
Fructose Consumption and Hepatocellular Carcinoma Promotion

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
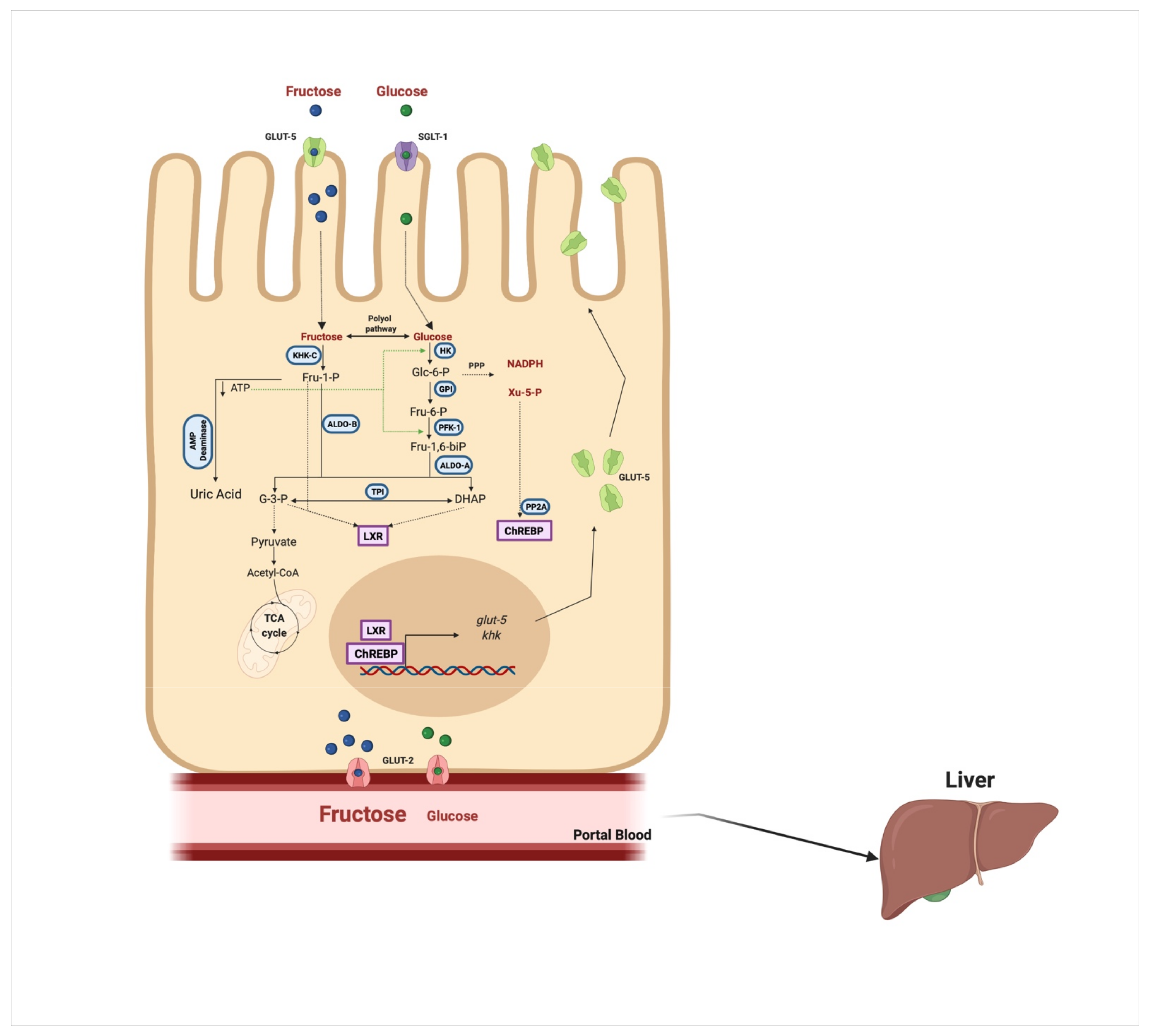
2. Fructose Consumption
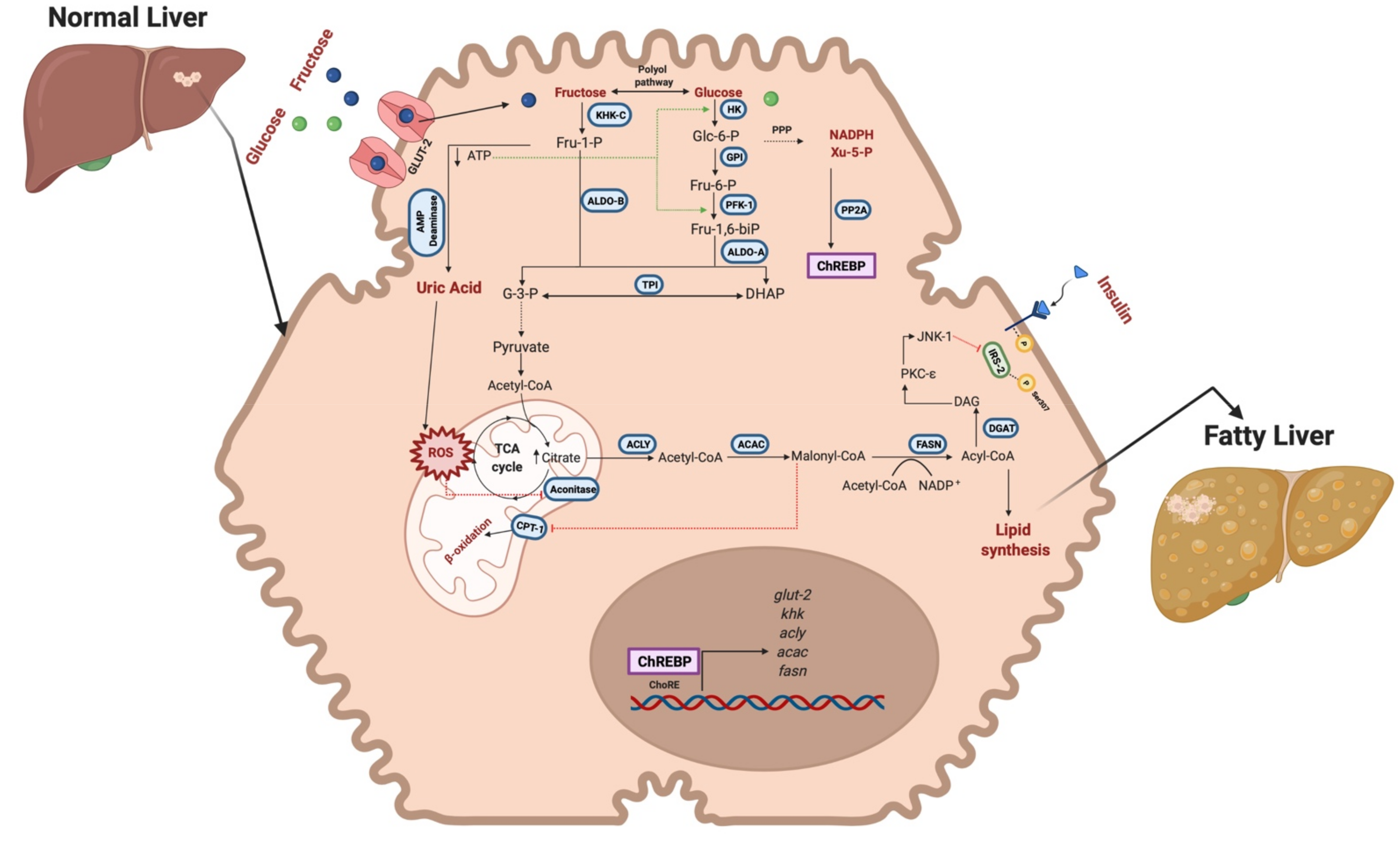
2.1. Fructose Consumption and MAFLD
2.1.1. Steatotic Stage
2.1.2. Inflammatory Stage
2.2. Fructose Consumption and HCC. Cancer Hallmarks
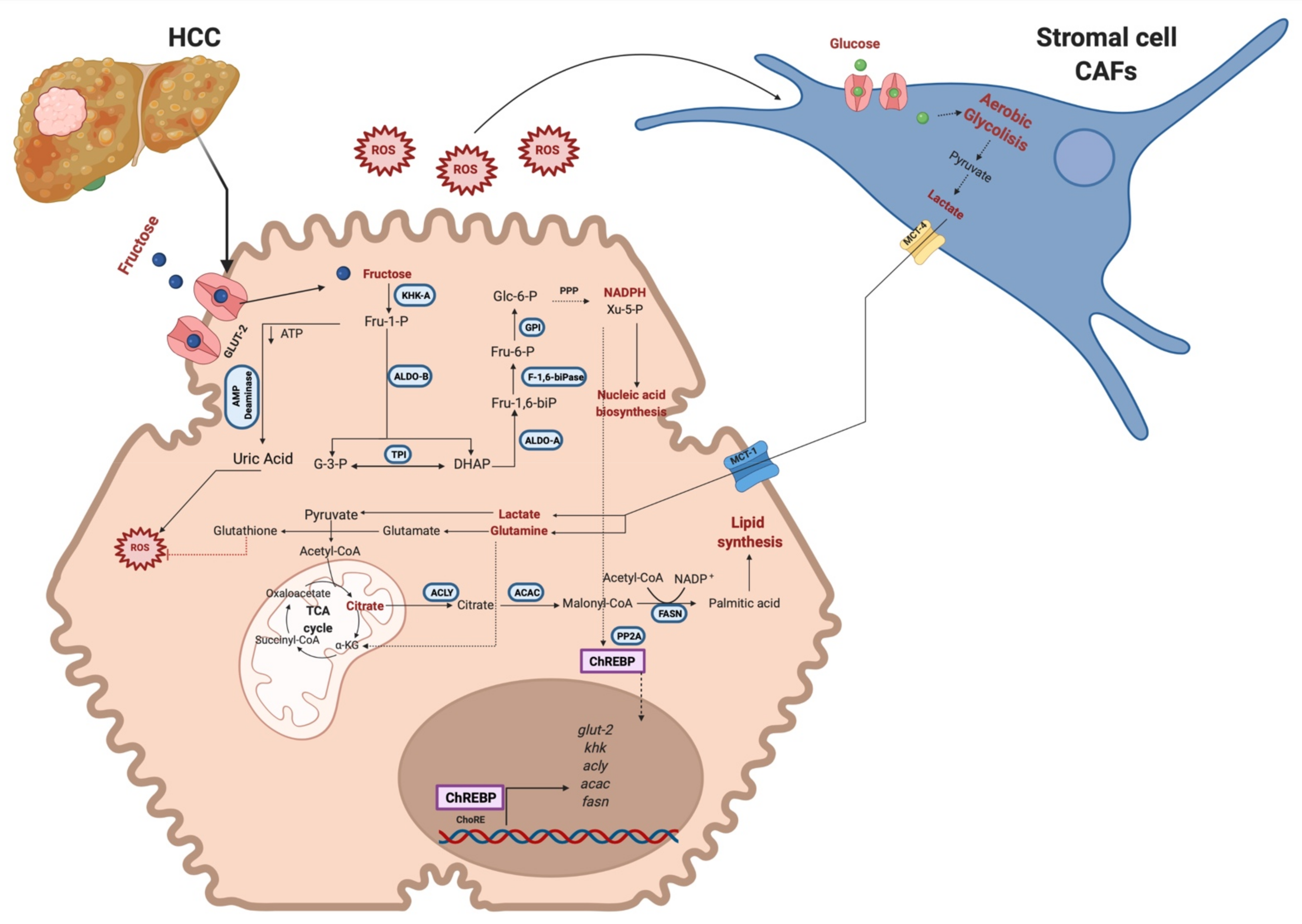
2.2.1. Reprogramming Energy Metabolism
2.2.2. Fructose Metabolism Promotes Invasion, Metastasis, and Sustaining Proliferative Signaling
2.2.3. Tumor-Promoting Inflammation
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
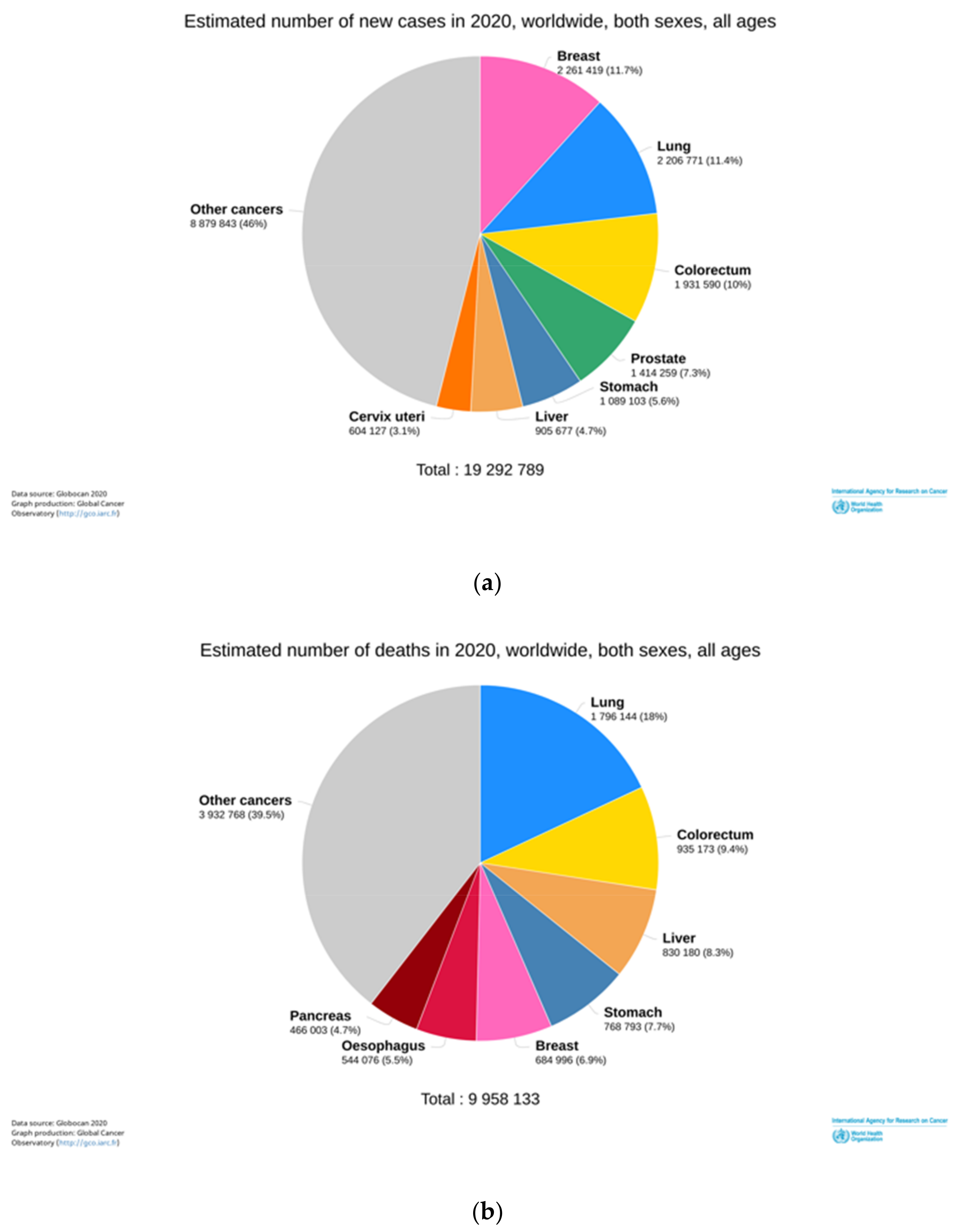
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. 2020, 40, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Mikolasevic, I.; Filipec-Kanizaj, T.; Mijic, M.; Jakopcic, I.; Milic, S.; Hrstic, I.; Sobocan, N.; Stimac, D.; Burra, P. Nonalcoholic fatty liver disease and liver transplantation—Where do we stand? World J. Gastroenterol. 2018, 24, 1491–1506. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; López-Otín, C.; Madeo, F.; de Cabo, R. Carbotoxicity-Noxious Effects of Carbohydrates. Cell 2018, 175, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windemuller, F.; Xu, J.; Rabinowitz, S.S.; Hussain, M.M.; Schwarz, S.M. Lipogenesis in Huh7 cells is promoted by increasing the fructose: Glucose mo.olar ratio. World J. Hepatol. 2016, 8, 838–843. [Google Scholar] [CrossRef]
- Sharma, S.P.; Chung, H.J.; Kim, H.J.; Hong, S.T. Paradoxical Effects of Fruit on Obesity. Nutrients 2016, 8, 633. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Alwahsh, S.M.; Gebhardt, R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch. Toxicol. 2017, 91, 1545–1563. [Google Scholar] [CrossRef]
- Ter Horst, K.W.; Serlie, M.J. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 981. [Google Scholar] [CrossRef] [Green Version]
- Basaranoglu, M.; Basaranoglu, G.; Sabuncu, T.; Sentürk, H. Fructose as a key player in the development of fatty liver disease. World J. Gastroenterol. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Li, X.; Qian, X.; Peng, L.X.; Jiang, Y.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Lee, J.H.; Cote, G.; Wang, H.; et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat. Cell Biol. 2016, 18, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Riveros, M.J.; Parada, A.; Pettinelli, P. Fructose consumption and its health implications; fructose malabsorption and nonalcoholic fatty liver disease. Nutr. Hosp. 2014, 29, 491–499. [Google Scholar] [CrossRef]
- Laughlin, M.R. Normal roles for dietary fructose in carbohydrate metabolism. Nutrients 2014, 6, 3117–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwarts, I.; van Zutphen, T.; Kruit, J.K.; Liu, W.; Oosterveer, M.H.; Verkade, H.J.; Uhlenhaut, N.H.; Jonker, J.W. Identification of the fructose transporter GLUT5 (SLC2A5) as a novel target of nuclear receptor LXR. Sci. Rep. 2019, 9, 9299. [Google Scholar] [CrossRef] [PubMed]
- Charrez, B.; Qiao, L.; Hebbard, L. The role of fructose in metabolism and cancer. Horm. Mol. Biol. Clin. Investig. 2015, 22, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, R.P.; Choe, J.Y.; Patel, C.R. Intestinal Absorption of Fructose. Annu. Rev. Nutr. 2018, 38, 41–67. [Google Scholar] [CrossRef]
- Patel, C.; Douard, V.; Yu, S.; Gao, N.; Ferraris, R.P. Transport, metabolism, and endosomal trafficking-dependent regulation of intestinal fructose absorption. FASEB J. 2015, 29, 4046–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020, 32, 117–127.e113. [Google Scholar] [CrossRef]
- Lee, H.J.; Cha, J.Y. Recent insights into the role of ChREBP in intestinal fructose absorption and metabolism. BMB Rep. 2018, 51, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, J.A.; Liu, W.; Perez, S.; Xing, G.; Sonnenberg, G.; Kou, K.; Blatnik, M.; Allen, R.; Weng, Y.; Vera, N.B.; et al. Pharmacologic inhibition of ketohexokinase prevents fructose-induced metabolic dysfunction. Mol. Metab. 2021, 48, 101196. [Google Scholar] [CrossRef] [PubMed]
- Merino, B.; Fernández-Díaz, C.M.; Cózar-Castellano, I.; Perdomo, G. Intestinal Fructose and Glucose Metabolism in Health and Disease. Nutrients 2019, 12, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.C.; Yang, Y.C.; Tien, C.P.; Yang, C.J.; Hsiao, M. Roles of Aldolase Family Genes in Human Cancers and Diseases. Trends Endocrinol. Metab. 2018, 29, 549–559. [Google Scholar] [CrossRef]
- DiStefano, J.K. Fructose-mediated effects on gene expression and epigenetic mechanisms associated with NAFLD pathogenesis. Cell Mol. Life Sci. 2020, 77, 2079–2090. [Google Scholar] [CrossRef]
- Dowman, J.K.; Hopkins, L.J.; Reynolds, G.M.; Nikolaou, N.; Armstrong, M.J.; Shaw, J.C.; Houlihan, D.D.; Lalor, P.F.; Tomlinson, J.W.; Hübscher, S.G.; et al. Development of hepatocellular carcinoma in a murine model of nonalcoholic steatohepatitis induced by use of a high-fat/fructose diet and sedentary lifestyle. Am. J. Pathol. 2014, 184, 1550–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef]
- Jegatheesan, P.; De Bandt, J.P. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef] [Green Version]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Fernando, D.H.; Forbes, J.M.; Angus, P.W.; Herath, C.B. Development and Progression of Non-Alcoholic Fatty Liver Disease: The Role of Advanced Glycation End Products. Int. J. Mol. Sci. 2019, 20, 5037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.H.; Chen, X.Y.; Hu, Q.H.; Wang, M.X.; Jin, R.; Zhang, Q.Y.; Wang, W.; Wang, R.; Kang, L.L.; et al. Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid Redox Signal. 2015, 22, 848–870. [Google Scholar] [CrossRef] [Green Version]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Sil, R.; Chakraborti, A.S. Oxidative Inactivation of Liver Mitochondria in High Fructose Diet-Induced Metabolic Syndrome in Rats: Effect of Glycyrrhizin Treatment. Phytother. Res. 2016, 30, 1503–1512. [Google Scholar] [CrossRef]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Berumen, C.I.; Ortiz-Avila, O.; Vargas-Vargas, M.A.; Del Rosario-Tamayo, B.A.; Guajardo-López, C.; Saavedra-Molina, A.; Rodríguez-Orozco, A.R.; Cortés-Rojo, C. The severity of rat liver injury by fructose and high fat depends on the degree of respiratory dysfunction and oxidative stress induced in mitochondria. Lipids Health Dis. 2019, 18, 78. [Google Scholar] [CrossRef] [Green Version]
- Mastrocola, R.; Collino, M.; Rogazzo, M.; Medana, C.; Nigro, D.; Boccuzzi, G.; Aragno, M. Advanced glycation end products promote hepatosteatosis by interfering with SCAP-SREBP pathway in fructose-drinking mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G398–G407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. Intracellular Toxic Advanced Glycation End-Products Promote the Production of Reactive Oxygen Species in HepG2 Cells. Int. J. Mol. Sci. 2020, 21, 4861. [Google Scholar] [CrossRef]
- Shepherd, E.L.; Saborano, R.; Northall, E.; Matsuda, K.; Ogino, H.; Yashiro, H.; Pickens, J.; Feaver, R.E.; Cole, B.K.; Hoang, S.A.; et al. Ketohexokinase inhibition improves NASH by reducing fructose-induced steatosis and fibrogenesis. JHEP Rep. 2021, 3, 100217. [Google Scholar] [CrossRef]
- Tran, C. Inborn Errors of Fructose Metabolism. What Can We Learn from Them? Nutrients 2017, 9, 356. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiolet, T.; Srour, B.; Sellem, L.; Kesse-Guyot, E.; Allès, B.; Méjean, C.; Deschasaux, M.; Fassier, P.; Latino-Martel, P.; Beslay, M.; et al. Consumption of ultra-processed foods and cancer risk: Results from NutriNet-Santé prospective cohort. BMJ 2018, 360, k322. [Google Scholar] [CrossRef] [Green Version]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef] [Green Version]
- Hindupur, S.K.; Colombi, M.; Fuhs, S.R.; Matter, M.S.; Guri, Y.; Adam, K.; Cornu, M.; Piscuoglio, S.; Ng, C.K.Y.; Betz, C.; et al. The protein histidine phosphatase LHPP is a tumour suppressor. Nature 2018, 555, 678–682. [Google Scholar] [CrossRef]
- Li, L.; Pilo, G.M.; Li, X.; Cigliano, A.; Latte, G.; Che, L.; Joseph, C.; Mela, M.; Wang, C.; Jiang, L.; et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016, 64, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, L.; Paliogiannis, P.; Cigliano, A.; Pilo, M.G.; Chen, X.; Calvisi, D.F. Pathogenetic, Prognostic, and Therapeutic Role of Fatty Acid Synthase in Human Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 1412. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.F.; Yuan, S.X.; Yang, F.; Yang, S.; Yang, Y.; Yuan, J.H.; Wang, Z.G.; Xu, Q.G.; Lin, K.Y.; Cai, J.; et al. Aldolase B inhibits metastasis through Ten-Eleven Translocation 1 and serves as a prognostic biomarker in hepatocellular carcinoma. Mol. Cancer 2015, 14, 170. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Li, M.; Yu, H.; Liu, G.; Wang, N.; Yin, C.; Tu, Q.; Narla, G.; Tao, Y.; Cheng, S.; et al. Loss of hepatic aldolase B activates Akt and promotes hepatocellular carcinogenesis by destabilizing the Aldob/Akt/PP2A protein complex. PLoS Biol. 2020, 18, e3000803. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Lanaspa, M.A.; Millan, I.S.; Fini, M.; Rivard, C.J.; Sanchez-Lozada, L.G.; Andres-Hernando, A.; Tolan, D.R.; Johnson, R.J. Fructose contributes to the Warburg effect for cancer growth. Cancer Metab. 2020, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [Green Version]
- Dewdney, B.; Roberts, A.; Qiao, L.; George, J.; Hebbard, L. A Sweet Connection? Fructose’s Role in Hepatocellular Carcinoma. Biomolecules 2020, 10, 496. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Li, X.; Shao, F.; Lv, G.; Lv, H.; Lee, J.-H.; Qian, X.; Wang, Z.; Xia, Y.; Du, L.; et al. The protein kinase activity of fructokinase A specifies the antioxidant responses of tumor cells by phosphorylating p62. Sci. Adv. 2019, 5, eaav4570. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Huang, D.; McArthur, D.L.; Boros, L.G.; Nissen, N.; Heaney, A.P. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. 2010, 70, 6368–6376. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kang, J.; Kang, Y.L.; Woo, J.; Kim, Y.; Huh, J.; Park, J.W. Ketohexokinase-A acts as a nuclear protein kinase that mediates fructose-induced metastasis in breast cancer. Nat. Commun. 2020, 11, 5436. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Oxidative Stress and Antioxidants Balance in Fatty Liver Disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chávez-Rodríguez, L.; Escobedo-Calvario, A.; Salas-Silva, S.; Miranda-Labra, R.U.; Bucio, L.; Souza, V.; Gutiérrez-Ruiz, M.C.; Gomez-Quiroz, L.E. Fructose Consumption and Hepatocellular Carcinoma Promotion. Livers 2021, 1, 250-262. https://doi.org/10.3390/livers1040020
Chávez-Rodríguez L, Escobedo-Calvario A, Salas-Silva S, Miranda-Labra RU, Bucio L, Souza V, Gutiérrez-Ruiz MC, Gomez-Quiroz LE. Fructose Consumption and Hepatocellular Carcinoma Promotion. Livers. 2021; 1(4):250-262. https://doi.org/10.3390/livers1040020
Chicago/Turabian StyleChávez-Rodríguez, Lisette, Alejandro Escobedo-Calvario, Soraya Salas-Silva, Roxana U. Miranda-Labra, Leticia Bucio, Verónica Souza, María Concepción Gutiérrez-Ruiz, and Luis E. Gomez-Quiroz. 2021. "Fructose Consumption and Hepatocellular Carcinoma Promotion" Livers 1, no. 4: 250-262. https://doi.org/10.3390/livers1040020