
Molecular Insights into the Genetic Variability of ORF Virus in a Mediterranean Region (Sardinia, Italy)

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
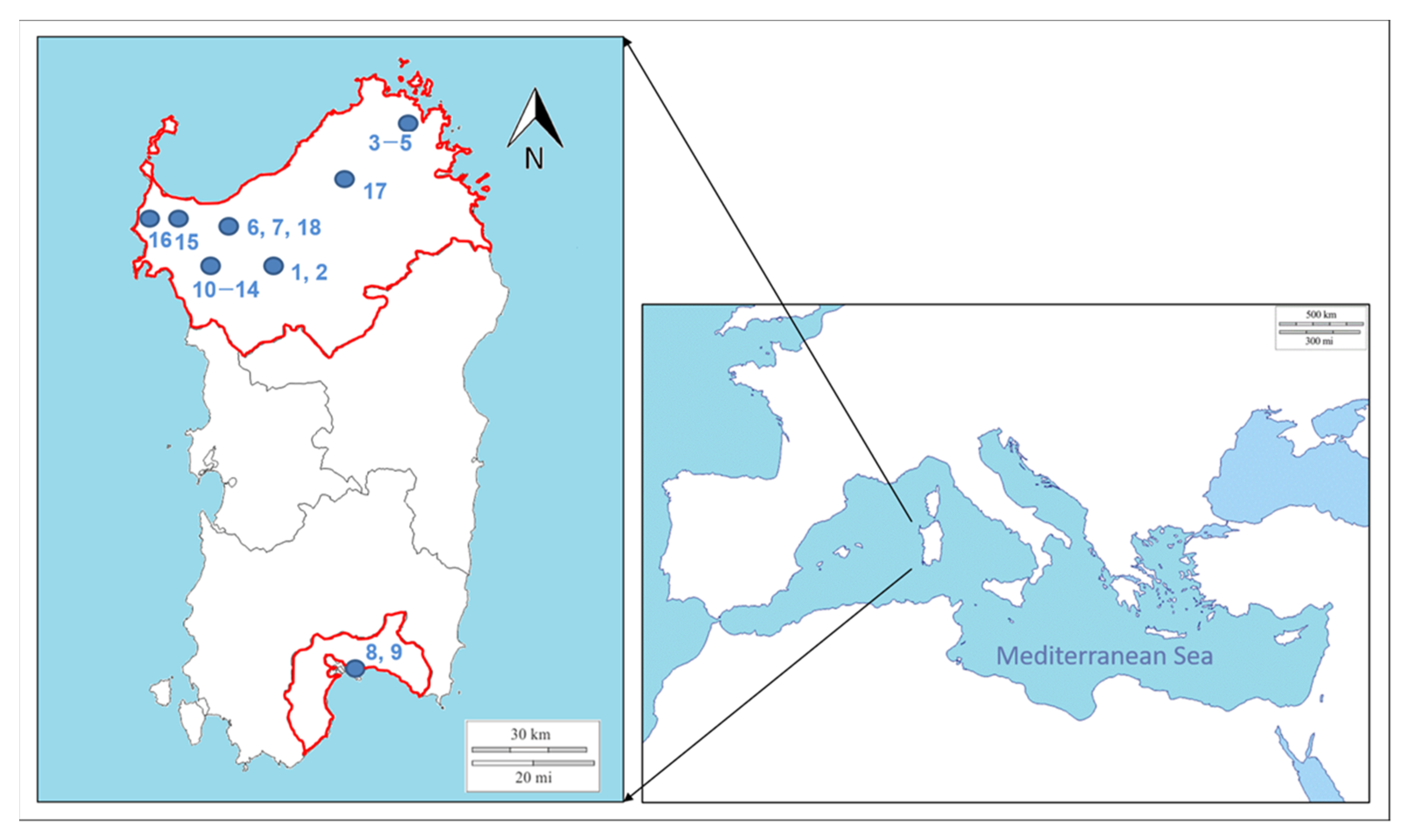
2.1. Sampling
2.2. Histopathology
2.3. Virus Isolation
2.4. ORFV DNA Extraction, PCR and Sequencing
2.5. Phylogenetic and Phylogeographic Analysis
3. Results
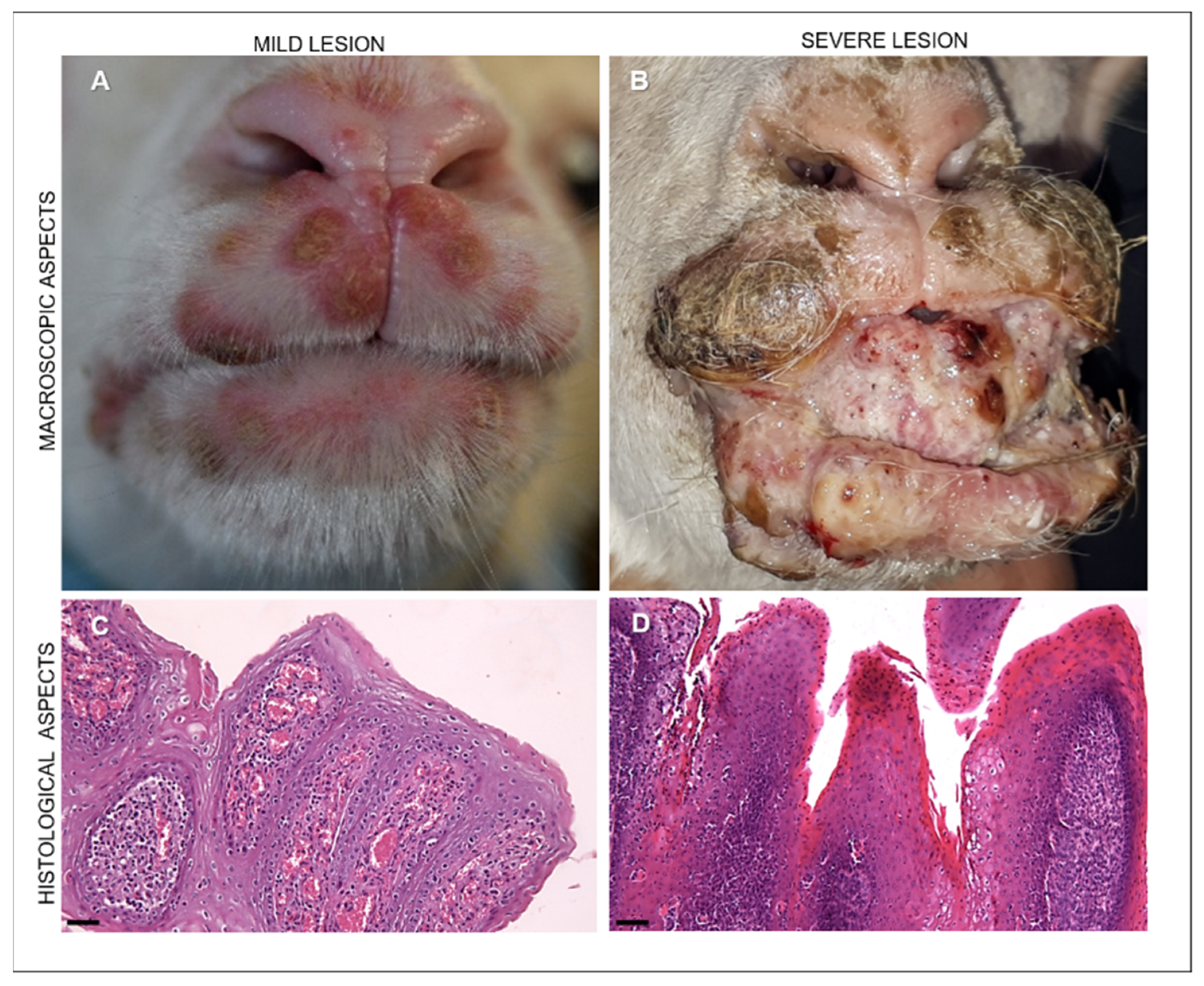
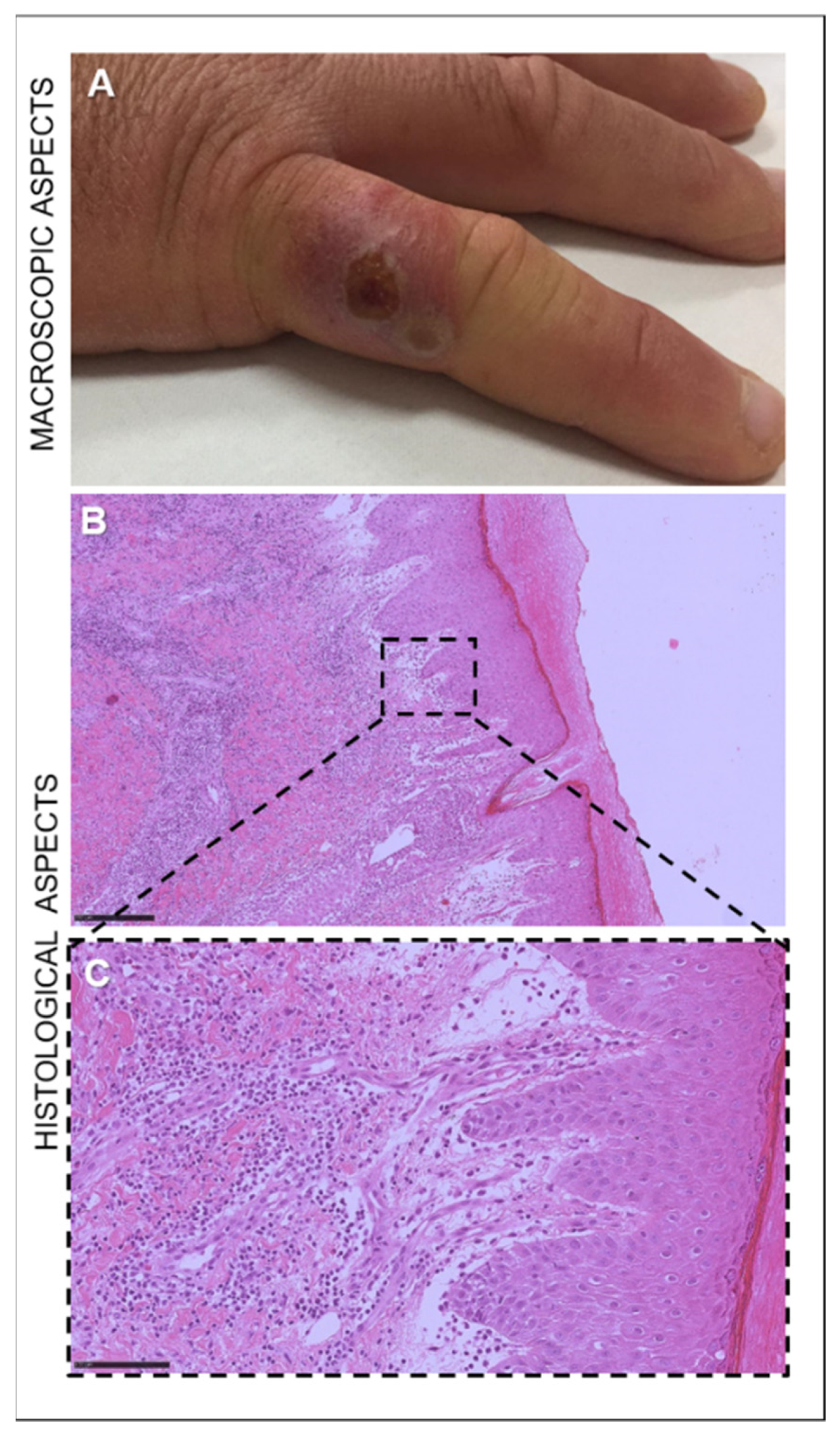
3.1. Clinical Findings
3.2. Histopathology
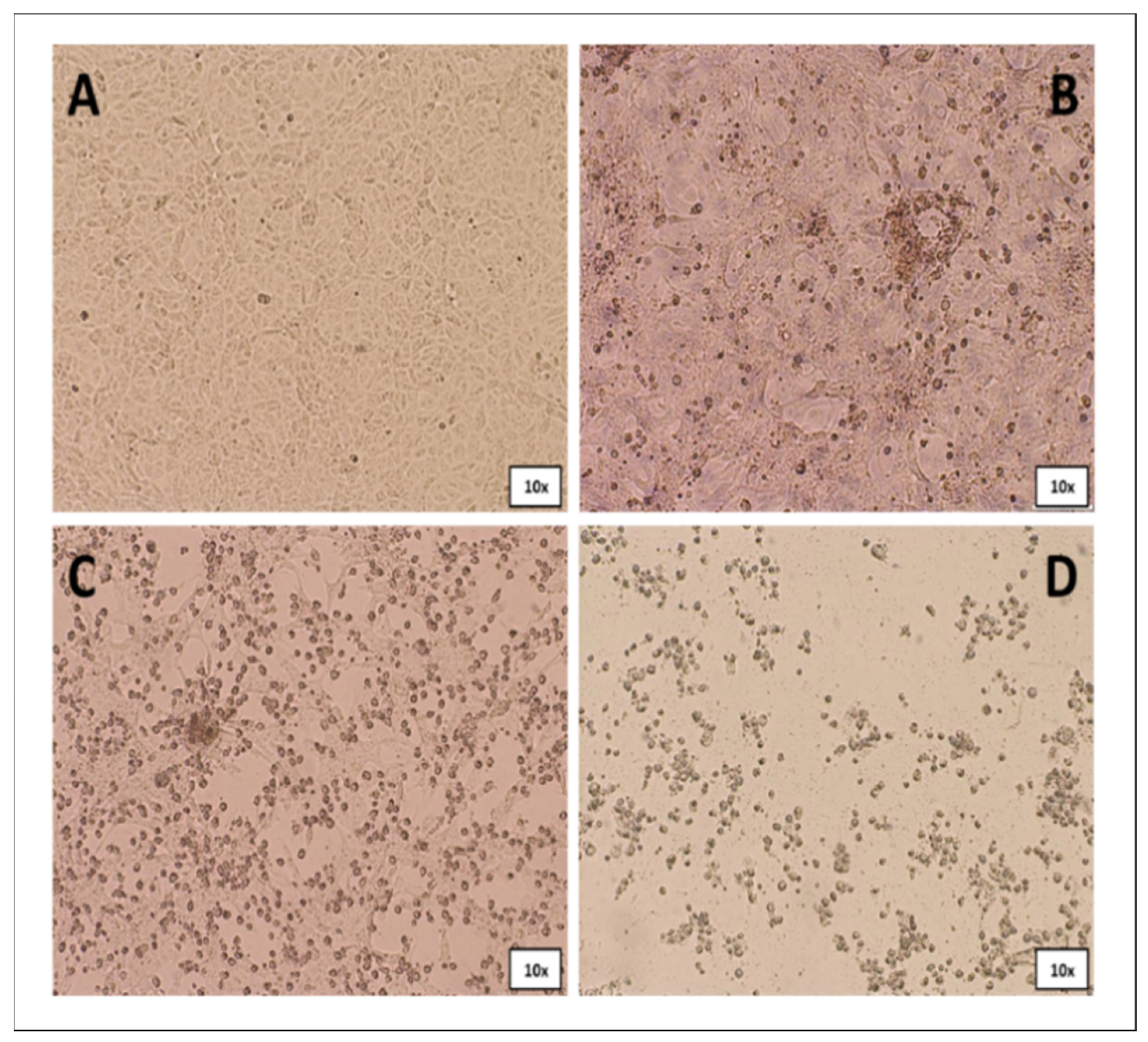
3.3. Virus Isolation
3.4. Molecular Virus Identification
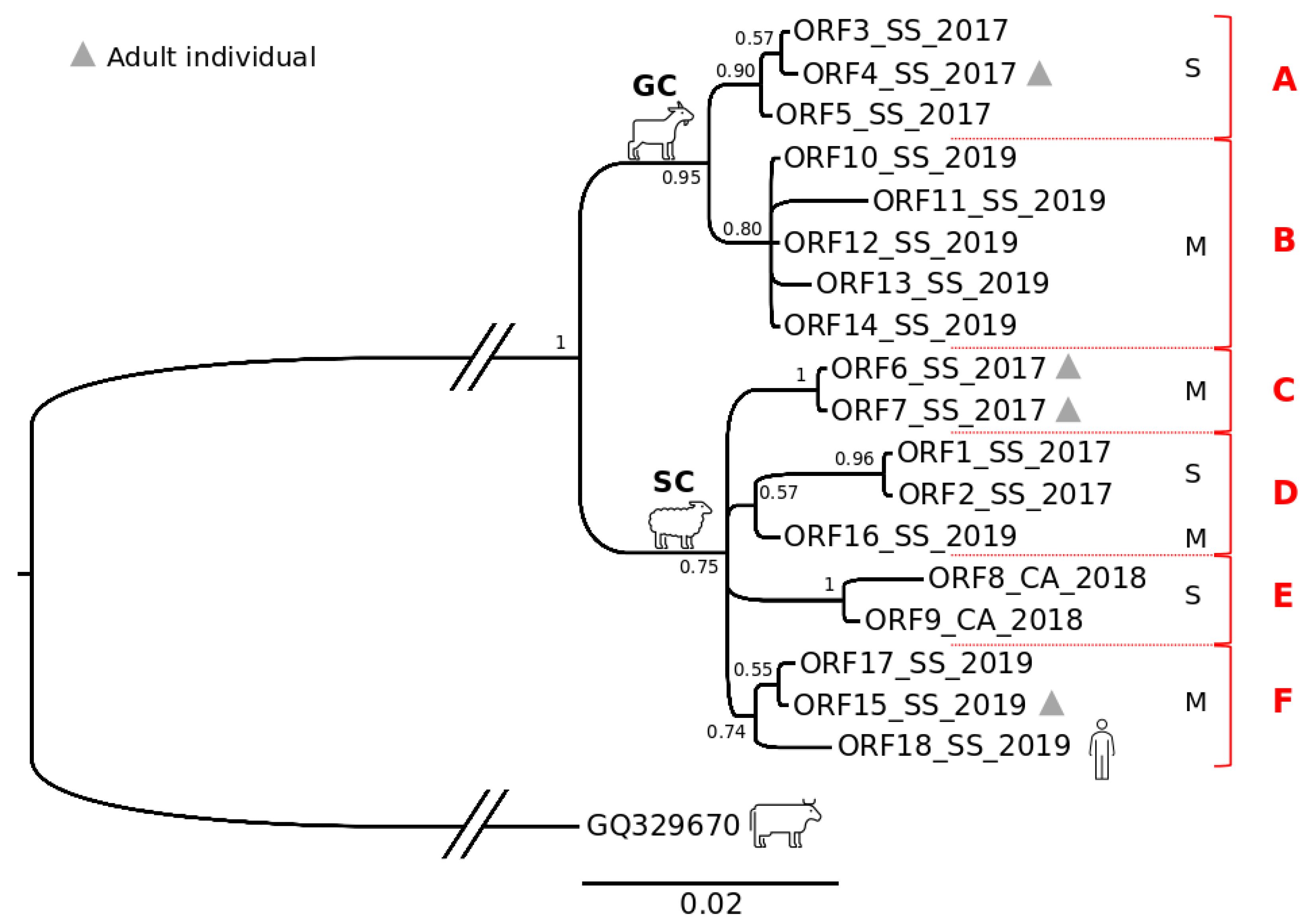
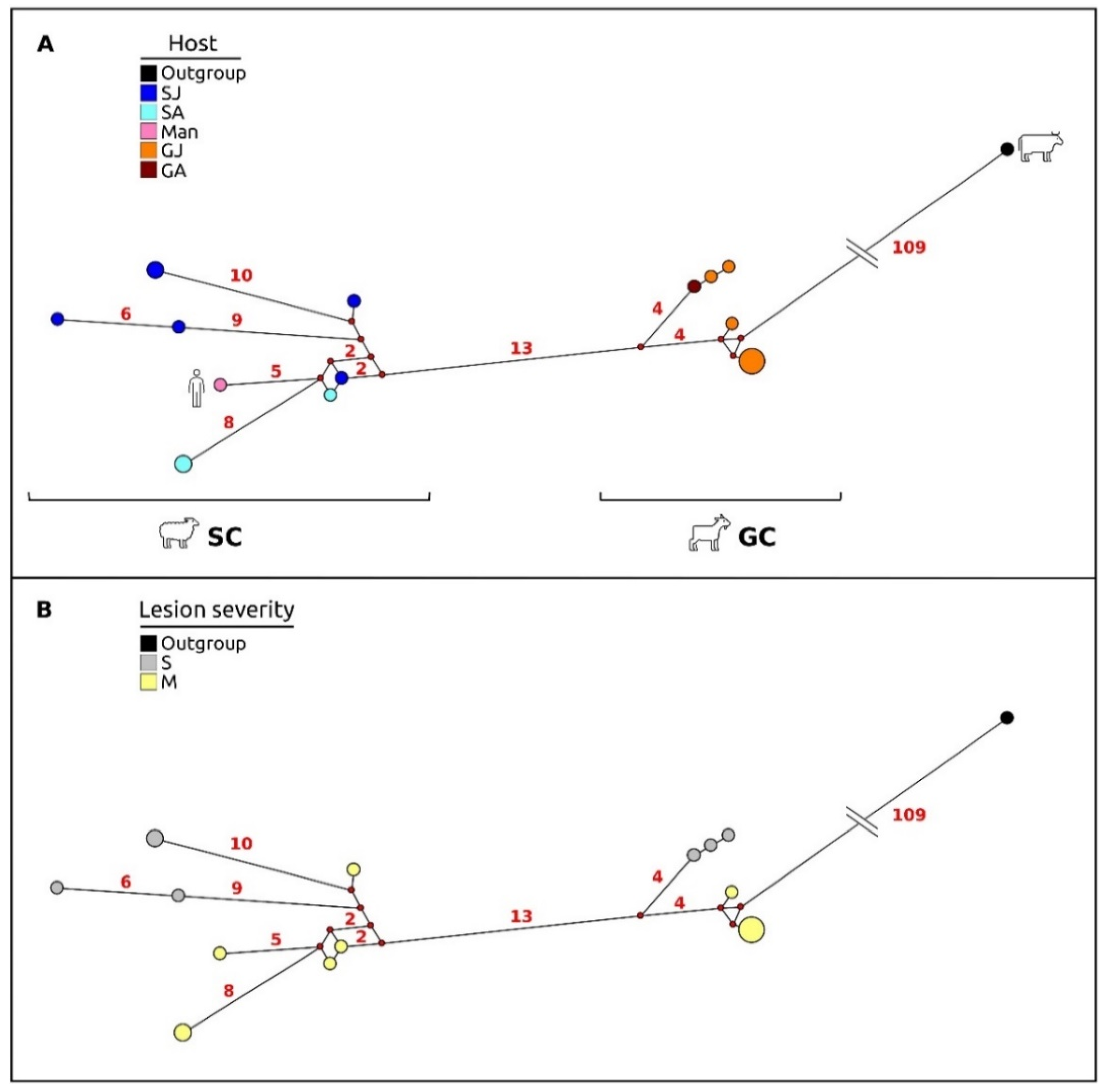
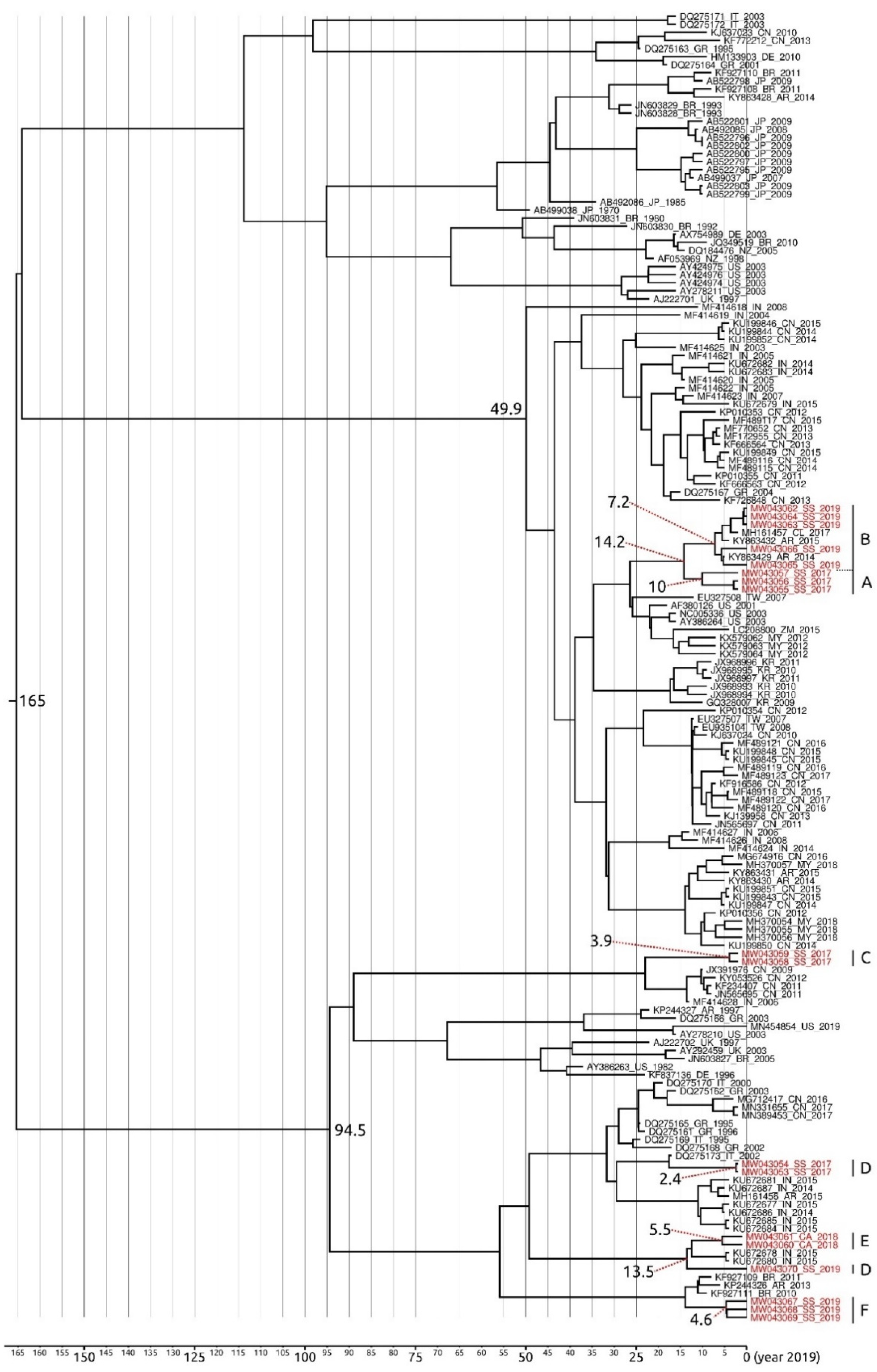
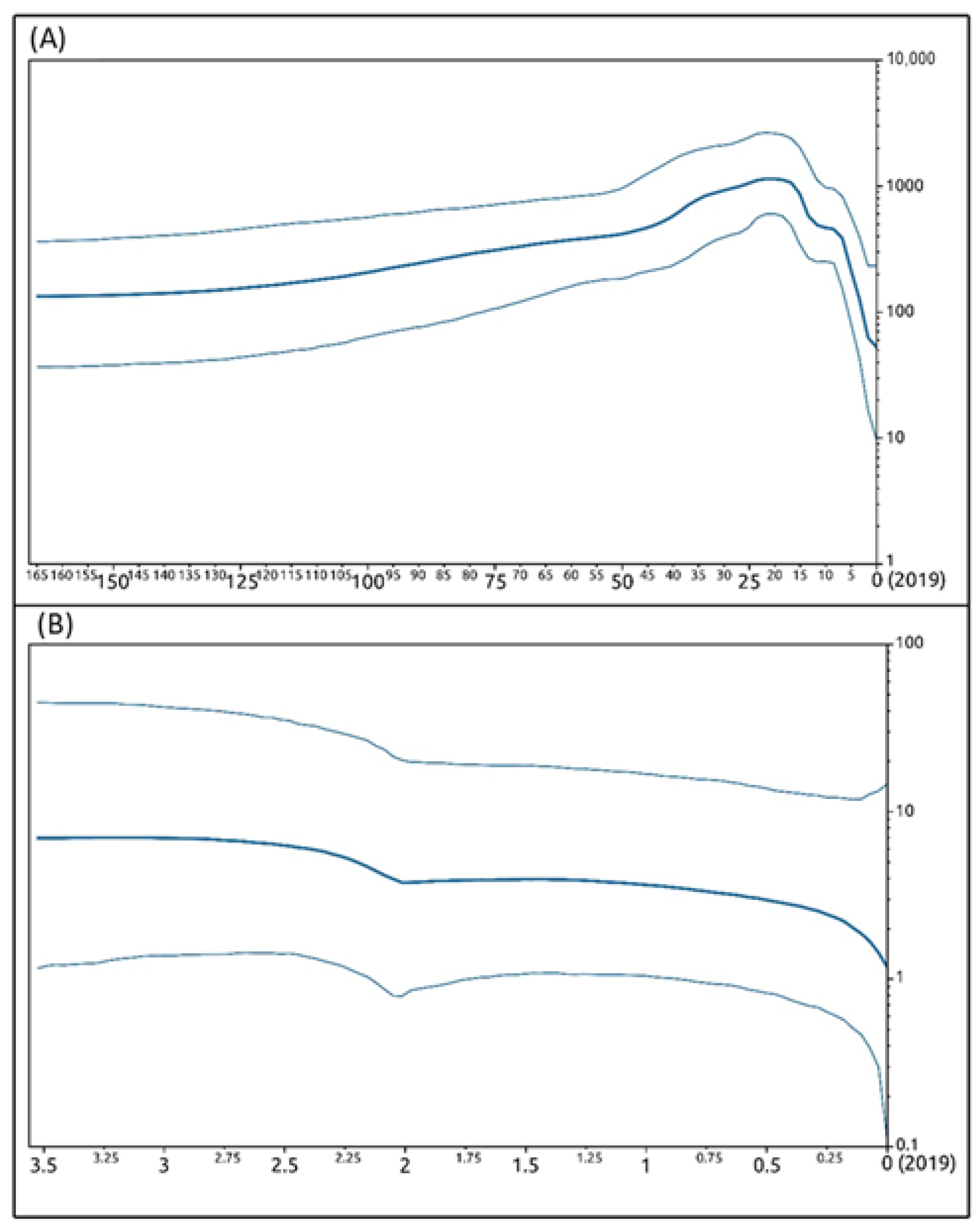
3.5. Phylogenetic and Phylogeographic Inferences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergqvist, C.; Kurban, M.; Abbas, O. Orf virus infection. Rev. Med. Virol. 2017, 27, 1–9. [Google Scholar] [CrossRef]
- Hosamani, M.; Scagliarini, A.; Bhanuprakash, V.; McInnes, C.J.; Singh, R.K. Orf: An update on current research and future perspectives. Expert Rev. Anti Infect. Ther. 2009, 7, 879–893. [Google Scholar] [CrossRef]
- Vaccari, F. Evolutionary Mechanisms of Parapoxviruses: Genomic Characterization of Pseudocowpoxviruses and Development of Systems for the Study of Recombinations. Ph.D. Thesis, University of Bologna, Bologna, Italy, 2009. [Google Scholar]
- Zeller, H. South-West African Goat Pox. Arb. Reichsgesundheitsamte 1920, 52, 501–537. [Google Scholar]
- Kumar, R.; Trivedi, R.N.; Bhatt, P.; Khan, S.H.; Khurana, S.K.; Tiwari, R.; Karthik, K.; Malik, Y.S.; Dhama, K.; Chandra, R. Contagious pustular dermatitis (orf disease)—Epidemiology, diagnosis, control and public health concerns. Adv. Anim. Vet. Sci. 2015, 3, 649–676. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhu, X.; Zheng, Y.; Wang, S.; Liu, Z.; Dou, Y.; Li, H.; Cai, X.; Luo, X. Phylogenetic analysis of two Chinese orf virus isolates based on sequences of B2L and VIR genes. Arch. Virol. 2013, 158, 1477–1485. [Google Scholar] [CrossRef]
- Gumbrell, R.C.; McGregor, D.A. Outbreak of severe fatal orf in lambs. Vet. Rec. 1997, 141, 150–151. [Google Scholar] [CrossRef]
- Mazur, C.; Machado, R.D. The isolation and identification of the contagious ecthyma virus of caprines in cell cultures. Rev. Microbiol. Sao Paulo 1990, 21, 127–130, ISSN: 0001-3714. [Google Scholar]
- Mazur, C.; Machado, R.D. Detection of contagious pustular dermatitis virus of goats in a severe outbreak. Vet. Rec. 1998, 125, 419–420. [Google Scholar] [CrossRef]
- Nandi, S.; De, U.K.; Chowdhury, S. Current status of contagious ecthyma or orf disease in goat and sheep—A global perspective. Small Rumin Res. 2011, 96, 73–82. [Google Scholar] [CrossRef]
- McInnes, C.J. Orf. Vet. Dermatol. 2014, 25, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Bala, J.A.; Balakrishnan, K.N.; Abdullah, A.A.; Mohamed, R.; Haron, A.W.; Jesse, F.F.A.; Noordin, M.M.; Mohd-Azmi, M.L. The re-emerging of orf virus infection: A call for surveillance, vaccination and effective control measures. Microb. Pathog. 2018, 120, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, V.; Valiakos, G. Orf virus infection in sheep or goats. Vet. Microbiol. 2015, 181, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hao, W.; Peng, Y.; Duan, C.; Tong, C.; Song, D.; Gao, F.; Li, M.; Rock, D.L.; Luo, S. Comparative genomic sequence analysis of Chinese orf virus strain NA1/11 with other parapoxviruses. Arch. Virol. 2015, 160, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Khalafalla, A.I.; Elhag, A.E.; Ishag, A.Z.A. Field investigation and phylogenetic characterization of orf virus (ORFV) circulating in small ruminants and Pseudocowpoxvirus (PCPV) in dromedary camels of eastern Sudan. Helyion 2020, 6, e03595. [Google Scholar] [CrossRef] [PubMed]
- Kottaridi, C.; Nomikou, K.; Teodori, L.; Savini, G.; Lelli, R.; Markoulatos, P.; Mangana, O. Phylogenetic correlation of Greek and Italian orf virus isolates based on VIR gene. Vet. Microbiol. 2006, 116, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Scagliarini, A.; Dal Pozzo, F.; Gallina, L.; Guercio, A.; Vaccari, F.; Battilani, M.; Ciulli, S.; Prosperi, S. In vitro activity of VEGF-E produced by orf virus strains isolated from classical and severe persistent contagious ecthyma. Vet. Microbiol. 2006, 114, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Veraldi, S.; Esposito, L.; Pontini, P.; Vaira, F.; Nazzaro, G. Feast of Sacrifice and Orf, Milan, Italy, 2015–2018. Emerg. Infect. Dis. 2019, 25, 1585. [Google Scholar] [CrossRef] [Green Version]
- Gallina, L.; Savini, F.; Casà, G.; Bertoletti, I.; Bianchi, A.; Gibelli, L.R.; Lelli, D.; Lavazza, A.; Scagliarini, A. Epitheliotropic Infections in Wildlife Ruminants From the Central Alps and Stelvio National Park. Front. Vet. Sci. 2020, 7, 229. [Google Scholar] [CrossRef] [PubMed]
- Franzoni, G.; Dei Giudici, S.; Loi, F.; Sanna, D.; Floris, M.; Fiori, M.; Sanna, M.L.; Madrau, P.; Scarpa, F.; Zinellu, S.; et al. African Swine Fever Circulation among Free-Ranging Pigs in Sardinia: Data from the Eradication Program. Vaccines 2020, 8, 549. [Google Scholar] [CrossRef]
- Guo, J.; Rasmussen, A.; Wunschmann, A.; De la Concha-Bermejillo, A. Genetic characterization of orf viruses isolated from various ruminant species of a zoo. Vet. Microbiol. 2004, 99, 81–92. [Google Scholar] [CrossRef]
- De la Concha-Bermejillo, A.; Guo, J.; Zhang, Z.; Waldron, D. Severe persistent orf in young goats. J. Vet. Diagn. Investig. 2003, 15, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoshima, Y.; Morooka, A.; Sentsui, H. Detection and diagnosis of parapoxvirus by the polymerase chain reaction. J. Virol. Methods 2000, 84, 201–208. [Google Scholar] [CrossRef]
- Kottaridi, C.; Nomikou, K.; Lelli, R.; Markoulatos, P.; Mangana, O. Laboratory diagnosis of contagious ecthyma: Comparison of different PCR protocols with virus isolation in cell culture. J. Virol. Methods 2006, 134, 119–124. [Google Scholar] [CrossRef]
- Delhon, G.; Tulman, E.R.; Afonso, C.L.; Lu, Z.; De la Concha-Bermejillo, A.; Lehmkuhl, H.D.; Piccone, M.E.; Kutish, G.F.; Rock, D.L. Genomes of the Parapoxviruses Orf Virus and Bovine Papular Stomatitis Virus. J. Virol. 2004, 78, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Higgins, D.G. Clustal Omega. Curr. Protoc. Bioinform. 2014, 48, 1.25.1–1.25.33. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; UGENE team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.A.; Von Haeseler, A. Phylogenetic inference using maximum likelihood methods. In The Phylogenetic Handbook, 5th ed.; Lemey, P., Salemi, M., Vandamme, A.M., Eds.; Cambridge University Press: Cambridge, UK, 2012; pp. 181–209. [Google Scholar]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; Von Haeseler, A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Teslenko, M.; Van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gelman, A.; Rubin, D.B. Inference from iterative simulation using multiple sequences. Stat. Sci. 1992, 7, 457–472. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Cossu, P.; Lai, T.; Casu, M.; Curini-Galletti, M. How to achieve internal fertilization without a vagina: The study case of the genus Archilina Ax, 1959 (Platyhelminthes, Proseriata) from Canary Islands. Mar. Biodiv. 2019, 49, 2057–2073. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, e214. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Shackelton, L.A.; Parrish, C.R.; Truyen, U.; Holmes, E.C. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc. Natl. Acad. Sci. USA 2005, 102, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.J.; Woo, P.C.; Lau, S.K.; Smith, D.K.; Yuen, K.Y. Accelerated evolutionary rate may be responsible for the emergence of lineage-specific genes in ascomycota. J. Mol. Evol. 2006, 63, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakov, T.; Beaulieu, J.M.; Alverson, A.J. Accelerated diversification is related to life history and locomotion in a hyperdiverse lineage of microbial eukaryotes (Diatoms, Bacillariophyta). New Phytol. 2018, 219, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Relethford, J.H. Genetics of Human Populations; Casa Editrice Ambrosiana: Milano, Italy, 2013; ISBN 9788808184405. [Google Scholar]
- Rodero, A.; Delgado, J.V.; Rodero, E. Primitive Andalusian Livestock and their implications in the discovery of America. Arch. Zootec. 1992, 41, 383–400. [Google Scholar]
- Amills, M.; Ramírez, O.; Tomàs, A.; Badaoui, B.; Marmi, J.; Acosta, J.; Sànchez, A.; Capote, J. Mitochondrial DNA diversity and origins of South and Central American goats. Anim. Genet. 2009, 40, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Paim, T.D.P.; Faria, D.A.; Hay, E.H.; McManus, C.; Lanari, M.R.; Esquivel, L.C.; Cascante, M.I.; Alfaro, E.J.; Mendez, A.; Faco, O.; et al. New world goat populations are a genetically diverse reservoir for future use. Nat. Res. 2019, 9, 1476. [Google Scholar] [CrossRef]
- Peralta, A.; Robles, C.A.; Micheluod, J.F.; Rossanigo, C.E.; Martinez, A.; Carosio, A.; König, G.A. Phylogenetic Analysis of ORF Viruses From Five Contagious Ecthyma Outbreaks in Argentinian Goats. Front. Vet. Sci. 2018, 5, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida da Costa, R.; Felipetto Cargnelutti, J.; Schild, C.O.; Furtado Flores, E.; Riet-Correa, F.; Giannitti, F. Outbreak of contagious ecthyma caused by Orf virus (Parapoxvirusovis) in a vaccinated sheep flock in Uruguay. Braz. J. Microbiol. 2019, 50, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Nadin-Davis, S.A.; Torres, G.; Ribas, M.D.L.A.; Guzman, M.; De La Paz, R.C.; Morales, M.; Wandeler, A.I. A molecular epidemiological study of rabies in Cuba. Epidemiol. Infect. 2006, 134, 1313–1324. [Google Scholar] [CrossRef]
- Bouznach, A.; Hahn, S.; Stram, Y.; Menasherov, S.; Edery, N.; Shicaht, N.; Kenigswald, G.; Perl, S. Case report: Contagious ecthyma—Deviations in the anatomical appearance of lesions in an outbreak in lambs in Israel. Isr. J. Vet. Med. 2013, 68, 246–251. [Google Scholar]
- Demiraslan, H.; Dinc, G.; Doganay, M. An Overwiev of ORF Virus Infection in Humans and Animals. Recent Pat. Anti Infect Drug. Discov. 2017, 12, 21–30. [Google Scholar] [CrossRef]
- Westphal, D.; Ledgerwood, E.C.; Hibma, M.H.; Fleming, S.B.; Whelan, E.M.; Mercer, A.A. A Novel Bcl-2-Like Inhibitor of Apoptosis Is Encoded by the Parapoxvirus Orf Virus. J. Virol. 2007, 81, 7178–7188. [Google Scholar] [CrossRef] [Green Version]
- Harvey, R.; McCaughan, C.; Wise, L.M.; Mercer, A.A.; Fleming, S.B. Orf virus inhibits interferon stimulated gene expression and modulates the JAK/STAT signalling pathway. Virus Res. 2015, 208, 180–188. [Google Scholar] [CrossRef]
- Bennett, J.R.; Lateef, Z.; Fleming, S.B.; Mercer, A.A.; Wise, L.M. Orf virus IL-10 reduces monocyte, dendritic cell and mast cell recruitment to inflamed skin. Virus Res. 2016, 213, 230–237. [Google Scholar] [CrossRef]
- Hong, T.; Yan, C.; Jinyan, W.; Tong, L.; Xiangtao, L. Identification and function analysis of the host cell protein that interacted with Orf virus Bcl-2-like protein ORFV125. Res. Vet. Sci. 2016, 108, 93–97. [Google Scholar] [CrossRef]
- Nagendraprabhu, P.; Khatiwada, S.; Chaulagain, S.; Delhon, G.; Rock, D.L. A parapoxviral virion protein targets the retinoblastoma protein to inhibit NF-κB signaling. PLoS Pathog. 2017, 13, e1006779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, S.B.; Wise, L.M.; Mercer, A.A. Molecular Genetic Analysis of Orf Virus: A Poxvirus That Has Adapted to Skin. Viruses 2015, 7, 1505–1539. [Google Scholar] [CrossRef] [PubMed]
- McKeever, D.J.; Jenkinson, D.M.; Hutchison, G.; Reid, H.W. Studies on the pathogenesis of orf virus infection in sheep. J. Comp. Pathol. 1988, 99, 317–328. [Google Scholar] [CrossRef]
- McInnes, C.J.; Wood, A.R.; Mercer, A.A. Orf Virus Encodes a Homolog of the Vaccinia Virus Interferon-Resistance Gene E3L. Virus Genes 1998, 17, 107–115. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Host | Age | Site and Type of Lesions | Sampling Year | Municipality |
|---|---|---|---|---|---|
| 1 | Sheep | Lamb | Muzzle and gum (proliferative lesions) | 2017 | Putifigari |
| 2 | 2017 | ||||
| 3 | Goat | Kid | Muzzle (proliferative lesion) | 2017 | Arzachena |
| 4 | Lip (proliferative lesion) | 2017 | |||
| 5 | Adult | Udder (fibroproliferative lesion) | 2017 | ||
| 6 | Sheep | Adult | Lip (crusted pustules) | 2017 | Sassari |
| 7 | 2017 | ||||
| 8 | Lamb | Limb | 2018 | Cagliari | |
| Eye | |||||
| Ear | |||||
| Lip | |||||
| Nose | |||||
| Submandibular lymph nodes | |||||
| (proliferative lesions) | |||||
| 9 | Tail | 2018 | |||
| Submandibular lymph node | |||||
| (proliferative lesions) | |||||
| 10 | Goat | Kid | Gum (crusted pustules) | 2019 | Mores |
| 11 | 2019 | ||||
| 12 | 2019 | ||||
| 13 | Lip (crusts) | 2019 | |||
| 14 | Tongue (crusted pustules), lower lip (crust), gum (crusted pustules) | 2019 | |||
| 15 | Sheep | Adult | Crusts | 2019 | Campanedda |
| 16 | Lamb | Muzzle and paraorbital region (crusts) | 2019 | Palmadula | |
| 17 | Lip (crusts) | 2019 | Tempio Pausania | ||
| 18 | Man | Adult | Hand (proliferative lesion) | 2019 | Sassari |
| Gene | Primer | Strand | Sequence | Size (bp) | Ta (°C) | Reference |
|---|---|---|---|---|---|---|
| B2L | PPP1 | Forward | 5′-gtcgtccacgatgcagct-3′ | 570 | 55 | [23] |
| PPP4 | Reverse | 5′-tacgtgggaagcgcctcgct-3′ | ||||
| PPP3 | Forward | 5′-gcgagtccgagaagaatacg-3′ | 235 | |||
| O45 | 045 F | Forward | 5′-cctacttctcggagttcagc-3′ | 400 | 47 | [24] |
| 045R | Reverse | 5′-gcagcacttctcctcgtag-3′ | ||||
| VIR | VIR1 | Forward | 5′-acaatggcctgcgagtg-3′ | 617 | 55 | [21] |
| VIR2 | Reverse | 5′-ttagaactgatgccgcag-3′ | ||||
| VIR 3 | Forward | 5′-tgatcaagtcgcctgca-3′ | 817 | 56 | Present study | |
| VIR 4 | Reverse | 5′-acaaatctcttgagcagct-3′ |
| N | bp | S | H | hd | π | |
|---|---|---|---|---|---|---|
| VIR | 18 | 431 | 45 | 13 | 0.948 ± 0.039 | 0.03797 |
| B2L | 18 | 215 | 6 | 7 | 0.837 ± 0.057 | 0.00945 |
| O45 | 18 | 336 | 6 | 6 | 0.810 ± 0.057 | 0.00550 |
| B2L-VIR-O45 | 18 | 933 | 49 | 13 | 0.948 ± 0.039 | 0.01828 |
| VIR global dataset | 162 | 382 | 76 | 79 | 0.978 ± 0.004 | 0.04005 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coradduzza, E.; Sanna, D.; Rocchigiani, A.M.; Pintus, D.; Scarpa, F.; Scivoli, R.; Bechere, R.; Dettori, M.A.; Montesu, M.A.; Marras, V.; et al. Molecular Insights into the Genetic Variability of ORF Virus in a Mediterranean Region (Sardinia, Italy). Life 2021, 11, 416. https://doi.org/10.3390/life11050416
Coradduzza E, Sanna D, Rocchigiani AM, Pintus D, Scarpa F, Scivoli R, Bechere R, Dettori MA, Montesu MA, Marras V, et al. Molecular Insights into the Genetic Variability of ORF Virus in a Mediterranean Region (Sardinia, Italy). Life. 2021; 11(5):416. https://doi.org/10.3390/life11050416
Chicago/Turabian StyleCoradduzza, Elisabetta, Daria Sanna, Angela M. Rocchigiani, Davide Pintus, Fabio Scarpa, Rosario Scivoli, Roberto Bechere, Maria A. Dettori, Maria A. Montesu, Vincenzo Marras, and et al. 2021. "Molecular Insights into the Genetic Variability of ORF Virus in a Mediterranean Region (Sardinia, Italy)" Life 11, no. 5: 416. https://doi.org/10.3390/life11050416