
Oxidative Stress, Mitochondrial Function and Adaptation to Exercise: New Perspectives in Nutrition

 ,
,  , , , , ,
, , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
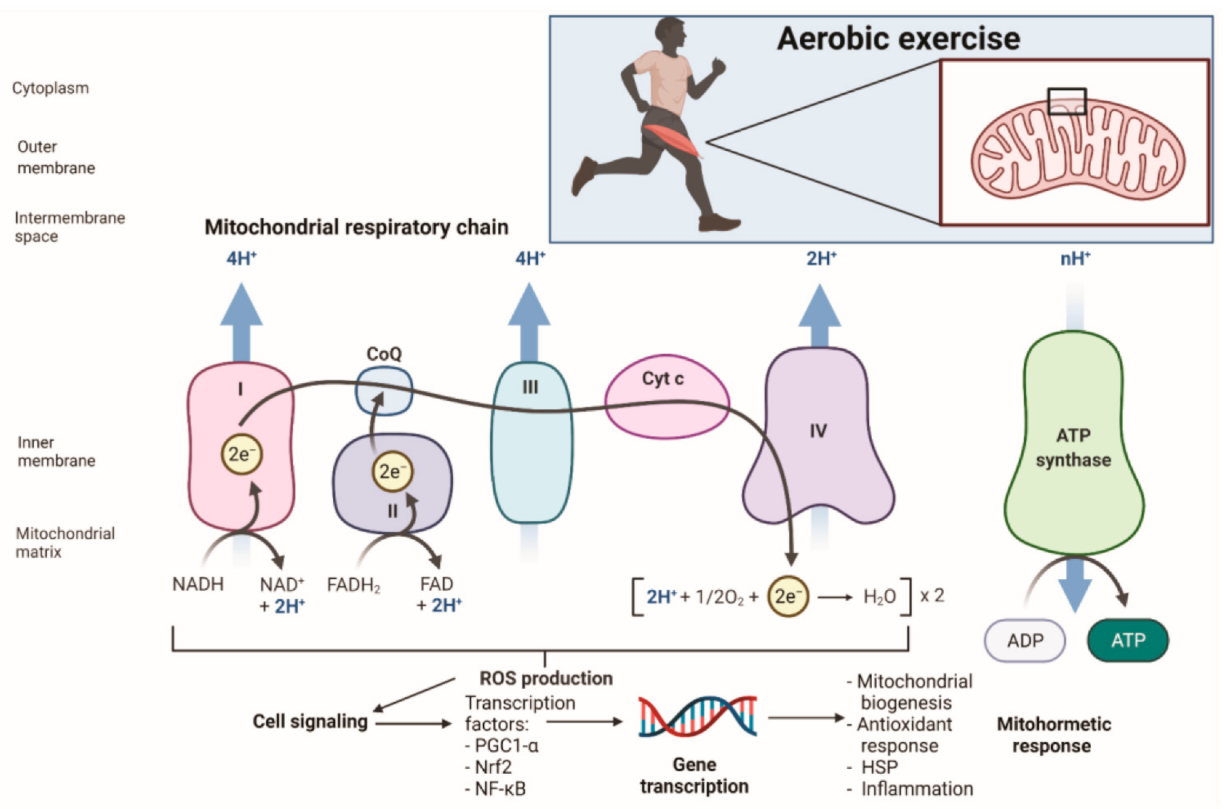
2. Oxidative Stress in Exercise
2.1. Impact of Oxidative Stress on Muscle Contraction
2.2. Mitochondrial ROS Production and Antioxidant Defense
3. Mitochondrial Function and Adaptation
3.1. Mitochondrial Adaptation to Exercise in the Presence of ROS
3.2. Mitohormesis and Exercise
3.3. Mitochondrial Uncoupling and Exercise
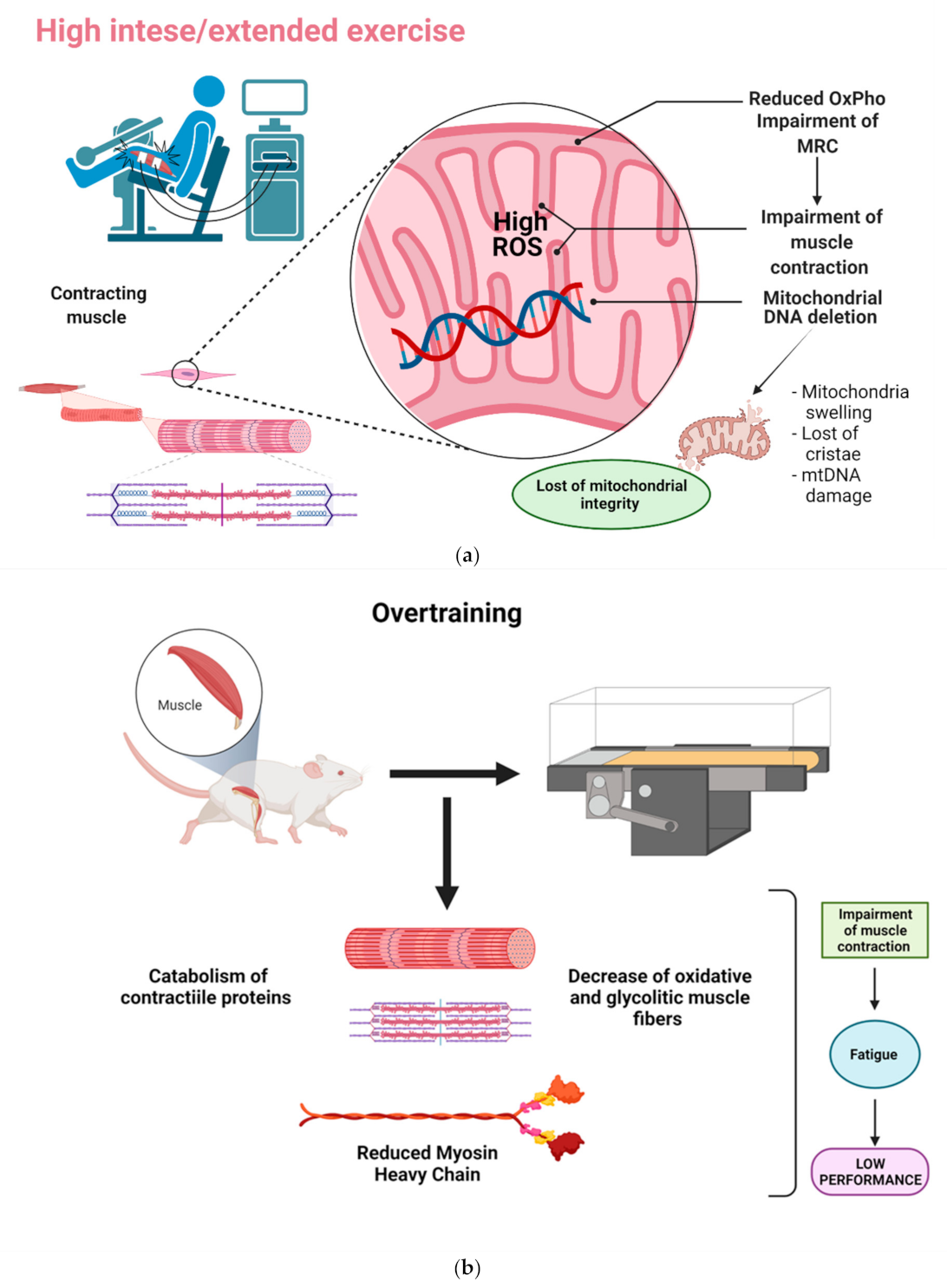
4. Mitochondrial Damage in Exercise Performance
5. Nutrition Insights into Mitochondrial Protection
5.1. General View of Antioxidants, Oxidative Stress, and Exercise Performance
5.2. Mitochondria as Targets for Protection from Oxidative Damage
5.3. Target Mitochondrial Antioxidants in Exercise-Induced Oxidative Damage
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free. Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.F.; Laitano, O. Regulation of NADPH oxidases in skeletal muscle. Free Radic. Biol. Med. 2016, 98, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, M.C.; Arbogast, S.; Guo, Z.; Mathenia, J.; Su, W.; Reid, M.B. Calcium-independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J. Appl. Physiol. 2006, 100, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nethery, D.; Stofan, D.; Callahan, L.; DiMarco, A.; Supinski, G. Formation of reactive oxygen species by the contracting diaphragm is PLA2dependent. J. Appl. Physiol. 1999, 87, 792–800. [Google Scholar] [CrossRef]
- Cheng, A.J.; Jude, B.; Lanner, J.T. Intramuscular mechanisms of overtraining. Redox Biol. 2020, 35, 101480. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.-J.; Qin, Z.; Wang, P.-Y.; Sun, Y.; Liu, X. Muscle fatigue: General understanding and treatment. Exp. Mol. Med. 2017, 49, e384. [Google Scholar] [CrossRef]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509 Pt 2, 565–575. [Google Scholar] [CrossRef]
- Cheng, A.J.; Yamada, T.; Rassier, D.E.; Andersson, D.C.; Westerblad, H.; Lanner, J.T. Reactive oxygen/nitrogen species and contractile function in skeletal muscle during fatigue and recovery. J. Physiol. 2016, 594, 5149–5160. [Google Scholar] [CrossRef] [Green Version]
- Debold, E.P. Potential molecular mechanisms underlying muscle fatigue mediated by reactive oxygen and nitrogen species. Front. Physiol. 2015, 6, 239. [Google Scholar] [CrossRef]
- Hargreaves, M.; Spriet, L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Jackson, M.J.; Vasilaki, A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic. Res. 2014, 48, 12–29. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef] [Green Version]
- Culotta, V.C.; Yang, M.; O’Halloran, T.V. Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta 2006, 1763, 747–758. [Google Scholar] [CrossRef] [Green Version]
- Criswell, D.; Powers, S.; Dodd, S.; Lawler, J.; Edwards, W.; Renshler, K.; Grinton, S. High intensity training-induced changes in skeletal muscle antioxidant enzyme activity. Med. Sci. Sports Exerc. 1993, 25, 1135–1140. [Google Scholar] [CrossRef]
- Powers, S.K.; Criswell, D.; Lawler, J.; Ji, L.L.; Martin, D.; Herb, R.A.; Dudley, G. Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am. J. Physiol. 1994, 266, R375–R380. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermingham, E.N.; Hesketh, J.E.; Sinclair, B.R.; Koolaard, J.P.; Roy, N.C. Selenium-enriched foods are more effective at increasing glutathione peroxidase (GPx) activity compared with selenomethionine: A meta-analysis. Nutrients 2014, 6, 4002–4031. [Google Scholar] [CrossRef]
- Li, R.; Fan, X.; Zhang, T.; Song, H.; Bian, X.; Nai, R.; Li, J.; Zhang, J. Expression of selenium-independent glutathione peroxidase 5 (GPx5) in the epididymis of Small Tail Han sheep. Asian-Australas. J. Anim. Sci. 2018, 31, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Y.; Lv, Y.; Tian, M.; You, J.; Chen, F.; Zhang, S.; Guan, W. Effects of Selenomethionine on Cell Viability, Selenoprotein Expression and Antioxidant Function in Porcine Mammary Epithelial Cells. Front. Nutr. 2021, 8, 665855. [Google Scholar] [CrossRef]
- Chen, Y.I.; Wei, P.C.; Hsu, J.L.; Su, F.Y.; Lee, W.H. NPGPx (GPx7): A novel oxidative stress sensor/transmitter with multiple roles in redox homeostasis. Am. J. Transl. Res. 2016, 8, 1626–1640. [Google Scholar]
- Khatib, A.; Solaimuthu, B.; Ben Yosef, M.; Abu Rmaileh, A.; Tanna, M.; Oren, G.; Schlesinger Frisch, M.; Axelrod, J.H.; Lichtenstein, M.; Shaul, Y.D. The glutathione peroxidase 8 (GPX8)/IL-6/STAT3 axis is essential in maintaining an aggressive breast cancer phenotype. Proc. Natl. Acad. Sci. USA 2020, 117, 21420–21431. [Google Scholar] [CrossRef]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [Green Version]
- Jastrząb, A.; Skrzydlewska, E. Thioredoxin-dependent system. Application of inhibitors. J. Enzym. Inhib. Med. Chem. 2021, 36, 362–371. [Google Scholar] [CrossRef]
- Zhong, L.; Arnér, E.S.; Holmgren, A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. USA 2000, 97, 5854–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnér, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kang, D. The Role of Peroxiredoxins in the Transduction of H(2)O(2) Signals. Antioxid. Redox Signal. 2018, 28, 537–557. [Google Scholar] [CrossRef]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-induced skeletal muscle remodeling and metabolic adaptation: Redox signaling and role of autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Carretero, A.; Millan-Domingo, F.; Garcia-Dominguez, E.; Correas, A.G.; Olaso-Gonzalez, G.; Viña, J. Redox-related biomarkers in physical exercise. Redox Biol. 2021, 42, 101956. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Spiegelman, B.M. Transcriptional control of energy homeostasis through the PGC1 coactivators. Novartis Found. Symp. 2007, 286, 3–6; discussion 6–12, 162–163, 196–203. [Google Scholar] [PubMed]
- Baar, K. Nutrition and the adaptation to endurance training. Sports Med. 2014, 44 (Suppl. 1), S5–S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinsomboon, J.; Ruas, J.; Gupta, R.K.; Thom, R.; Shoag, J.; Rowe, G.C.; Sawada, N.; Raghuram, S.; Arany, Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2009, 106, 21401–21406. [Google Scholar] [CrossRef] [Green Version]
- Arany, Z.; Lebrasseur, N.; Morris, C.; Smith, E.; Yang, W.; Ma, Y.; Chin, S.; Spiegelman, B.M. The transcriptional coactivator PGC-1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 2007, 5, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Eggler, A.L.; Small, E.; Hannink, M.; Mesecar, A.D. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem. J. 2009, 422, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Kang, D.; Kim, S.H.; Hamasaki, N. Mitochondrial transcription factor A (TFAM): Roles in maintenance of mtDNA and cellular functions. Mitochondrion 2007, 7, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [Green Version]
- Garza-Lombó, C.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Redox homeostasis, oxidative stress and mitophagy. Mitochondrion 2020, 51, 105–117. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Murata, D.; Adachi, Y.; Itoh, K.; Kameoka, S.; Igarashi, A.; Kato, T.; Araki, Y.; Huganir, R.L.; Dawson, T.M.; et al. Mitochondrial Stasis Reveals p62-Mediated Ubiquitination in Parkin-Independent Mitophagy and Mitigates Nonalcoholic Fatty Liver Disease. Cell Metab. 2018, 28, 588–604. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulos, N.D.; Frison, M.; Alvarez, M.S.; Bertrand, H.; Wells, G.; Campanella, M. Reversible Keap1 inhibitors are preferential pharmacological tools to modulate cellular mitophagy. Sci. Rep. 2017, 7, 10303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef]
- Li, H.; Miao, W.; Ma, J.; Xv, Z.; Bo, H.; Li, J.; Zhang, Y.; Ji, L.L. Acute Exercise-Induced Mitochondrial Stress Triggers an Inflammatory Response in the Myocardium via NLRP3 Inflammasome Activation with Mitophagy. Oxidative Med. Cell. Longev. 2016, 2016, 1987149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, L.L.; Gomez-Cabrera, M.C.; Vina, J. Exercise and hormesis: Activation of cellular antioxidant signaling pathway. Ann. N. Y. Acad. Sci. 2006, 1067, 425–435. [Google Scholar] [CrossRef]
- Lawler, J.M.; Rodriguez, D.A.; Hord, J.M. Mitochondria in the middle: Exercise preconditioning protection of striated muscle. J. Physiol. 2016, 594, 5161–5183. [Google Scholar] [CrossRef] [Green Version]
- Alleman, R.J.; Katunga, L.A.; Nelson, M.A.; Brown, D.A.; Anderson, E.J. The “Goldilocks Zone” from a redox perspective-Adaptive vs. deleterious responses to oxidative stress in striated muscle. Front. Physiol. 2014, 5, 358. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Hung, M.C. Beyond NF-κB activation: Nuclear functions of IκB kinase α. J. Biomed. Sci. 2013, 20, 3. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Engelhardt, J.F. Interleukin-1beta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J. Biol. Chem. 2006, 281, 1495–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109 (Suppl. 1), S81–S96. [Google Scholar] [CrossRef] [Green Version]
- Obata, T.; Brown, G.E.; Yaffe, M.B. MAP kinase pathways activated by stress: The p38 MAPK pathway. Crit. Care Med. 2000, 28, N67–N77. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [Green Version]
- Cuschieri, J.; Maier, R.V. Mitogen-activated protein kinase (MAPK). Crit. Care Med. 2005, 33, S417–S419. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.L.; Ristow, M. Mitohormesis in exercise training. Free. Radic. Biol. Med. 2016, 98, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Musci, R.V.; Hamilton, K.L.; Linden, M.A. Exercise-Induced Mitohormesis for the Maintenance of Skeletal Muscle and Healthspan Extension. Sports 2019, 7, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Li, C.G.; Qi, Z.; Cui, D.; Ding, S. Acute exercise stress promotes Ref1/Nrf2 signalling and increases mitochondrial antioxidant activity in skeletal muscle. Exp. Physiol. 2016, 101, 410–420. [Google Scholar] [CrossRef]
- Merry, T.L.; Ristow, M. Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J. Physiol. 2016, 594, 5195–5207. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Galeotti, T. Role of MnSOD and p66shc in mitochondrial response to p53. Antioxid. Redox Signal. 2011, 15, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Koch, O.R.; Galeotti, T. The p53-p66shc-Manganese Superoxide Dismutase (MnSOD) network: A mitochondrial intrigue to generate reactive oxygen species. Int. J. Biochem. Cell Biol. 2009, 41, 1002–1005. [Google Scholar] [CrossRef]
- Wang, P.; Li, C.G.; Qi, Z.; Cui, D.; Ding, S. Acute exercise induced mitochondrial H2O2 production in mouse skeletal muscle: Association with p(66Shc) and FOXO3a signaling and antioxidant enzymes. Oxidative Med. Cell. Longev. 2015, 2015, 536456. [Google Scholar] [CrossRef]
- Iversen, N.; Krustrup, P.; Rasmussen, H.N.; Rasmussen, U.F.; Saltin, B.; Pilegaard, H. Mitochondrial biogenesis and angiogenesis in skeletal muscle of the elderly. Exp. Gerontol. 2011, 46, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [Green Version]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. Slip and leak in mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta 1989, 977, 123–141. [Google Scholar] [CrossRef]
- Crichton, P.G.; Lee, Y.; Kunji, E.R. The molecular features of uncoupling protein 1 support a conventional mitochondrial carrier-like mechanism. Biochimie 2017, 134, 35–50. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, R.C.; Muñoz, V.R.; Kuga, G.K.; Nakandakari, S.; Minuzzi, L.G.; Botezelli, J.D.; da Silva, A.S.R.; Cintra, D.E.; de Moura, L.P.; Ropelle, E.R.; et al. Acute physical exercise increases leptin-induced hypothalamic extracellular signal-regulated kinase1/2 phosphorylation and thermogenesis of obese mice. J. Cell. Biochem. 2019, 120, 697–704. [Google Scholar] [CrossRef] [Green Version]
- Berardi, M.J.; Chou, J.J. Fatty acid flippase activity of UCP2 is essential for its proton transport in mitochondria. Cell Metab. 2014, 20, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S.; Buckingham, J.A.; Samec, S.; Seydoux, J.; Din, N.; Dulloo, A.G.; Brand, M.D. UCP2 and UCP3 rise in starved rat skeletal muscle but mitochondrial proton conductance is unchanged. FEBS Lett. 1999, 462, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Anedda, A.; López-Bernardo, E.; Acosta-Iborra, B.; Saadeh Suleiman, M.; Landázuri, M.O.; Cadenas, S. The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free Radic. Biol. Med. 2013, 61, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.A.; Stuart, J.A.; Jekabsons, M.B.; Roussel, D.; Brindle, K.M.; Dickinson, K.; Jones, R.B.; Brand, M.D. Artifactual uncoupling by uncoupling protein 3 in yeast mitochondria at the concentrations found in mouse and rat skeletal-muscle mitochondria. Biochem. J. 2002, 361, 49–56. [Google Scholar] [CrossRef]
- Nabben, M.; Shabalina, I.G.; Moonen-Kornips, E.; van Beurden, D.; Cannon, B.; Schrauwen, P.; Nedergaard, J.; Hoeks, J. Uncoupled respiration, ROS production, acute lipotoxicity and oxidative damage in isolated skeletal muscle mitochondria from UCP3-ablated mice. Biochim. Biophys. Acta 2011, 1807, 1095–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusuhara, K.; Tobe, T.; Negoro, T.; Abe, T. A rapid up-regulation in UCP3 transcriptional activity in response to moderate intensity exercise in rat skeletal muscle. J. Sports Sci. Med. 2005, 4, 170–178. [Google Scholar]
- Jiang, N.; Zhang, G.; Bo, H.; Qu, J.; Ma, G.; Cao, D.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Upregulation of uncoupling protein-3 in skeletal muscle during exercise: A potential antioxidant function. Free. Radic. Biol. Med. 2009, 46, 138–145. [Google Scholar] [CrossRef]
- Bo, H.; Wang, Y.H.; Li, H.Y.; Zhao, J.; Zhang, H.Y.; Tong, C.Q. Endurance training attenuates the bioenergetics alterations of rat skeletal muscle mitochondria submitted to acute hypoxia: Role of ROS and UCP3. Sheng Li Xue Bao [Acta physiologica Sinica] 2008, 60, 767–776. [Google Scholar]
- Peake, J.M.; Markworth, J.F.; Nosaka, K.; Raastad, T.; Wadley, G.D.; Coffey, V.G. Modulating exercise-induced hormesis: Does less equal more? J. Appl. Physiol. 2015, 119, 172–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadaja, L.; Eimre, M.; Paju, K.; Roosimaa, M.; Põdramägi, T.; Kaasik, P.; Pehme, A.; Orlova, E.; Mudist, M.; Peet, N.; et al. Impaired oxidative phosphorylation in overtrained rat myocardium. Exp. Clin. Cardiol. 2010, 15, e116–e127. [Google Scholar]
- Urhausen, A.; Gabriel, H.H.; Weiler, B.; Kindermann, W. Ergometric and psychological findings during overtraining: A long-term follow-up study in endurance athletes. Int. J. Sports Med. 1998, 19, 114–120. [Google Scholar] [CrossRef]
- Huang, C.C.; Lin, T.J.; Chen, C.C.; Lin, W.T. Endurance training accelerates exhaustive exercise-induced mitochondrial DNA deletion and apoptosis of left ventricle myocardium in rats. Eur. J. Appl. Physiol. 2009, 107, 697–706. [Google Scholar] [CrossRef]
- King, D.W.; Gollnick, P.D. Ultrastructure of rat heart and liver after exhaustive exercise. Am. J. Physiol. 1970, 218, 1150–1155. [Google Scholar] [CrossRef] [Green Version]
- Venditti, P.; Di Meo, S. Antioxidants, tissue damage, and endurance in trained and untrained young male rats. Arch. Biochem. Biophys. 1996, 331, 63–68. [Google Scholar] [CrossRef]
- Seene, T.; Kaasik, P.; Alev, K.; Pehme, A.; Riso, E.M. Composition and turnover of contractile proteins in volume-overtrained skeletal muscle. Int. J. Sports Med. 2004, 25, 438–445. [Google Scholar] [CrossRef]
- Ferraresso, R.L.; de Oliveira, R.; Macedo, D.V.; Nunes, L.A.; Brenzikofer, R.; Damas, D.; Hohl, R. Interaction between overtraining and the interindividual variability may (not) trigger muscle oxidative stress and cardiomyocyte apoptosis in rats. Oxidative Med. Cell. Longev. 2012, 2012, 935483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, W.; Li, Z.; Liu, X.H.; Liu, Z.M.; Dun, Y.; Feng, H. Effects of exhaustive exercise on the expression of PHB1 and the function of mitochondria in rats. Zhongguo Yingyong Shenglixue Zazhi (Chin. J. Appl. Physiol.) 2017, 33, 544–549. [Google Scholar] [CrossRef]
- Ross, J.A.; Nagy, Z.S.; Kirken, R.A. The PHB1/2 phosphocomplex is required for mitochondrial homeostasis and survival of human T cells. J. Biol. Chem. 2008, 283, 4699–4713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meo, S.; Venditti, P. Mitochondria in exercise-induced oxidative stress. Biol. Signals Recept. 2001, 10, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Joseph, A.M.; Saleem, A.; Uguccioni, G.; Collu-Marchese, M.; Lai, R.Y.; Nguyen, L.M.; Hood, D.A. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: Effects of exercise and aging. Biochim. Biophys. Acta 2010, 1800, 223–234. [Google Scholar] [CrossRef]
- Hood, D.A. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle. Appl. Physiol. Nutr. Metab. (Physiologie appliquee Nutrition et Metabolisme) 2009, 34, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Laguens, R.P.; Gómez-Dumm, C.L. Fine structure of myocardial mitochondria in rats after exercise for one-half to two hours. Circ. Res. 1967, 21, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merry, T.L.; Ristow, M. Do antioxidant supplements interfere with skeletal muscle adaptation to exercise training? J. Physiol. 2016, 594, 5135–5147. [Google Scholar] [CrossRef]
- Peternelj, T.T.; Coombes, J.S. Antioxidant supplementation during exercise training: Beneficial or detrimental? Sports Med. 2011, 41, 1043–1069. [Google Scholar] [CrossRef]
- Slattery, K.; Bentley, D.; Coutts, A.J. The role of oxidative, inflammatory and neuroendocrinological systems during exercise stress in athletes: Implications of antioxidant supplementation on physiological adaptation during intensified physical training. Sports Med. 2015, 45, 453–471. [Google Scholar] [CrossRef]
- Antonioni, A.; Fantini, C.; Dimauro, I.; Caporossi, D. Redox homeostasis in sport: Do athletes really need antioxidant support? Res. Sports Med. (Print) 2019, 27, 147–165. [Google Scholar] [CrossRef]
- Margaritis, I.; Rousseau, A.S. Does physical exercise modify antioxidant requirements? Nutr. Res. Rev. 2008, 21, 3–12. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, D.C.X.; Rosa, F.T.; Simões-Ambrósio, L.; Jordao, A.A.; Deminice, R. Antioxidant vitamin supplementation prevents oxidative stress but does not enhance performance in young football athletes. Nutrition 2019, 63–64, 29–35. [Google Scholar] [CrossRef]
- Ryan, M.J.; Dudash, H.J.; Docherty, M.; Geronilla, K.B.; Baker, B.A.; Haff, G.G.; Cutlip, R.G.; Alway, S.E. Vitamin E and C supplementation reduces oxidative stress, improves antioxidant enzymes and positive muscle work in chronically loaded muscles of aged rats. Exp. Gerontol. 2010, 45, 882–895. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.; Hughes, J.; Della Gatta, P.A.; Mason, S.; Lamon, S.; Russell, A.P.; Wadley, G.D. Vitamin C and E supplementation prevents some of the cellular adaptations to endurance-training in humans. Free. Radic. Biol. Med. 2015, 89, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, G.; Cumming, K.T.; Holden, G.; Hallén, J.; Rønnestad, B.R.; Sveen, O.; Skaug, A.; Paur, I.; Bastani, N.E.; Østgaard, H.N.; et al. Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: A double-blind, randomised, controlled trial. J. Physiol. 2014, 592, 1887–1901. [Google Scholar] [CrossRef] [PubMed]
- Cory, H.; Passarelli, S.; Szeto, J.; Tamez, M.; Mattei, J. The Role of Polyphenols in Human Health and Food Systems: A Mini-Review. Front. Nutr. 2018, 5, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myburgh, K.H. Polyphenol supplementation: Benefits for exercise performance or oxidative stress? Sports Med. 2014, 44 (Suppl. 1), S57–S70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elejalde, E.; Villarán, M.C.; Alonso, R.M. Grape polyphenols supplementation for exercise-induced oxidative stress. J. Int. Soc. Sports Nutr. 2021, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Salehi, M.; Mashhadi, N.S.; Esfahani, P.S.; Feizi, A.; Hadi, A.; Askari, G. The Effects of Curcumin Supplementation on Muscle Damage, Oxidative Stress, and Inflammatory Markers in Healthy Females with Moderate Physical Activity: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Int. J. Prev. Med. 2021, 12, 94. [Google Scholar] [CrossRef]
- Vargas-Mendoza, N.; Ángeles-Valencia, M.; Madrigal-Santillán, E.O.; Morales-Martínez, M.; Tirado-Lule, J.M.; Solano-Urrusquieta, A.; Madrigal-Bujaidar, E.; Álvarez-González, I.; Fregoso-Aguilar, T.; Morales-González, Á.; et al. Effect of Silymarin Supplementation on Physical Performance, Muscle and Myocardium Histological Changes, Bodyweight, and Food Consumption in Rats Subjected to Regular Exercise Training. Int. J. Mol. Sci. 2020, 21, 7724. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Komine, S.; Warabi, E.; Akiyama, K.; Ishii, A.; Ishige, K.; Mizokami, Y.; Kuga, K.; Horie, M.; Miwa, Y.; et al. Nuclear factor (erythroid derived 2)-like 2 activation increases exercise endurance capacity via redox modulation in skeletal muscles. Sci. Rep. 2017, 7, 12902. [Google Scholar] [CrossRef] [Green Version]
- Ruhee, R.T.; Suzuki, K. The Integrative Role of Sulforaphane in Preventing Inflammation, Oxidative Stress and Fatigue: A Review of a Potential Protective Phytochemical. Antioxidants 2020, 9, 521. [Google Scholar] [CrossRef]
- Vargas-Mendoza, N.; Morales-González, Á.; Morales-Martínez, M.; Soriano-Ursúa, M.A.; Delgado-Olivares, L.; Sandoval-Gallegos, E.M.; Madrigal-Bujaidar, E.; Álvarez-González, I.; Madrigal-Santillán, E.; Morales-Gonzalez, J.A. Flavolignans from Silymarin as Nrf2 Bioactivators and Their Therapeutic Applications. Biomedicines 2020, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Farzaei, M.H.; Zobeiri, M.; Parvizi, F.; El-Senduny, F.F.; Marmouzi, I.; Coy-Barrera, E.; Naseri, R.; Nabavi, S.M.; Rahimi, R.; Abdollahi, M. Curcumin in Liver Diseases: A Systematic Review of the Cellular Mechanisms of Oxidative Stress and Clinical Perspective. Nutrients 2018, 10, 855. [Google Scholar] [CrossRef] [Green Version]
- Hassan, F.U.; Rehman, M.S.; Khan, M.S.; Ali, M.A.; Javed, A.; Nawaz, A.; Yang, C. Curcumin as an Alternative Epigenetic Modulator: Mechanism of Action and Potential Effects. Front. Genet. 2019, 10, 514. [Google Scholar] [CrossRef] [Green Version]
- Tryfidou, D.V.; McClean, C.; Nikolaidis, M.G.; Davison, G.W. DNA Damage Following Acute Aerobic Exercise: A Systematic Review and Meta-analysis. Sports Med. 2020, 50, 103–127. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [Green Version]
- Yavari, A.; Javadi, M.; Mirmiran, P.; Bahadoran, Z. Exercise-induced oxidative stress and dietary antioxidants. Asian J. Sports Med. 2015, 6, e24898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pingitore, A.; Lima, G.P.; Mastorci, F.; Quinones, A.; Iervasi, G.; Vassalle, C. Exercise and oxidative stress: Potential effects of antioxidant dietary strategies in sports. Nutrition 2015, 31, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, O.; Yfanti, C. Antioxidants in Athlete’s Basic Nutrition: Considerations towards a Guideline for the Intake of Vitamin C and Vitamin E. In Antioxidants in Sport Nutrition; Lamprecht, M., Ed.; Taylor & Francis Group, LLC.: Boca Raton, FL, USA, 2015. [Google Scholar]
- Fogarty, M.C.; Devito, G.; Hughes, C.M.; Burke, G.; Brown, J.C.; McEneny, J.; Brown, D.; McClean, C.; Davison, G.W. Effects of α-lipoic acid on mtDNA damage after isolated muscle contractions. Med. Sci. Sports Exerc. 2013, 45, 1469–1477. [Google Scholar] [CrossRef]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018, 10, 1887–1899. [Google Scholar]
- Mao, G.; Kraus, G.A.; Kim, I.; Spurlock, M.E.; Bailey, T.B.; Zhang, Q.; Beitz, D.C. A mitochondria-targeted vitamin E derivative decreases hepatic oxidative stress and inhibits fat deposition in mice. J. Nutr. 2010, 140, 1425–1431. [Google Scholar] [CrossRef]
- Mao, G.; Kraus, G.A.; Kim, I.; Spurlock, M.E.; Bailey, T.B.; Beitz, D.C. Effect of a mitochondria-targeted vitamin E derivative on mitochondrial alteration and systemic oxidative stress in mice. Br. J. Nutr. 2011, 106, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Hu, X.-H.; Jia, Z.-G.; Xu, M.-H.; Guo, Z.-Y.; Gao, F.-H. Tiron protects against UVB-induced senescence-like characteristics in human dermal fibroblasts by the inhibition of superoxide anion production and glutathione depletion. Australas. J. Dermatol. 2012, 53, 172–180. [Google Scholar] [CrossRef]
- Finichiu, P.G.; Larsen, D.S.; Evans, C.; Larsen, L.; Bright, T.P.; Robb, E.L.; Trnka, J.; Prime, T.A.; James, A.M.; Smith, R.A.J.; et al. A mitochondria-targeted derivative of ascorbate: MitoC. Free. Radic. Biol. Med. 2015, 89, 668–678. [Google Scholar] [CrossRef] [Green Version]
- Williamson, J.; Davison, G. Targeted Antioxidants in Exercise-Induced Mitochondrial Oxidative Stress: Emphasis on DNA Damage. Antioxidants 2020, 9, 1142. [Google Scholar] [CrossRef]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.A.J.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Cochemé, H.M.; Kelso, G.F.; James, A.M.; Ross, M.F.; Trnka, J.; Mahendiran, T.; Asin-Cayuela, J.; Blaikie, F.H.; Manas, A.R.; Porteous, C.M.; et al. Mitochondrial targeting of quinones: Therapeutic implications. Mitochondrion 2007, 7 (Suppl.), S94–S102. [Google Scholar] [CrossRef]
- Ross, M.F.; Kelso, G.F.; Blaikie, F.H.; James, A.M.; Cochemé, H.M.; Filipovska, A.; Da Ros, T.; Hurd, T.R.; Smith, R.A.; Murphy, M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochem. Biokhimiia 2005, 70, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta 2008, 1777, 1028–1031. [Google Scholar] [CrossRef] [Green Version]
- James, A.M.; Sharpley, M.S.; Manas, A.R.; Frerman, F.E.; Hirst, J.; Smith, R.A.; Murphy, M.P. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718. [Google Scholar] [CrossRef] [Green Version]
- James, A.M.; Cochemé, H.M.; Smith, R.A.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shill, D.D.; Southern, W.M.; Willingham, T.B.; Lansford, K.A.; McCully, K.K.; Jenkins, N.T. Mitochondria-specific antioxidant supplementation does not influence endurance exercise training-induced adaptations in circulating angiogenic cells, skeletal muscle oxidative capacity or maximal oxygen uptake. J. Physiol. 2016, 594, 7005–7014. [Google Scholar] [CrossRef]
- Williamson, J.; Hughes, C.M.; Cobley, J.N.; Davison, G.W. The mitochondria-targeted antioxidant MitoQ, attenuates exercise-induced mitochondrial DNA damage. Redox Biol. 2020, 36, 101673. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Understanding and preventing mitochondrial oxidative damage. Biochem. Soc. Trans. 2016, 44, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef]
- Schlame, M.; Greenberg, M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2017, 1862, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Pekas, E.J.; Headid, R.J., 3rd; Son, W.M.; Wooden, T.K.; Song, J.; Layec, G.; Yadav, S.K.; Mishra, P.K.; Pipinos, I.I. Acute mitochondrial antioxidant intake improves endothelial function, antioxidant enzyme activity, and exercise tolerance in patients with peripheral artery disease. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H456–H467. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, J.; Li, G.; Wu, J.; Wu, X.; Wang, G.; Gu, G.; Ren, H.; Hong, Z.; Li, J. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018, 9, 403. [Google Scholar] [CrossRef]
- Boris, A.F.; Vladimir, P.S. Cellular and Molecular Mechanisms of Action of Mitochondria-Targeted Antioxidants. Curr. Aging Sci. 2017, 10, 41–48. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [Green Version]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. JASN 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [Green Version]
- Min, K.; Smuder, A.J.; Kwon, O.S.; Kavazis, A.N.; Szeto, H.H.; Powers, S.K. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 2011, 111, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.J.; Rothnagel, J.A. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014, 15, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Xiao, M.H.; Chen, H.X.; Meng, Y.; Zhao, N.; Yang, L.; Tang, H.; Wang, J.L.; Liu, X.; Zhu, Y.; et al. A novel mitochondrial micropeptide MPM enhances mitochondrial respiratory activity and promotes myogenic differentiation. Cell Death Dis. 2019, 10, 528. [Google Scholar] [CrossRef]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and Other Nutrigenomic Nrf2 Activators: Can the Clinician’s Expectation Be Matched by the Reality? Oxidative Med. Cell. Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Dashwood, R.H. Epigenetic Regulation of NRF2/KEAP1 by Phytochemicals. Antioxidants 2020, 9, 865. [Google Scholar] [CrossRef] [PubMed]
- Nocella, C.; Cammisotto, V.; Pigozzi, F.; Borrione, P.; Fossati, C.; D’Amico, A.; Cangemi, R.; Peruzzi, M.; Gobbi, G.; Ettorre, E.; et al. Impairment between Oxidant and Antioxidant Systems: Short- and Long-term Implications for Athletes’ Health. Nutrients 2019, 11, 1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vargas-Mendoza, N.; Angeles-Valencia, M.; Morales-González, Á.; Madrigal-Santillán, E.O.; Morales-Martínez, M.; Madrigal-Bujaidar, E.; Álvarez-González, I.; Gutiérrez-Salinas, J.; Esquivel-Chirino, C.; Chamorro-Cevallos, G.; et al. Oxidative Stress, Mitochondrial Function and Adaptation to Exercise: New Perspectives in Nutrition. Life 2021, 11, 1269. https://doi.org/10.3390/life11111269
Vargas-Mendoza N, Angeles-Valencia M, Morales-González Á, Madrigal-Santillán EO, Morales-Martínez M, Madrigal-Bujaidar E, Álvarez-González I, Gutiérrez-Salinas J, Esquivel-Chirino C, Chamorro-Cevallos G, et al. Oxidative Stress, Mitochondrial Function and Adaptation to Exercise: New Perspectives in Nutrition. Life. 2021; 11(11):1269. https://doi.org/10.3390/life11111269
Chicago/Turabian StyleVargas-Mendoza, Nancy, Marcelo Angeles-Valencia, Ángel Morales-González, Eduardo Osiris Madrigal-Santillán, Mauricio Morales-Martínez, Eduardo Madrigal-Bujaidar, Isela Álvarez-González, José Gutiérrez-Salinas, César Esquivel-Chirino, Germán Chamorro-Cevallos, and et al. 2021. "Oxidative Stress, Mitochondrial Function and Adaptation to Exercise: New Perspectives in Nutrition" Life 11, no. 11: 1269. https://doi.org/10.3390/life11111269