
Novel [FeFe]-Hydrogenase Mimics: Unexpected Course of the Reaction of Ferrocenyl α-Thienyl Thioketone with Fe3(CO)12 †

 , , , , , and
, , , , , and
Abstract
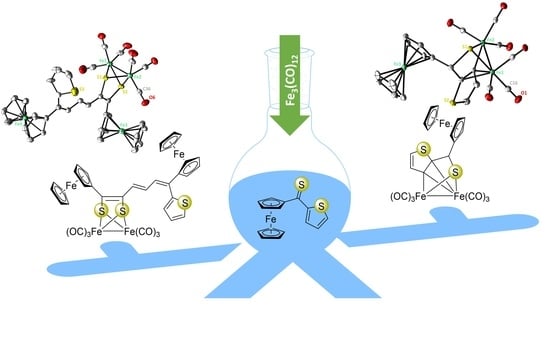
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Synthesis of Complexes 2 and 3
3. Results and Discussion
3.1. Synthesis and Characterization of the Binuclear Complexes
3.2. Molecular Structures of Complexes 2 and 3
3.3. Electrochemical Investigation
3.4. Electrocatalysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abu-Rumman, G.; Khdair, A.I.; Khdair, S.I. Current status and future investment potential in renewable energy in Jordan: An overview. Heliyon 2020, 6, e03346. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.R.; Dogutan, D.K.; Reece, S.Y.; Surendranath, Y.; Teets, T.S.; Nocera, D.G. Solar energy supply and storage for the legacy and nonlegacy worlds. Chem. Rev. 2010, 110, 6474–6502. [Google Scholar] [CrossRef] [PubMed]
- Frey, M. Hydrogenases: Hydrogen-activating enzymes. ChemBioChem 2002, 3, 153–160. [Google Scholar] [CrossRef]
- Adams, M.W. The structure and mechanism of iron hydrogenases. Biochim. Biophys. Acta Bioenerg. 1990, 1020, 115–145. [Google Scholar] [CrossRef]
- Vignais, P.M.; Billoud, B.; Meyer, J. Classification and phylogeny of hydrogenases. FEMS Microbiol. Rev. 2001, 25, 455–501. [Google Scholar] [CrossRef]
- Cammack, R. Bioinorganic Chemistry: Hydrogenase sophistication. Nature 1999, 397, 214–215. [Google Scholar] [CrossRef]
- Hatchikian, E.C.; Forget, N.; Fernandez, V.M.; Williams, R.; Cammack, R. Further characterization of the [Fe]-hydrogenase from desulfovibrio desulf uricans ATCC 7757. Eur. J. Biochem. 1992, 209, 357–365. [Google Scholar] [CrossRef]
- Peters, J.W.; Lanzilotta, W.N.; Lemon, B.J.; Seefeldt, L.C. X-ray crystal structure of the fe-only hydrogenase (cpi) from clostridium pasteurianum to 1.8 angstrom resolution. Science 1998, 282, 1853–1858. [Google Scholar] [CrossRef]
- Nicolet, Y.; de Lacey, A.L.; Vernede, X.; Fernandez, V.M.; Hatchikian, E.C.; Fontecilla-Camps, J.C. Crystallographic and FTIR spectroscopic evidence of changes in Fe coordination upon reduction of the active site of the Fe-only hydrogenase from desulfovibrio desulfuricans. J. Am. Chem. Soc. 2001, 123, 1596–1601. [Google Scholar] [CrossRef]
- Silakov, A.; Wenk, B.; Reijerse, E.; Lubitz, W. 14NHYSCORE investigation of the H-cluster of [FeFe] hydrogenase: Evidence for a nitrogen in the dithiol bridge. Phys. Chem. Chem. Phys. 2009, 11, 6592–6599. [Google Scholar] [CrossRef]
- Berggren, G.; Adamska, A.; Lambertz, C.; Simmons, T.R.; Esselborn, J.; Atta, M.; Gambarelli, S.; Mouesca, J.M.; Reijerse, E.; Lubitz, W.; et al. Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature 2013, 499, 66–69. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Liu, Y.; Shao, Y.; Jiang, D.; Duan, Q. Iron carbonyl compounds with aromatic dithiolate bridges as organometallic mimics of [FeFe] hydrogenases. Coord. Chem. Rev. 2020, 402, 213081, and references cited therein. [Google Scholar] [CrossRef]
- Li, Y.; Rauchfuss, T.B. Synthesis of diiron(i) dithiolato carbonyl complexes. Chem. Rev. 2016, 116, 7043–7077, and references cited therein. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Fan, W.; Liu, Y.; Jiang, D.; Duan, Q. Artificial water-soluble systems inspired by [FeFe]-hydrogenases for electro- and photocatalytic hydrogen production. Int. J. Hydrogen Energy 2020, 45, 4305–4327. [Google Scholar] [CrossRef]
- Almazahreh, L.R.; Arrigoni, F.; Abul-Futouh, H.; El-khateeb, M.; Görls, H.; Elleouet, C.; Schollhammer, P.; Bertini, L.; De Gioia, L.; Rudolph, M.; et al. Proton shuttle mediated by (SCH2)2P = O moiety in [fefe]-hydrogenase mimics: Electrochemical and DFT studies. ACS Catal. 2021, 11, 7080–7098. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Almazahreh, L.R.; Abaalkhail, S.J.; Görls, H.; Stripp, S.T.; Weigand, W. Ligand effects on structural, protophilic and reductive features of stannylated dinuclear iron dithiolato complexes. New J. Chem. 2021, 45, 36–44. [Google Scholar] [CrossRef]
- Amaro-Gahete, J.; Pavliuk, M.V.; Tian, H.; Esquivel, D.; Romero-Salguero, F.J.; Ott, S. Catalytic systems mimicking the [FeFe]-hydrogenase active site for visible-light-driven hydrogen production. Coord. Chem. Rev. 2021, 448, 214172. [Google Scholar] [CrossRef]
- Tschierlei, S.; Ott, S.; Lomoth, R. Spectroscopically characterized intermediates of catalytic H2 formation by [FeFe] hydrogenase models. Energy Environ. Sci. 2011, 4, 2340–2352. [Google Scholar] [CrossRef]
- Simmons, T.R.; Berggren, G.; Bacchi, M.; Fontecave, M.; Artero, V. Mimicking hydrogenases: From biomimetics to artificial enzymes. Coord. Chem. Rev. 2014, 270, 127–150. [Google Scholar] [CrossRef]
- Wu, L.Z.; Chen, B.; Li, Z.J.; Tung, C.H. Enhancement of the efficiency of photocatalytic reduction of protons to hydrogen via molecular assembly. Acc. Chem. Res. 2014, 47, 2177–2185. [Google Scholar] [CrossRef]
- Schilter, D.; Camara, J.M.; Huynh, M.T.; Hammes-Schiffer, S.; Rauchfuss, T.B. Hydrogenase enzymes and their synthetic models: The role of metal hydrides. Chem. Rev. 2016, 116, 8693–8749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abul-Futouh, H.; Daraosheh, A.Q.; Windhager, J.; Görls, H.; Weigand, W. Synthesis and characterization of [FeFe]-hydrogenase models mediated by a 1,2,4-trithiolane derivative: Insight into molecular structures and electrochemical characteristics. Polyhedron 2019, 174, 114155. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Abaalkhail, S.J.; Harb, M.K.; Görls, H.; Weigand, W. Structural studies and electrochemical catalysis investigation of [FeFe]-hydrogenase H-cluster mimics mediated by monophosphane ligands. Polyhedron 2021, 207, 115382. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Mebi, C.A.; Petro, B.J.; Vannucci, A.K.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Review of electrochemical studies of complexes containing the Fe2S2 core characteristic of [FeFe]-hydrogenases including catalysis by these complexes of the reduction of acids to form dihydrogen. J. Organomet. Chem. 2009, 694, 2681–2699, and references cited therein. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Skabeev, A.; Botteri, D.; Zagranyarski, Y.; Görls, H.; Weigand, W.; Peneva, K. Toward a tunable synthetic [FeFe]-hydrogenase H-cluster mimic mediated by perylene monoimide model complexes: Insight into molecular structures and electrochemical characteristics. Organometallics 2018, 37, 3278–3285. [Google Scholar] [CrossRef]
- Daraosheh, A.Q.; Görls, H.; El-khateeb, M.; Mloston, G.; Weigand, W. Reactions of selected aromatic thioketones with dodecarbonyltriiron. Eur. J. Inorg. Chem. 2011, 349–355. [Google Scholar] [CrossRef]
- Daraosheh, A.Q.; Apfel, U.-P.; Görls, H.; Friebe, C.; Schubert, U.S.; El-khateeb, M.; Mloston, G.; Weigand, W. New approach to [FeFe]-hydrogenase models using aromatic thioketones. Eur. J. Inorg. Chem. 2012, 318–326. [Google Scholar] [CrossRef]
- Mlostoń, G.; Grzelak, P.; Hamera-Fałdyga, R.; Jasiński, M.; Pipiak, P.; Urbaniak, K.; Albrecht, Ł.; Heimgartner, H. Aryl, hetaryl, and ferrocenyl thioketones as versatile building blocks for exploration in the organic chemistry of sulfur. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 204–211. [Google Scholar] [CrossRef]
- Mlostoń, G.; Urbaniak, K.; Utecht, G.; Lentz, D.; Jasiński, M. Trifluoromethylated 2,3-dihydro-1,3,4-thiadiazoles via the regioselective [3+2]-cycloadditions of fluorinated nitrile imines with aryl, hetaryl, and ferrocenyl thioketones. J. Fluorine Chem. 2016, 192, 147–154. [Google Scholar] [CrossRef]
- Ali, K.A.; Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. [3+2]-Cycloadditions of nitrilimines with heteroaryl thioketones. J. Sulfur Chem. 2017, 38, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Pipiak, P.; Linden, A.; Heimgartner, H. Studies on the reactions of thiocarbonyl S-methanides with hetaryl thioketones. Helv. Chim. Acta 2015, 98, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Selenophen-2-yl-substituted thiocarbonyl ylides—At the borderline of dipolar and biradical reactivity. Helv. Chim. Acta 2015, 98, 453–461. [Google Scholar] [CrossRef] [Green Version]
- McKee, M.L.; Mlostoń, G.; Urbaniak, K.; Heimgartner, H. Dimerization reactions of aryl selenophen-2-yl-substituted thiocarbonyl S-methanides as diradical processes: A computational study. Beilstein J. Org. Chem. 2017, 13, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlostoń, G.; Pipiak, P.; Heimgartner, H. Diradical reaction mechanisms in [3 + 2]-cycloadditions of hetaryl thioketones with alkyl- or trimethylsilyl-substituted diazomethanes. Beilstein J. Org. Chem. 2016, 12, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hejmanowska, J.; Jasiński, M.; Mlostoń, G.; Albrecht, Ł. Taming of thioketones: The first asymmetric thia-Diels–Alder reaction with thioketones as heterodienophiles. Eur. J. Org. Chem. 2017, 850–954. [Google Scholar] [CrossRef]
- Mloston, G.; Grzelak, P.; Linden, A.; Heimgartner, H. Thia-Diels–Alder reactions of hetaryl thioketones with nonactivated 1,3-dienes leading to 3,6-dihydro-2H-thiopyrans: Evidence for a diradical mechanism. Chem. Heterocycl. Chem. 2021, 53, 518–525, Correction: Chem. Heterocycl. Chem. 2021, 57, 610. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Hamera, R.; Heimgartner, H. Synthesis of ferrocenyl thioketones and their reactions with diphenyldiazomethane. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 2125–2133. [Google Scholar] [CrossRef] [Green Version]
- Alper, H.; Chan, A.S.K. Sulfur-donor ligand ortho-metalated complexes derived from thiobenzophenones. A Simple approach to isobenzothiophene heterocycles. J. Am. Chem. Soc. 1973, 95, 4905–4913. [Google Scholar] [CrossRef]
- Mlostoń, G.; Heimgartner, H. Generation and typical reactions of thiocarbonyl ylides. Pol. J. Chem. 2000, 74, 1503–1533. [Google Scholar]
- Mlostoń, G.; Heimgartner, H. Thiocarbonyl ylides. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A., Pearson, W.H., Eds.; John Wiley & Sons: New York, NY, USA, 2002; pp. 315–360. [Google Scholar]
- Wright, R.J.; Lim, C.; Tilley, T.D. Diiron Proton Reduction Catalysts Possessing Electron-Rich and Electron-Poor Naphthalene-1,8-dithiolate Ligands. Chem. Eur. J. 2009, 15, 8518–8525. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; El-Khateeb, M.; Görls, H.; Asali, K.J.; Weigand, W. Selenium makes the difference: Protonation of [FeFe]-hydrogenase mimics with diselenolato ligands. Dalton Trans. 2017, 46, 2937–2947. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.; Zhong, W.; Wei, Z.; Wang, H.; Xiao, Z.; Long, L.; Liu, X. Diiron hexacarbonyl complexes bearing naphthalene-1,8-dithiolate bridge moiety as mimics of the sub-unit of [FeFe]-hydrogenase: Synthesis, characterisation and electrochemical Investigations. New J. Chem. 2015, 39, 9752–9760. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Almazahreh, L.R.; Harb, M.K.; Görls, H.; El-khateeb, M.; Weigand, W. [FeFe]-Hydrogenase H-cluster mimics with various − S(CH2)nS− linker lengths (n = 2−8): A systematic study. Inorg. Chem. 2017, 56, 10437–10451. [Google Scholar] [CrossRef] [PubMed]
- Bertini, L.; Fantucci, P.; De Gioia, L. On the photochemistry of the low-lying excited state of Fe2(CO)6S2. A DFT and QTAIM investigation. Organometallics 2011, 30, 487–498. [Google Scholar] [CrossRef]
- Samuel, A.P.S.; Co, D.T.; Stern, C.L.; Wasielewski, M.R. Ultrafast Photodriven intramolecular electron transfer from a zinc porphyrin to a readily reduced diiron hydrogenase model complex. J. Am. Chem. Soc. 2010, 132, 8813–8815. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Amirjalayer, S.; Hartl, F.; Lutz, M.; Bruin, B.; Becker, R.; Woutersen, S.; Reek, J.N.H. Direct probing of photoinduced electron transfer in a self-assembled biomimetic [2Fe2S]-hydrogenase complex using ultrafast vibrational spectroscopy. Inorg. Chem. 2014, 53, 5373–5383. [Google Scholar] [CrossRef] [PubMed]
- Abul-Futouh, H.; Zagranyarski, Y.; Müller, C.; Schulz, M.; Kupfer, S.; Görls, H.; Elkhateeb, M.; Gräfe, S.; Dietzek, B.; Peneva, K.; et al. [FeFe]-Hydrogenase H-cluster mimics mediated by naphthalene monoimide derivatives of peri-substituted dichalcogenides. Dalton Trans. 2017, 46, 11180–11191. [Google Scholar] [CrossRef]
- Nicholson, R.S.; Shain, I. Theory of stationary electrode polarography for a chemical reaction coupled between two charge transfers. Anal. Chem. 1965, 37, 178–190. [Google Scholar] [CrossRef]
- Gloaguen, F.; Morvan, D.; Capon, J.-F.; Schollhammer, P.; Talarmin, J. Electrochemical proton reduction at mild potentials by monosubstituted diiron organometallic complexes bearing a benzenedithiolate bridge. J. Electroanal. Chem. 2007, 603, 15–20. [Google Scholar] [CrossRef]
- Schwartz, L.; Singh, P.S.; Eriksson, L.; Lomoth, R.; Ott, S. Tuning the electronic properties of Fe2(μ-arenedithiolate)-(CO)6−n(PMe3)n (n = 0, 2) complexes related to the [Fe−Fe]-hydrogenase active site. Comptes Rendus Chim. 2008, 11, 875–889. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Görls, H.; Weigand, W. Synthesis and electrochemical investigation of mono- and di-phosphite Substituted [FeFe]-hydrogenase H-cluster mimics. Z. Anorg. Allg. Chem. 2017, 643, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Vannucci, A.K.; Mebi, C.A.; Okumura, N.; Borowski, S.C.; Swenson, M.; Lockett, L.T.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Synthesis of diiron hydrogenase mimics bearing hydroquinone and related ligands. Electrochemical and computational studies of the mechanism of hydrogen production and the Role of O−H·S hydrogen bonding. Organometallics 2010, 29, 5330–5340. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2001. [Google Scholar]
- Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. Electrochemical proton reduction by thiolate-bridged hexacarbonyldiiron clusters. J. Electroanal. Chem. 2004, 566, 241–247. [Google Scholar] [CrossRef]
- Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. Activation of proton by the two-electron reduction of a di-iron organometallic complex. J. Electroanal. Chem. 2006, 595, 47–52. [Google Scholar] [CrossRef]
- Daraosheh, A.Q.; Abul-Futouh, H.; Görls, H.; Weigand, W. Synthesis and electrochemical investigations of the ortho-metalated complexes [Fe2(CO)6{к,μ-S,η2-(R)}] and their substitution reactions. Inorg. Chim. Acta 2020, 503, 119377. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Vannucci, A.K.; Chen, J.; Lockett, L.T.; Okumura, N.; Petro, B.J.; Zakai, U.I.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Hydrogen generation from weak acids: Electrochemical and computational studies of a diiron hydrogenase Mimic. J. Am. Chem. Soc. 2007, 129, 12521–12530. [Google Scholar] [CrossRef]
- Borg, S.J.; Ibrahim, S.K.; Pickett, C.J.; Best, S.P. Electrocatalysis of hydrogen evolution by synthetic diiron units using weak acids as the proton source: Pathways of doubtful relevance to enzymic catalysis by the diiron subsite of [FeFe] hydrogenase. Comptes Rendus Chim. 2008, 11, 852–860. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Beck, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H., Jr. Gaussian basis functions for use in molecular calculations. III. Contraction of (10s6p) atomic basis sets for the first-row atoms. J. Chem. Phys. 1971, 55, 716–723. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. A binitio pseudopotential study of the first-row transition metal monoxides and iron monohydride. J. Chem. Phys. 1987, 86, 2123–2131. [Google Scholar] [CrossRef]
- COLLECT Data Collection Software; Nonius B.V.: Delft, The Netherlands, 1998.
- Otwinowski, Z.; Minor, W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kratzert, D.; Krossing, I. Recent improvements in DSR. J. Appl. Cryst. 2018, 51, 928–934. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daraosheh, A.Q.; Abul-Futouh, H.; Murakami, N.; Ziems, K.M.; Görls, H.; Kupfer, S.; Gräfe, S.; Ishii, A.; Celeda, M.; Mlostoń, G.; et al. Novel [FeFe]-Hydrogenase Mimics: Unexpected Course of the Reaction of Ferrocenyl α-Thienyl Thioketone with Fe3(CO)12. Materials 2022, 15, 2867. https://doi.org/10.3390/ma15082867
Daraosheh AQ, Abul-Futouh H, Murakami N, Ziems KM, Görls H, Kupfer S, Gräfe S, Ishii A, Celeda M, Mlostoń G, et al. Novel [FeFe]-Hydrogenase Mimics: Unexpected Course of the Reaction of Ferrocenyl α-Thienyl Thioketone with Fe3(CO)12. Materials. 2022; 15(8):2867. https://doi.org/10.3390/ma15082867
Chicago/Turabian StyleDaraosheh, Ahmad Q., Hassan Abul-Futouh, Natsuki Murakami, Karl Michael Ziems, Helmar Görls, Stephan Kupfer, Stefanie Gräfe, Akihiko Ishii, Małgorzata Celeda, Grzegorz Mlostoń, and et al. 2022. "Novel [FeFe]-Hydrogenase Mimics: Unexpected Course of the Reaction of Ferrocenyl α-Thienyl Thioketone with Fe3(CO)12" Materials 15, no. 8: 2867. https://doi.org/10.3390/ma15082867