
Evaluation of Pharmacological Rescue of Melanocortin-4 Receptor Nonsense Mutations by Aminoglycoside
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ligands and Plasmids
2.2. Cell Culture
2.3. Transfection
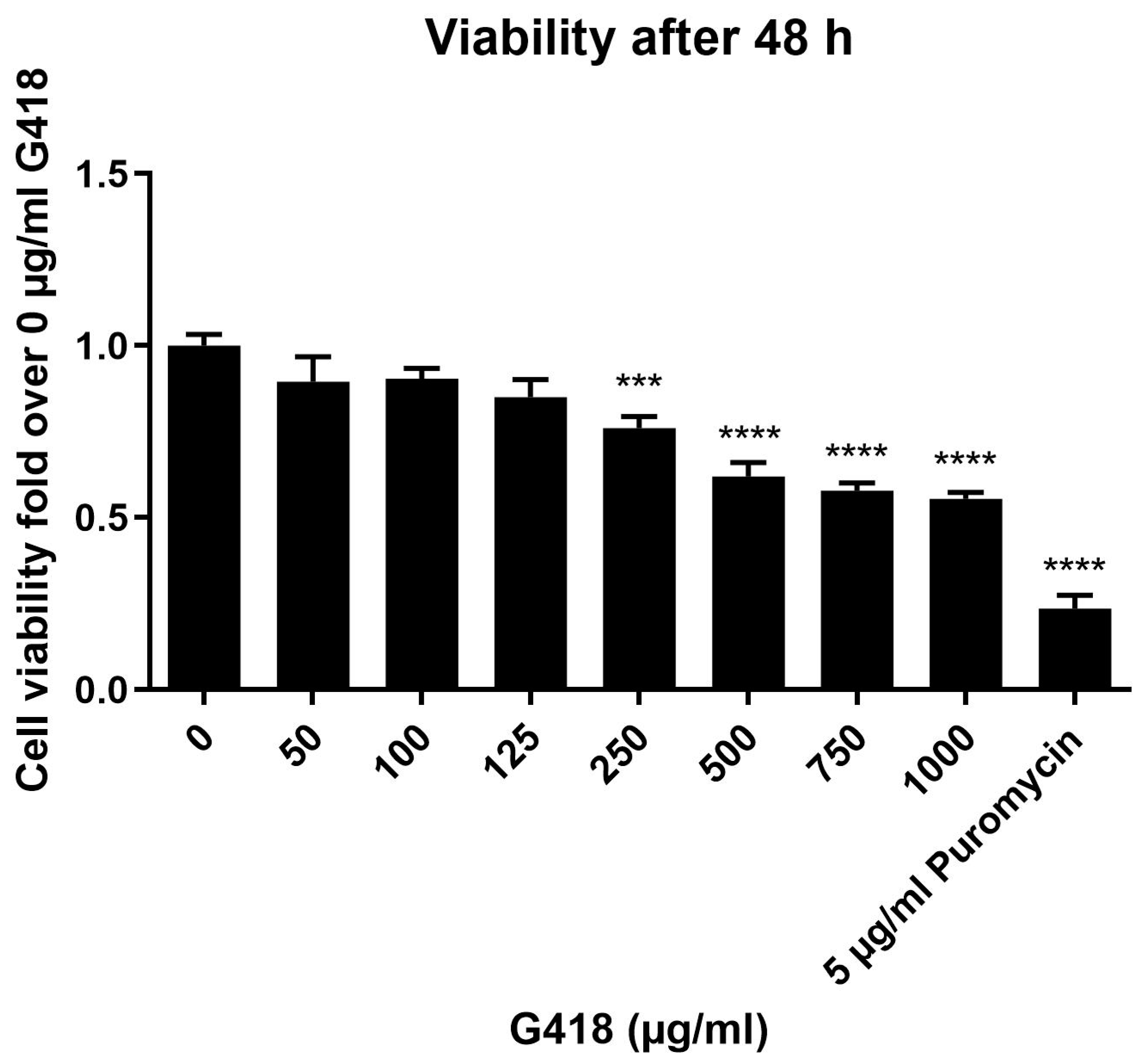
2.4. Antibiotic Kill Curve for Evaluation of Cell Viability
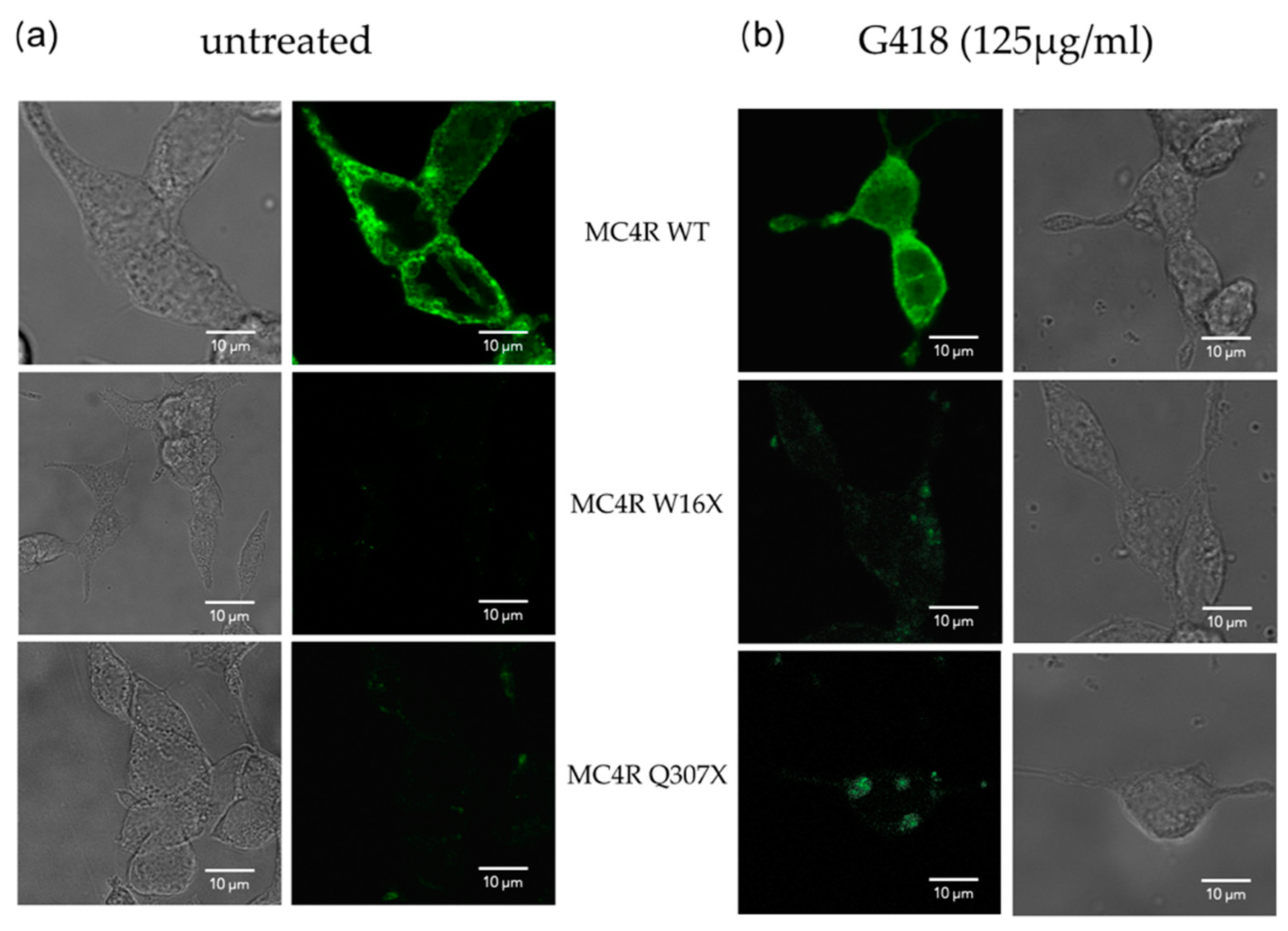
2.5. Confocal Microscopy
2.6. Analysis of Cell Surface and Total Protein Expression
2.7. Measurement of cAMP Increase via GloSensorTM
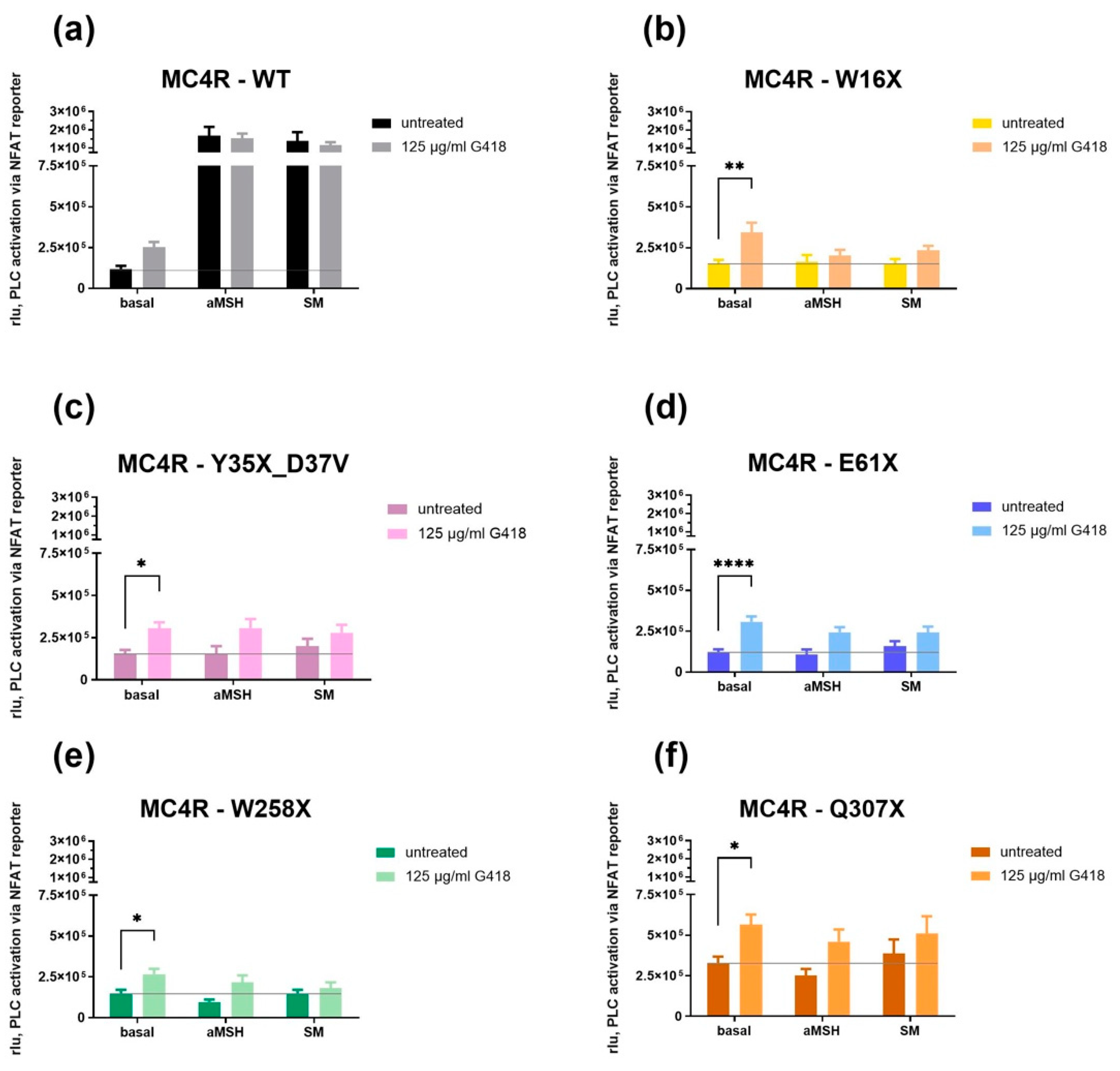
2.8. Reporter Gene Assays for the Determination of PLC Activation
2.9. Statistical Analysis
3. Results
3.1. Evaluation of G418 Cytotoxicity in HEK-293 Cells
3.2. Determination of MC4R Cell Surface and Total Expression
3.2.1. Confocal Microscopy Showed Successful Readthrough Activity
3.2.2. HiBiT Assay for the Determination of Cell Surface and Total Receptor Expression
3.3. G418 Did Not Restore Intracellular Gs Signaling of MC4R Stop Mutants
3.4. G418 Only Increased Basal Gq/11 Signaling in MC4R Stop Mutants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Theilade, S.; Christensen, M.B.; Vilsbøll, T.; Knop, F.K. An overview of obesity mechanisms in humans: Endocrine regulation of food intake, eating behaviour and common determinants of body weight. Diabetes Obes. Metab. 2021, 23, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, M.; Bentham, J.; Stevens, G.A.; Zhou, B.; Danaei, G.; Lu, Y.; Bixby, H.; Cowan, M.J.; Riley, L.M.; Hajifathalian, K.; et al. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef] [Green Version]
- Flegal, K.M.; Graubard, B.I.; Williamson, D.F.; Gail, M.H. Cause-specific excess deaths associated with underweight, overweight, and obesity. JAMA 2007, 298, 2028–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Guh, D.P.; Zhang, W.; Bansback, N.; Amarsi, Z.; Birmingham, C.L.; Anis, A.H. The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health 2009, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Pasquali, R.; Patton, L.; Gambineri, A. Obesity and infertility. Curr. Opin. Endocrinol. Diabetes Obes. 2007, 14, 482–487. [Google Scholar] [CrossRef]
- World Health Organisation. Obesity and Overweight—Key Facts. 2018. Available online: http://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 26 November 2018).
- Hinney, A.; Körner, A.; Fischer-Posovszky, P. The promise of new anti-obesity therapies arising from knowledge of genetic obesity traits. Nat. Rev. Endocrinol. 2022, 18, 623–637. [Google Scholar] [CrossRef]
- Liu, T.; Ji, R.-L.; Tao, Y.-X. Naturally occurring mutations in G protein-coupled receptors associated with obesity and type 2 diabetes mellitus. Pharmacol. Ther. 2022, 234, 108044. [Google Scholar] [CrossRef]
- Kühnen, P.; Krude, H.; Biebermann, H. Melanocortin-4 Receptor Signalling: Importance for Weight Regulation and Obesity Treatment. Trends Mol. Med. 2019, 25, 136–148. [Google Scholar] [CrossRef]
- Vollbach, H.; Brandt, S.; Lahr, G.; Denzer, C.; Von Schnurbein, J.; Debatin, K.-M.; Wabitsch, M. Prevalence and phenotypic characterization of MC4R variants in a large pediatric cohort. Int. J. Obes. 2016, 41, 13–22. [Google Scholar] [CrossRef]
- Paisdzior, S.; Dimitriou, I.M.; Schöpe, P.C.; Annibale, P.; Scheerer, P.; Krude, H.; Lohse, M.J.; Biebermann, H.; Kühnen, P. Differential Signaling Profiles of MC4R Mutations with Three Different Ligands. Int. J. Mol. Sci. 2020, 21, 1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, E.A.; Chai, B.-X.; Zhang, W.; Li, J.-Y.; Ammori, J.B.; Mulholland, M.W. Activation of the Melanocortin-4 Receptor Mobilizes Intracellular Free Calcium in Immortalized Hypothalamic Neurons. J. Surg. Res. 2006, 132, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Clément, K.; Biebermann, H.; Farooqi, I.S.; Van der Ploeg, L.; Wolters, B.; Poitou, C.; Puder, L.; Fiedorek, F.; Gottesdiener, K.; Kleinau, G.; et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat. Med. 2018, 24, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Q.; Shrestha, Y.B.; Pandey, M.; Chen, M.; Kablan, A.; Gavrilova, O.; Offermanns, S.; Weinstein, L.S. Gq/11α and Gsα mediate distinct physiological responses to central melanocortins. J. Clin. Investig. 2015, 126, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collet, T.-H.; Dubern, B.; Mokrosinski, J.; Connors, H.; Keogh, J.M.; de Oliveira, E.M.; Henning, E.; Poitou-Bernert, C.; Oppert, J.-M.; Tounian, P.; et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol. Metab. 2017, 6, 1321–1329. [Google Scholar] [CrossRef]
- Heyder, N.; Kleinau, G.; Szczepek, M.; Kwiatkowski, D.; Speck, D.; Soletto, L.; Cerdá-Reverter, J.M.; Krude, H.; Kühnen, P.; Biebermann, H.; et al. Signal Transduction and Pathogenic Modifications at the Melanocortin-4 Receptor: A Structural Perspective. Front. Endocrinol. 2019, 10, 515. [Google Scholar] [CrossRef] [Green Version]
- MC4R Gene. Available online: https://www.mc4r.org.uk/ (accessed on 29 August 2022).
- Wade, K.H.; Lam, B.Y.H.; Melvin, A.; Pan, W.; Corbin, L.J.; Hughes, D.A.; Rainbow, K.; Chen, J.-H.; Duckett, K.; Liu, X.; et al. Loss-of-function mutations in the melanocortin 4 receptor in a UK birth cohort. Nat. Med. 2021, 27, 1088–1096. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Keogh, J.M.; Yeo, G.S.H.; Lank, E.J.; Cheetham, T.; O’Rahilly, S. Clinical Spectrum of Obesity and Mutations in the Melanocortin 4 Receptor Gene. N. Engl. J. Med. 2003, 348, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2021, 23, 120–133. [Google Scholar] [CrossRef]
- Davies, J.; Gilbert, W.; Gorini, L. Streptomycin, Suppression, and the Code. Proc. Natl. Acad. Sci. USA 1964, 51, 883–890. [Google Scholar] [CrossRef]
- Lee, H.-L.R.; Dougherty, J.P. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol. Ther. 2012, 136, 227–266. [Google Scholar] [CrossRef] [PubMed]
- Martins-Dias, P.; Romão, L. Nonsense suppression therapies in human genetic diseases. Cell. Mol. Life Sci. 2021, 78, 4677–4701. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, D.-Y.; Tao, Y.-X. Therapeutic strategies for diseases caused by loss-of-function mutations in G protein-coupled receptors. Prog. Mol. Biol. Transl. Sci. 2018, 161, 181–210. [Google Scholar] [CrossRef] [PubMed]
- Schilff, M.; Sargsyan, Y.; Hofhuis, J.; Thoms, S. Stop Codon Context-Specific Induction of Translational Readthrough. Biomolecules 2021, 11, 1006. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Bebök, Z.; Ruiz, F.; King, C.; Jones, J.; Walker, L.; Greer, H.; Hong, J.; Wing, L.; Macaluso, M.; et al. Evidence that Systemic Gentamicin Suppresses Premature Stop Mutations in Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2001, 163, 1683–1692. [Google Scholar] [CrossRef]
- Sangkuhl, K.; Schulz, A.; Römpler, H.; Yun, J.; Wess, J.; Schöneberg, T. Aminoglycoside-mediated rescue of a disease-causing nonsense mutation in the V2 vasopressin receptor gene in vitro and in vivo. Hum. Mol. Genet. 2004, 13, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Keeling, K.M.; Wang, D.; Conard, S.E.; Bedwell, D.M. Suppression of premature termination codons as a therapeutic approach. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 444–463. [Google Scholar] [CrossRef] [Green Version]
- Brumm, H.; Mühlhaus, J.; Bolze, F.; Scherag, S.; Hinney, A.; Hebebrand, J.; Wiegand, S.; Klingenspor, M.; Grüters, A.; Krude, H.; et al. Rescue of Melanocortin 4 Receptor (MC4R) Nonsense Mutations by Aminoglycoside-Mediated Read-Through. Obesity 2012, 20, 1074–1081. [Google Scholar] [CrossRef]
- Tabatabaei, M.; Caetano, F.A.; Vedraine, S.; Norton, P.R.; Ferguson, S.S.; Lagugné-Labarthet, F. Directing GPCR-transfected cells and neuronal projections with nano-scale resolution. Biomaterials 2013, 34, 10065–10074. [Google Scholar] [CrossRef]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef]
- He, B.; Soderlund, D.M. Human embryonic kidney (HEK293) cells express endogenous voltage-gated sodium currents and Nav1.7 sodium channels. Neurosci. Lett. 2010, 469, 268–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooqi, I.S. Monogenic human obesity syndromes. Handb. Clin. Neurol. 2021, 181, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Reininghaus, N.; Paisdzior, S.; Höpfner, F.; Jyrch, S.; Cetindag, C.; Scheerer, P.; Kühnen, P.; Biebermann, H. A Setmelanotide-like Effect at MC4R Is Achieved by MC4R Dimer Separation. Biomolecules 2022, 12, 1119. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Heyder, N.A.; Kleinau, G.; Speck, D.; Schmidt, A.; Paisdzior, S.; Szczepek, M.; Bauer, B.; Koch, A.; Gallandi, M.; Kwiatkowski, D.; et al. Structures of active melanocortin-4 receptor–Gs-protein complexes with NDP-α-MSH and setmelanotide. Cell Res. 2021, 31, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Brasell, E.J.; Chu, L.; El Kares, R.; Seo, J.H.; Loesch, R.; Iglesias, D.M.; Goodyer, P. The aminoglycoside geneticin permits translational readthrough of the CTNS W138X nonsense mutation in fibroblasts from patients with nephropathic cystinosis. Pediatr. Nephrol. 2018, 34, 873–881. [Google Scholar] [CrossRef]
- Lombardi, S.; Ferrarese, M.; Marchi, S.; Pinton, P.; Pinotti, M.; Bernardi, F.; Branchini, A. Translational readthrough ofGLAnonsense mutations suggests dominant-negative effects exerted by the interaction of wild-type and missense variants. RNA Biol. 2019, 17, 254–263. [Google Scholar] [CrossRef]
- Salvatori, F.; Breveglieri, G.; Zuccato, C.; Finotti, A.; Bianchi, N.; Borgatti, M.; Feriotto, G.; Destro, F.; Canella, A.; Brognara, E.; et al. Production of β-globin and adult hemoglobin following G418 treatment of erythroid precursor cells from homozygous β039 thalassemia patients. Am. J. Hematol. 2009, 84, 720–728. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.H.; Lee, H.; Kim, J.-H.; Sim, D.Y.; Ahn, H.; Chang, S. p53-Dependent Apoptotic Effect of Puromycin via Binding of Ribosomal Protein L5 and L11 to MDM2 and Its Combination Effect with RITA or Doxorubicin. Cancers 2019, 11, 582. [Google Scholar] [CrossRef] [Green Version]
- Buhr, F.; Kohl-Landgraf, J.; Dieck, S.T.; Hanus, C.; Chatterjee, D.; Hegelein, A.; Schuman, E.M.; Wachtveitl, J.; Schwalbe, H. Design of Photocaged Puromycin for Nascent Polypeptide Release and Spatiotemporal Monitoring of Translation. Angew. Chem. Int. Ed. 2015, 54, 3717–3721. [Google Scholar] [CrossRef]
- FDA. FDA Approves First Treatment for Weight Management for People with Certain Rare Genetic Conditions. Available online: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-first-treatment-weight-management-people-certain-rare-genetic-conditions (accessed on 31 August 2022).
- Iepsen, E.W.; Zhang, J.; Thomsen, H.S.; Hansen, E.L.; Hollensted, M.; Madsbad, S.; Hansen, T.; Holst, J.J.; Holm, J.-C.; Torekov, S.S. Patients with Obesity Caused by Melanocortin-4 Receptor Mutations Can Be Treated with a Glucagon-like Peptide-1 Receptor Agonist. Cell Metab. 2018, 28, 23–32.e3. [Google Scholar] [CrossRef]
- Woodley, D.T.; Cogan, J.; Hou, Y.; Lyu, C.; Marinkovich, M.P.; Keene, D.; Chen, M. Gentamicin induces functional type VII collagen in recessive dystrophic epidermolysis bullosa patients. J. Clin. Investig. 2017, 127, 3028–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Translarna. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/translarna (accessed on 31 August 2022).
- Bolze, F.; Mocek, S.; Zimmermann, A.; Klingenspor, M. Aminoglycosides, but not PTC124 (Ataluren), rescue nonsense mutations in the leptin receptor and in luciferase reporter genes. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, B.S.; Mirshahi, T. Genetic variants help define the role of the MC4R C-terminus in signaling and cell surface stability. Sci. Rep. 2018, 8, 10397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.; Stupack, D.G.; Wilkinson, M.F. Nonsense-mediated RNA decay: An emerging modulator of malignancy. Nat. Cancer 2022, 22, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Hug, N.; Longman, D.; Cáceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Brocke, K.S.; Neu-Yilik, G.; Gehring, N.H.; Hentze, M.; Kulozik, A.E. The human intronless melanocortin 4-receptor gene is NMD insensitive. Hum. Mol. Genet. 2002, 11, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Schonnop, L.; Kleinau, G.; Herrfurth, N.; Volckmar, A.-L.; Cetindag, C.; Müller, A.; Peters, T.; Herpertz, S.; Antel, J.; Hebebrand, J.; et al. Decreased melanocortin-4 receptor function conferred by an infrequent variant at the human melanocortin receptor accessory protein 2 gene. Obesity 2016, 24, 1976–1982. [Google Scholar] [CrossRef]
- Sebag, J.A.; Hinkle, P.M. Melanocortin-2 receptor accessory protein MRAP forms antiparallel homodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 20244–20249. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, M.; Fu, Y.; Zhang, C.; Kuang, Z.; Bian, S.; Wan, R.; Qu, S.; Zhang, C. Reversion of MRAP2 Protein Sequence Generates a Functional Novel Pharmacological Modulator for MC4R Signaling. Biology 2022, 11, 874. [Google Scholar] [CrossRef]
- Soletto, L.; Hernández-Balfagó, S.; Rocha, A.; Scheerer, P.; Kleinau, G.; Cerdá-Reverter, J.M. Melanocortin Receptor Accessory Protein 2-Induced Adrenocorticotropic Hormone Response of Human Melanocortin 4 Receptor. J. Endocr. Soc. 2018, 3, 314–323. [Google Scholar] [CrossRef]
- Chan, L.F.; Webb, T.R.; Chung, T.-T.; Meimaridou, E.; Cooray, S.N.; Guasti, L.; Chapple, J.P.; Egertová, M.; Elphick, M.R.; Cheetham, M.E.; et al. MRAP and MRAP2 are bidirectional regulators of the melanocortin receptor family. Proc. Natl. Acad. Sci. USA 2009, 106, 6146–6151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Li, L.; Jin, X.; Xu, B.; Pi, L.; Liu, S.; Zhu, W.; Zhang, C.; Luan, B.; Gong, L.; et al. Pharmacological effect of human melanocortin-2 receptor accessory protein 2 variants on hypothalamic melanocortin receptors. Endocrine 2018, 61, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-L.; Tao, Y.-X. Regulation of Melanocortin-3 and -4 Receptors by Isoforms of Melanocortin-2 Receptor Accessory Protein 1 and 2. Biomolecules 2022, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- ATCC. COS-7 CRL-1651™. Available online: https://www.atcc.org/products/crl-1651 (accessed on 30 August 2022).
- Hamada, K.; Omura, N.; Taguchi, A.; Baradaran-Heravi, A.; Kotake, M.; Arai, M.; Takayama, K.; Taniguchi, A.; Roberge, M.; Hayashi, Y. New Negamycin-Based Potent Readthrough Derivative Effective against TGA-Type Nonsense Mutations. ACS Med. Chem. Lett. 2019, 10, 1450–1456. [Google Scholar] [CrossRef]
- Stäubert, C.; Tarnow, P.; Brumm, H.; Pitra, C.; Gudermann, T.; Grüters, A.; Schöneberg, T.; Biebermann, H.; Römpler, H. Evolutionary Aspects in Evaluating Mutations in the Melanocortin 4 Receptor. Endocrinology 2007, 148, 4642–4648. [Google Scholar] [CrossRef] [Green Version]
- Campofelice, A.; Lentini, L.; Di Leonardo, A.; Melfi, R.; Tutone, M.; Pace, A.; Pibiri, I. Strategies against Nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs). Int. J. Mol. Sci. 2019, 20, 3329. [Google Scholar] [CrossRef] [Green Version]
- Ko, W.; Porter, J.J.; Sipple, M.T.; Edwards, K.M.; Lueck, J.D. Efficient suppression of endogenous CFTR nonsense mutations using anticodon-engineered transfer RNAs. Mol. Ther. Nucleic Acids 2022, 28, 685–701. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MC4R Mutation | WT Sequence | Stop Mutation Sequence | Stop Codon Name | Localization of Mutation |
|---|---|---|---|---|
| W16X | CAC-CTC-TGG-AAC-CGC | CAC-CTC-TGA-AAC-CGC | opal | N-terminus |
| Y35X_D37V | AAA-GGC-TAC-TCT-GAT | AAA-GGC-TAA-TCT-GTT | ochre | N-terminus |
| E61X | TTG-TTG-GAG-AAT-ATC | TTG-TTG-TAG-AAT-ATC | amber | TMH 1 |
| W258X | GTC-TGC-TGG-GCC-CCA | GTC-TGC-TGA-GCC-CCA | opal | TMH 6 |
| Q307X | CGG-AGT-CAA-CAA-CTG | CGG-AGT-TAA-GAA-CTG | ochre | helix 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Höpfner, F.; Paisdzior, S.; Reininghaus, N.; Sohail, I.; Scheerer, P.; Annibale, P.; Biebermann, H.; Kühnen, P. Evaluation of Pharmacological Rescue of Melanocortin-4 Receptor Nonsense Mutations by Aminoglycoside. Life 2022, 12, 1793. https://doi.org/10.3390/life12111793
Höpfner F, Paisdzior S, Reininghaus N, Sohail I, Scheerer P, Annibale P, Biebermann H, Kühnen P. Evaluation of Pharmacological Rescue of Melanocortin-4 Receptor Nonsense Mutations by Aminoglycoside. Life. 2022; 12(11):1793. https://doi.org/10.3390/life12111793
Chicago/Turabian StyleHöpfner, Friederike, Sarah Paisdzior, Nanina Reininghaus, Iqra Sohail, Patrick Scheerer, Paolo Annibale, Heike Biebermann, and Peter Kühnen. 2022. "Evaluation of Pharmacological Rescue of Melanocortin-4 Receptor Nonsense Mutations by Aminoglycoside" Life 12, no. 11: 1793. https://doi.org/10.3390/life12111793