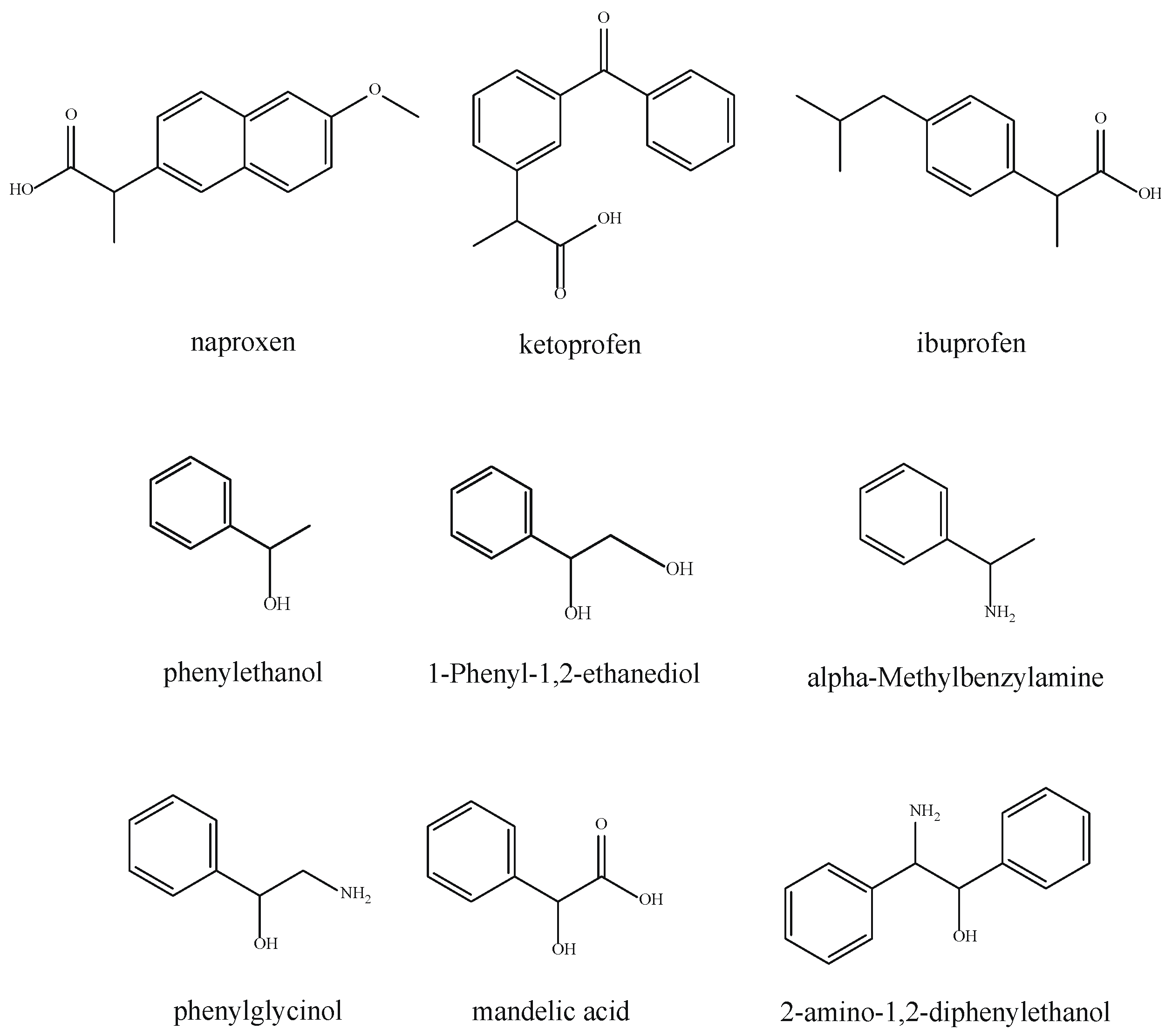
| Racemates | MIL−101−Ppa@SiO2 | NH2−MIL−101−Ppa@SiO2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mobile Phase (v/v) | Rs b | α c | Binding Affinity (kcal/mol) d | Mobile Phase (v/v) | Rsb | α c | Binding Affinity (kcal/mol) d | |||
| R | S | R | S | |||||||
| Naproxen | 40:60 | 1.61 | 35.71 | −8.28 ± 0.09 (−8.3) | −8.37 ± 0.07 (−8.4) | - | - | - | −7.19 ± 0.03 (−7.2) | −7.00 ± 0.00 (−7.0) |
| Ketoprofen | 60:40 | 0.92 | 10.02 | −9.08 ± 0.10 (−9.2) | −9.26 ± 0.27 (−9.4) | - | - | - | −8.36 ± 0.07 (−8.5) | −8.28 ± 0.04 (−8.3) |
| Ibuprofen | 40:60 | 1.62 | 21.98 | −7.42 ± 0.04 (−7.5) | −7.58 ± 0.07 (−7.6) | - | - | - | −6.61 ± 0.03 (−6.7) | −6.60 ± 0.01 (−6.6) |
| 1-Phenyl-1,2-ethanediol | - | - | - | −6.35 ± 0.11 (−6.6) | −6.45 ± 0.12 (−6.8) | - | - | - | −6.28 ± 0.36 (−6.7) | −6.25 ± 0.43 (−6.7) |
| Phenylethanol | - | - | - | −6.30 ± 0.02 (−6.4) | −6.39 ± 0.02 (−6.4) | - | - | - | −6.26 ± 0.39 (−6.5) | −6.42 ± 0.36 (−6.6) |
| α-Methylbenzylamine | 10:90 | 1.08 | 12.71 | −6.30 ± 0.00 (−6.3) | −6.40 ± 0.00 (−6.4) | 20:80 | 0.88 | 10.38 | −6.11 ± 0.41(−6.5) | −6.23 ± 0.36 (−6.5) |
| DL-Phenylglycinol | 10:90 | 0.97 | 6.26 | −6.36 ± 0.10 (−6.7) | −6.31 ± 0.10 (−6.8) | - | - | - | −6.04 ± 0.50 (−6.7) | −6.08 ± 0.51 (−6.7) |
| (±)-Mandelic acid | 20:80 | 0.93 | 3.02 | −6.71 ± 0.19 (−7.2) | −6.66 ± 0.16 (−6.9) | 20:80 | 1.09 | 9.81 | −6.25 ± 0.56 (−6.9) | −6.13 ± 0.49 (−6.9) |
| 2-Amino-1,2-diphenylethanol | 80:20 | 1.15 | 7.68 | −8.02 ± 0.05 (−8.2) | −8.37 ± 0.05 (−8.4) | 90:10 | 1.04 | 1.80 | −7.70 ± 0.00 (−7.7) | −7.90 ± 0.01 (−7.9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, R.; Bai, X.; Yang, W.; Fan, K.; Zhang, H. Grafting (S)-2-Phenylpropionic Acid on Coordinatively Unsaturated Metal Centers of MIL−101(Al) Metal–Organic Frameworks for Improved Enantioseparation. Materials 2022, 15, 8456. https://doi.org/10.3390/ma15238456
Zhao R, Bai X, Yang W, Fan K, Zhang H. Grafting (S)-2-Phenylpropionic Acid on Coordinatively Unsaturated Metal Centers of MIL−101(Al) Metal–Organic Frameworks for Improved Enantioseparation. Materials. 2022; 15(23):8456. https://doi.org/10.3390/ma15238456
Chicago/Turabian StyleZhao, Rui, Xueyan Bai, Wenhui Yang, Kun Fan, and Haiyang Zhang. 2022. "Grafting (S)-2-Phenylpropionic Acid on Coordinatively Unsaturated Metal Centers of MIL−101(Al) Metal–Organic Frameworks for Improved Enantioseparation" Materials 15, no. 23: 8456. https://doi.org/10.3390/ma15238456