
The Multifaceted Regulation of Mitochondria in Ferroptosis

1
State Key Laboratory of Agricultural Microbiology, College of Veterinary Medicine, Huazhong Agricultural University, Wuhan 430070, China
2
Institute of Reproductive Health, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China
3
State Key Laboratory of Membrane Biology, Institute of Zoology, Chinese Academy of Sciences, Beijing 100101, China
4
State Key Laboratory of Agricultural Microbiology, College of Animal Science and Technology, Huazhong Agricultural University, Wuhan 430070, China
*
Author to whom correspondence should be addressed.
†
These authors contribute equally.
Life 2021, 11(3), 222; https://doi.org/10.3390/life11030222
Submission received: 2 February 2021
/
Revised: 27 February 2021
/
Accepted: 5 March 2021
/
Published: 10 March 2021
(This article belongs to the Special Issue Cross-Talk between Mitochondria and Proteasomes in Diseases)
Abstract
:Ferroptosis is characterized as a novel form of regulated cell death, which is initiated by the lethal accumulation of lipid peroxidation catalyzed by cellular labile free iron. This iron driven cell death sharply differs from other well characterized forms of regulated cell death at morphological, genetic and biochemical levels. Increasing research has elaborated a high relevance between dysregulated ferroptosis and the pathogenesis of degenerative diseases and organs injury in human patients. Additionally, targeted induction of ferroptosis is considered as a potentially therapeutic design for the clinical intervention of other therapy-resistant cancers. It is well understood that mitochondria, the cellular powerhouse, determine several types of regulated cell death. Recently, compromised mitochondrial morphology and functionalities have been primarily formulated in ferroptosis. Several mitochondria associated proteins and metabolic processes have been elaborated to fine-tune ferroptotic program. Herein, we critically review the recent advances in this booming field, with focus on summarizing the multifaceted mitochondrial regulation of ferroptosis and providing a perspective on the potential biochemical basis. Finally, we are attempting to shed light on an integrative view on the possibility of mitochondria- and ferroptosis-targeting therapeutics as novel treatment designs for the intervention of ferroptosis related diseases.
Regulated cell death (RCD, or programmed cell death, PCD) is a fundamental process essential for maintaining cell and tissue homeostasis in metazoans. RCD is tightly organized by signaling cascades and successively executed by a series of unique molecules. Based on the morphological, biochemical, and genetic characteristics, RCD is classified into distinct types, including apoptosis, necroptosis, pyroptosis, ferroptosis and other forms of RCD, according to the formulated guidelines of the Nomenclature Committee on Cell Death (NCCD) [1,2]. Among them, ferroptosis is a newly characterized form of non-apoptotic cell death driven by iron-dependent lipid peroxidation, due to the pharmacological or pathological perturbation of lipophilic antioxidant systems, especially the GSH (glutathione)-GPX4 (glutathione peroxidase 4) axis. During this decade, it has been gradually recognized that dysregulated ferroptosis is potentially implicated in the cell degeneration and tissue damage during the pathogenesis of several human diseases, especially the neurodegenerative diseases. In addition, targeted induction of ferroptosis provides promising antitumor therapies by triggering lethal cytotoxicity of cancer cells, which are potently resistant to other therapies. Therefore, ferroptosis has attracted increasing attention. In the current review, we will summarize the recent advances in this field and provide a critical perspective on the biochemical and genetic basis, with focus on formulating the multifaceted regulation of ferroptosis by mitochondria. Finally, we will also deliberate the possibility of ferroptosis- and mitochondria-targeting therapeutics for the clinical intervention of ferroptosis- or iron overload-associated human diseases.
1. A Brief Introduction of Ferroptosis
1.1. Pioneering Studies Related to Ferroptosis
Although ferroptosis was characterized and the term was coined by Dr. Brent R. Stockwell and his colleagues in 2012 [3], it is worth noting that earlier studies enlightened the pioneering understanding. In 1955, extracellular cystine was identified as the essential nutrient for cultured carcinoma cell HeLa and fibroblasts in vitro [4]. Cystine deprivation triggers cell death with a unique microscopic change [5]. This cell death is manifested by GSH exhaustion [6], and could be mitigated by lipophilic antioxidant α-tocopherol [6] or iron chelator DFO (deferoxamine) [7]. Additionally, extracellular glutamate overload induces cytotoxicity of neuronal cells by restraining cystine transport, leading to GSH exhaustion and accumulation of intracellular peroxides [7]. Although this glutamate excitotoxicity was originally referred to oxytosis [8], and alternative mechanisms were reported to be involved in [9,10], it is still unknown whether oxytosis and ferroptosis are similar or even exactly identical form of RCD [11]. These earlier studies thus established the preliminary understanding of cysteine/cystine deprivation, glutamate overload and GSH exhaustion in the process of iron and oxidation dependent cell death, which are now considered as the fundamental features of ferroptosis.
1.2. The Discovery of Ferroptosis
In 2003, by using a synthetic lethal compound high-throughput screening, Dr. Stockwell and the colleagues identified a novel antitumor compound named erastin, which could trigger lethal cytotoxicity of the engineered tumorigenic cells expressing mutant Ras oncogene, but not their isogenic normal cell counterparts [12]. A later synthetic lethal screening assay in 2008 identified two oncogenic-Ras-selective lethal compounds, RSL3 and RSL5, which trigger apoptosis independent cell death in human foreskin fibroblasts (BeJLR cells) [13]. Erastin and RSL induced cell death differs sharply from other well characterized forms of RCD. Morphologically, the ballooning phenotype, mainly characterized by the formation of a clear rounded cell consisting of empty cytosol, can be microscopically observed during ferroptosis, but not other types of cell death [14]. Biochemically, neither cytochrome c release nor PARP1/Caspase 3 cleavage takes place upon erastin or RSL3 challenge [15]. Additionally, the pro-apoptotic proteins Bax and Bak, as well as the pro-necroptotic proteins RIP1 and RIP3, are dispensable for erastin or RSL3 induced cell death [16]. Chemical inhibition of apoptosis or necroptosis by the corresponding inhibitor fails to mitigate this cell death. In striking contrast, the iron chelators and ROS (reactive oxygen species) scavengers could almost completely block this lethal cytotoxicity, suggesting an iron- and oxidation-dependency of this non-apoptotic cell death.
1.3. The Core Regulatory Circuit of Ferroptosis
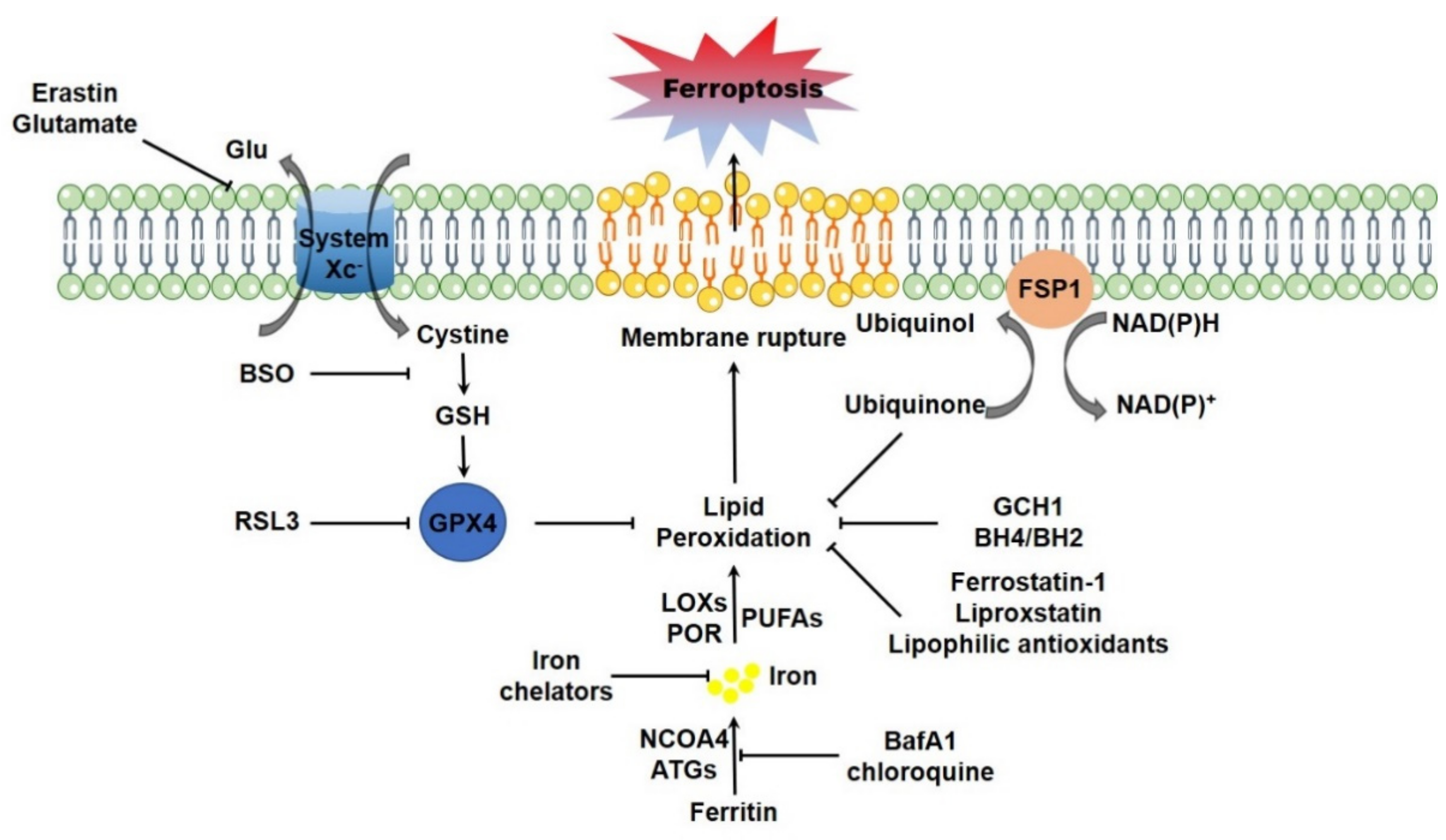
Erastin binds to the cystine/glutamate antiporter (system XC-, a member of the heteromeric amino acid transporter family, typically exchanges the extracellular cystine and intracellular glutamate, composed of a 12-pass transmembrane component SLC7A11 and a 1-pass transmembrane regulatory component SLC3A2 [17]), interferes the cystine uptake and thus disrupts the biosynthesis of GSH, the γ-L-glutamyl-L-cysteinyl-glycine tripeptide as a key determinant of cellular redox homeostasis [3]. RSL3 fails to impinge on the cystine uptake and GSH biosynthesis, but alternatively targets to GPX4 directly and restrains its enzymatic activity [18]. GPX4 is a member of GPXs family, which catalyzes the reduction of hydrogen peroxide and organic hydroperoxides by using GSH as the essential cofactor. It was reported that GPX4 is the unique antioxidant enzyme for the ability to directly reduce phospholipid hydroperoxides and oxidized lipoproteins to their respective lipid-alcohol within cellular bio-membranes [19]. Therefore, targeted deletion of GPX4 elevates lipid peroxidation and thus triggers ferroptotic cell death in cultured mammalian cells [20]. Cystine deprivation or chemical inhibition of GSH biosynthesis leading to GPX4 inactivation also initiates ferroptotic cell death [18,21,22,23]. Moreover, genetic ablation of GPX4 leads to early embryonic lethality [24,25], while inducible deletion of GPX4 results in acute renal failure and early death in mice [20]. By using T lymphocytes specific GPX4 knockout mice as a model, it was reported that GPX4 is vital for the homeostatic survival of CD8+ T cells in the periphery (spleen, peripheral lymph node and mesenteric lymph node), as well as for the expansion of both CD4+ and CD8+ T cells upon T cell receptor triggering in response to infection through preventing lipid peroxidation and counteracting ferroptosis [26]. Additionally, inducible depletion of GPX4 specifically in neuron causes rapid paralysis, muscle atrophy and early death. A ferroptotic degeneration of spinal cord motor neurons is observed in these mutant animals [27,28]. Other studies using tissue specific GPX4 knockout mice as models therewith demonstrated the critical importance of GPX4-GSH system in maintenance of hepatocellular homeostasis and normal liver function [29], survival of endothelial cells [30], as well as the maturation of photoreceptor cells [31]. Collaboratively, these studies thus highlight the core regulatory circuit composed of system XC- mediated cystine uptake-GSH biosynthesis-GPX4-lipid peroxidation in navigating ferroptotic program. More recently, two independent groups identified a novel regulatory circuit parallel to GPX4-GSH system in ferroptosis regulation. Flavoprotein AIFM2 (apoptosis-inducing factor mitochondria-associated 2), which was initially identified as a pro-apoptotic and P53-responsive protein associated with mitochondrial outer membrane [32,33], was recently characterized as a novel ferroptosis modulator. Myristoylation of AIFM2 leads to a plasma membrane translocation, where AIFM2 serves as a NADPH (β-nicotinamide adenine dinucleotide 2′-phosphate, reduced) dependent oxidoreductase to mediate the reduction of ubiquinone (also known as coenzyme Q10, CoQ10), trap lipid peroxyl radicals and thus suppress lipid peroxidation. AIFM2 is thus renamed FSP1 (ferroptosis suppressor protein 1) [34,35]. Unlike the embryonic lethality of GPX4 knockout in mice, FSP1 knockout mice are produced normally and are without any overt abnormal phenotypes up to 1 year old [36]. This is possibly due to the compensatory detoxification of lipid peroxidation by GPX4. Additionally, it was just recently reported GCH1 (GTP cyclohydrolase-1), the rate-limiting enzyme catalyzing the biosynthesis of BH4 (tetrahydrobiopterin) and BH2 (dihydrobiopterin), as another highly potent endogenous inhibitory protein for ferroptosis. Specifically, GCH1 expression leads to the enhanced synthesis of BH4 and BH2, which facilitate lipid remodeling by selectively preventing the depletion of phospholipids with two polyunsaturated fatty acyl tails, and thus antagonize ferroptosis, without any overt impact on other ferroptosis regulators [37] (Figure 1).
2. Lipid Peroxidation Driving Ferroptosis
Accumulation of lipid peroxides directly executes ferroptotic cell death. Although erastin and RSL3 do elevate the cytosolic superoxide, but not limited to the lipid peroxides, it seems that the cytosolic superoxide dose not contribute to ferroptosis as the compounds elevating cytosolic superoxide fail to trigger ferroptosis [38].
Lipidomic studies revealed that PUFAs (polyunsaturated fatty acids)-containing PLs (phospholipids), especially PEs (phosphatidylethanolamines), are the most susceptible lipids to peroxidation at the bis-allylic position during ferroptosis [39,40]. Oxygenated PEs are accumulated in GPX4 inactivated cells and in GPX4 ablated kidney tissue. It is worth noting that free PUFAs, such as AA (arachidonic acid) and AdA (adrenaline), must be esterified into PLs for the peroxidation. ACSL4 (Acyl-CoA synthetase long-chain family member 4) and LPCAT3 (lysophosphatidylcholine acyltransferase 3) have been identified for this PUFAs incorporation and lipid remodeling during ferroptosis [41,42] (Figure 2). Genetic or pharmacological inhibition of ACSL4 leads to a substantial ferroptotic insensitivity [40,41]. Unlike to the PUFAs, exogenous MUFAs (monounsaturated fatty acids) potently restrain ferroptotic cell death by decreasing oxidizable PUFAs-containing PLs and thus remodeling membrane lipidome [43].
2.1. Lipid Peroxidation Enzymatically Catalyzed by LOXs and POR
Oxidation of PUFA-PLs could be enzymatically catalyzed by LOXs (lipoxygenases), COXs (cyclooxygenases), and CYPs (cytochrome P450), as well as non-enzymatically catalyzed by free iron via Fenton reaction [44] (Figure 2). LOXs, the non-heme iron-containing dioxygenases encoded by the ALOXs genes, are identified as the most critical molecules for lipid peroxidation. Transfection of the siRNA pools targeting all ALOXs genes or general pharmacological inhibition of LOXs strikingly prevents erastin induced lipid peroxidation and ferroptosis [39]. Among them, it is still elusive whether any unique LOX functions dominantly or they catalyze lipid peroxidation redundantly, due to the different expression patterns in different tissues and cells. The siRNA pools targeting ALOX15B and ALOXE3 could efficiently prevent the lethal cytotoxicity of erastin in BJeLR and HT-1080 cells [39]. Another study showed that 12/15-LOX is indispensable for ferroptosis in GPX4 knockout or inactivated cells [28]. PEBP1 (PE-binding protein 1), which was previously identified as the RKIP1 (Raf1 kinase inhibitory protein) to interact and inhibit the Raf1 kinase cascade, is a master regulatory molecule for the LOXs, especially 15-LOX. PEBP1 modulates the substrate specificity of 15-LOX to PUFA-PEs, leading to generation of HpETE-PEs [45]. However, only pharmacological inhibition of LOXs by the 12/15-LOX inhibitor has been evidenced to efficiently protect GPX4 deficient cells from death [28], while specific ablation of ALOX15 gene does not rescue homozygous knock-in mice expressing catalytically inactivated GPX4 mutant Sec46Ala from embryonic lethality [46]. It was reported that the ferroptosis inhibitor liproxstatin-1 could suppress the production of pro-ferroptotic oxygenated PEs by inhibiting the enzymatic activity of 15-LOX [40]. However, other study reported that liproxstatin-1 and ferrostatin-1 perform poor inhibition directly on 15-LOX, but alternatively present as radical-trapping antioxidants to suppress lipid peroxidation [47]. These evidences thus indicate that it is reasonable additional molecules may exist to catalyze the lipid peroxidation bypass LOXs [48].
POR-CYP system is typically composed of a POR (cytochrome P450 oxidoreductase) and a CYP (cytochrome P450) isoenzyme that contains a protoporphyrin IX heme-iron prosthetic group [49]. It is known that POR-CYP plays key roles in maintaining cellular redox homeostasis and detoxification of xenobiotic chemicals. Just recently, by using genome-wide CRISPR-Cas9 screening, Zou and colleagues reported POR as a critical modulator of ferroptotic cell death. Redox lipidomics assay showed that POR depletion significantly reduces both hydroperoxyl- and hydroxyl-PUFA-PEs, suggesting that POR facilitates lipid peroxidation [50] (Figure 2). CYP accepts electrons from POR, and simultaneously catalyzes the peroxidation of PUFA lipids, while POR facilitates lipid peroxidation by accelerating the switch between Fe2+ and Fe3+ in the heme prosthetic group of CYP [51,52]. However, the CRISPR-Cas9 screening was not able to nominate any specific CYP as the major POR partner in catalyzing lipid peroxidation and facilitating ferroptosis [50]. Just recently, Yan and colleagues reported that POR-CYP interactions are dispensable to POR-mediated ferroptosis [53].
In addition, there is no direct evidence presenting a causal link between COXs mediated lipid peroxidation and ferroptosis execution. COX-2, encoded by Ptgs2 gene, is transcriptionally upregulated during ferroptosis, which is now considered as one of the major hallmarks of ferroptosis. However, chemical inhibition of COX-2 fails to affect ferroptotic cell death [18].
2.2. Lipid Peroxidation Non-Enzymatically Catalyzed by Iron
The non-enzymatic lipid peroxidation refers to Fenton chemistry, which is accomplished by the cellular labile free ferrous iron. The cellular iron is always ligated by heme or ISC (iron-sulfur cluster) in the heme- or ISC-containing proteins, respectively, or tightly chelated by ferritin [54]. There is a small portion of cellular ferrous iron (labile iron pool) that is loosely ligated and could catalyze lipid peroxidation via Fenton chemistry [55,56]. In cells, the labile iron pool is highly dynamic. The iron-containing ferritin could be selectively degraded via proteasome dependent manner [57] or autophagy dependent manner (which is termed ferritinophagy) [58,59]. Free iron is thus released for the cellular utilization. It was reported that ferritinophagy is initiated and free iron is thus elevated during ferroptosis. Knockdown of core autophagy components including ATG3, ATG13, ATG5 and ATG7, or the specific ferritinophagy receptor NCOA4, or pharmacologically inhibition of autophagy by the lysosomal inhibitor BafA1 or chloroquine, significantly mitigates ferroptotic cell death [60,61] (Figure 1). Likewise, activation of HO-1 (heme oxygenase-1), which enhances heme catabolism and iron release, is expectable to accelerate ferroptotic program. The specific HO-1 inhibitor and genetic ablation of HO-1 could markedly combat ferroptosis [62,63,64]. However, the other study raised an opposite argument that HO-1 protects cells from ferroptosis [65]. The redox active ferrous iron mediates the initiation step for the generation of reactive hydroxyl radical in Fenton reaction [66]. Additionally, the switch between Fe2+ and Fe3+ redox states also supports the catalytic reaction of LOXs and CYPs for the corresponding lipid peroxidation [67,68].
2.3. Lipid Peroxidation Driving Ferroptotic Cell Death
It was well understood that certain type of lipid peroxidation could drive certain form of RCD, including apoptosis [69] and pyroptosis [70,71]. However, it may be more complicated for lipid peroxidation to drive ferroptotic cell death. The lipid peroxidation may kill cells directly by damaging the bio-membranes or indirectly through the metabolic derivatives [72]. On one hand, lipid peroxidation perturbs the integrity of the bio-membrane bilayer structure. More specifically, the oxidized phospholipids confined in bio-membrane could change the bio-membrane fluidity, bio-membrane permeability, as well as bio-membrane associated signaling and metabolic processes leading to cell death [73]. The lipid peroxidation alters the local shape and curvature of lipid membranes, promotes accessibility to oxidants, which expedites membrane destruction and triggers ultimate cell death [74]. Additionally, phospholipids peroxidation would generate a variety of oxidized phospholipids containing a shortened fatty acyl chain by the rearrangement of alkoxyl radicals. These short-chain phospholipids perform a detergent-like activity [75] and secondarily damage bio-membrane [76,77]. On the other hand, lipid peroxidation-derived aldehydes including 4-HNE (4-hydroxy-2-nonenal), 4-HHE (4-hydroxy-2-hexenal), and MDA (malondialdehyde), could diffuse and react with proteins, DNA, and phospholipid molecules to generate a variety of intra-molecular and inter-molecular covalent adducts [44,78]. Specifically, MDA could covalently bind multiple proteins including enzymatic proteins, carrier proteins, cytoskeletal proteins, mitochondrial proteins as well as antioxidant proteins [79]. A recent study by using a quantitative chemoproteomic method to profile protein carbonylation showed that more than 400 proteins are carbonylated during ferroptosis, which is potently driven by these reactive aldehydes. Among them, several ferroptosis associated molecules have been identified [80]. In addition, these aldehydes could act as signaling molecules to drive cell death [81]. Nevertheless, the exact mechanism for which lipid peroxidation drives ferroptotic cell death is to be investigated [72] (Figure 2).
3. Disrupted Mitochondrial Integrity during Ferroptosis
The cellular powerhouse mitochondria control a set of critical biological processes, including energy production via electron transport coupled with oxidative phosphorylation, fatty acid β-oxidation, TCA (tricarboxylic acid) cycle, iron metabolism, as well as calcium homeostasis. It is well understood that mitochondria also function as a signaling hub to orchestrate intracellular or extracellular clues and communicate to other cellular compartments [82,83]. In addition to such diverse functions, mitochondria also determine several types of cell death, including apoptosis [84,85], pyroptosis [86], and necroptosis [87].
Mitochondria are highly dynamic organelle with constant fusion, fission and turnover. The mitochondrial quality and quantity are systematically coordinated by the mitochondrial biogenesis and mitochondrial clearance [88]. The mitochondrial architecture is mainly determined by the dynamin-related GTPases MFN1/MFN2 (which mediate fusion of mitochondrial outer membrane) and OPA1 (which mediates fusion of mitochondrial inner membrane) driven fusion, as well as by the cytosolic protein DRP1 and its mitochondrially localized receptors driven fission [89,90]. It has been well understood that mitochondrial dynamics are substantial for compensating mitochondrial damage through fusion and eliminating mitochondria with unrecoverable damage through fission. Increasing number of studies have elaborated a tight association between disturbed mitochondrial dynamics and the occurrence or development of diverse human diseases [91,92]. Mitochondrial biogenesis is composed of multiple steps including replication, transcription and translation of mtDNA, transcription and translation of nuclear-encoded proteins, biosynthesis and transport of mitochondria associated lipid, which are mainly orchestrated by several transcriptional factors, including PGC-1α, NFR1, and TFAM [93,94]. Mitochondrial clearance is mainly achieved by mitophagy, which refers to a highly selective recognition and removal of mitochondria by the autophagosome-lysosome machinery [95,96]. To data, PINK1-Parkin dependent mitophagy and receptors mediated mitophagy have been dissected in mammalian cells [97].
3.1. The Engagement of Mitochondria in Iron Overload Associated Cell Damage
Trace elemental iron is one of the essential nutrients for cell growth. However, iron overload, one of the most substantial causes and typical hallmarks of ferroptosis, facilitates mitochondrial damage at distinct levels [98,99]. Iron overload leads to the abnormal mitochondrial morphology, resulting in a fragmented architecture in several types of cultured mammalian cells [100,101]. Excess iron supplementation also compromises mitochondrial functionalities by restricting mitochondrial oxidative phosphorylation and antioxidant response [102]. The mtDNA is vulnerable to iron deposition. It was reported that double strand breaks of mtDNA are observed in iron-exposed mitochondria. Iron overload also leads to progressive loss of intact mtDNA, as well as reduced mtDNA transcription and decreased expression of respiratory chain subunits encoded by mitochondrial genome [103,104,105,106]. Conversely, preserving mitochondrial architecture and functionalities by utilizing distinct approaches was evidenced to protect cells from iron toxicity in multiple models. Specifically, inhibition of DRP1 mediated mitochondrial fragmentation prevents ferric ammonium citrate-induced neuronal cell loss [107]. Injection of the mitochondrially targeting antioxidant mito-TEMPOL [108] could alleviate hepatotoxicity and lower hepatocytic death induced by excess dietary iron [109]. These studies thus enlightened a causal role of mitochondrial dysfunction in iron overload mediated cell damage, which is now considered to highly relate to ferroptosis.
3.2. Disrupted Mitochondrial Morphology in Ferroptosis
As previous study demonstrated a potential binding to the multifunctional mitochondrial proteins VDAC2/3 (voltage-dependent anion channel 2/3) of erastin [15], a plausible involvement of mitochondria in ferroptosis regulation was hypothesized. Upon exposure of erastin, shrunken mitochondria appear smaller with increased membrane density in BJeLR cells as imaged by the transmission electronic microscope [3]. A similar phenotype was observed in HT-22, a mouse hippocampal neuronal cell Line. Erastin induces mitochondrial fragmentation and accumulation around the nucleus as shown by the MitoTracker DeepRed staining [110]. Similarly, exposure to BSO (buthionine sulfoximine), the chemical inhibitor of γ-GCS (γ-glutamylcysteine synthetase) essential for GSH biosynthesis, leads to lamellar and tubular structured mitochondria with diminished cristae [20]. GPX4 ablated cells contain swollen mitochondria with a reduced number of cristae and a lamellar architecture. Furthermore, exposure of RSL3 leads to a rupture of mitochondrial outer membrane in Pfa1 cells in a time-dependent manner [20]. Currently, abnormal mitochondrial architecture including mitochondrial fragmentation, shrunken mitochondria and rupture of mitochondrial outer membrane, as well as vanished mitochondrial cristae, is regarded as the typical morphological characteristic of ferroptotic cell death [111].
3.3. Mitochondrial ROS Burst in Ferroptosis
No increase in mitochondrial ROS was observed in erastin treated HT-1080 cells as evidenced by mitoSOX (the specific mitochondrial ROS probe) staining, although erastin significantly elevates both cytosolic and lipid ROS [3]. However, Carsten Culmsee and colleagues demonstrated a substantial increase of mitochondrial ROS in erastin challenged HT-22 and MEF cells [110]. This group also reported that exposure of RSL3 similarly triggers a burst of mitochondrial ROS in MEF and HT-22 cells [112]. This discrepancy may be due to the different cells used or different exposure time. Mitochondria-targeted ROS scavenger MitoQ (mitoquinone) can protect neuronal cells from RSL3 induced ferroptosis [112]. Compared to a more broadly distributed antioxidant, the specifically mitochondria-targeted nitroxide performs a more potent effect on protecting cells from ferroptotic cell death [113]. These studies thus collaboratively indicate the possibility of an engagement of mitochondrial ROS in navigating ferroptotic program.
3.4. Altered Mitochondrial Membrane Potential in Ferroptosis
The mitochondrial membrane potential (ΔΨm) generated by proton pumps is essential for ATP production by coupling with oxidative phosphorylation [114]. It was reported by Dr. Xuejun Jiang’s lab that diverse ferroptosis inducers, including cystine starvation, amino acid-free medium plus full serum (induces ferroptosis that was reported in [115]), erastin and glutamate can all induce mitochondrial hyper-polarization by using TMRE (tetramethylrhodamine ethyl ester) staining, while the mitochondrial uncoupler CCCP could disrupt mitochondrial membrane potential and almost completely block cystine starvation induced lipid ROS accumulation and ferroptosis [116]. Furthermore, previous study demonstrated a potential binding to VDAC2 and VDAC3 of erastin [15], while RSL3 exposure leads to the carbonylation of VDAC2 [80]. It is reasonable that the direct binding to VDACs of erastin or the carbonylation of VDAC2 by RSL3 may result in the conformational change of VDACs, and thus lead to mitochondrial hyper-polarization [117,118], as VDACs serve as a gatekeeper to facilitate transmembrane exchange of ions and metabolites [117,119,120]. This is confirmed by a NADH oxidation assay, and erastin treatment yields a decrease in the rate of NADH oxidation, suggesting a decreased permeability of mitochondria to NADH [15]. Depolarization of mitochondria could restrain cystine starvation induced, but not GPX4 knockout induced ferroptosis [20]. This is consistent with the recent study from Dr. Xuejun Jiang’s lab. ETC inhibitors including the mitochondrial uncoupler CCCP sharply suppress lipid peroxidation and ferroptosis induced by erastin or cystine deprivation [116]. However, these ETC inhibitors fail to impinge on lipid ROS accumulation and ferroptosis upon GPX4 inhibition, neither induced by RSL3 nor by GPX4 knockout [116]. This is possible that mitochondria function upstream of GPX4 to result in an exhaustion of GSH. In striking contrast, the studies presented by Carsten Culmsee group reported a profound loss of mitochondrial membrane potential upon the exposure of glutamate, erastin or RSL3 in MEF and HT-22 cells by using the identical TMRE staining [110,112].
3.5. Elevated Mitochondrial Lipid Peroxidation in Ferroptosis
GPX4 knockout kidneys show an accumulation of oxidized cardiolipin [20]. Examination of subcellular localization of the lipid ROS by confocal imaging showed that the lipid peroxidation first appears in a distribution significantly co-localized with mitochondria, then trans-localizes with the plasma membrane at later time points [116]. This is consistent with the pattern of doxorubicin-induced myocardial ferroptosis. Fang et al. showed that lipid peroxidation is specifically increased inside the mitochondria, but not in the cytoplasm by utilizing a mitochondria fractionation assay. Moreover, mitochondria-targeted antioxidant mito-TEMPOL [108], but not the non-mitochondria targeted antioxidant TEMPO, could significantly abolish doxorubicin induced lipid peroxidation and cardiac ferroptosis, thus highlighting a critical importance of mitochondrial lipid peroxidation in navigating ferroptosis [64]. However, other report suggested that ferroptosis is initiated by extra-mitochondrial lipid peroxidation [20].
Altogether, the current research demonstrates disrupted mitochondrial integrity, including abnormal mitochondrial architecture, burst of mitochondrial ROS, alteration of mitochondrial membrane potential, as well as elevated mitochondrial lipid peroxidation during ferroptosis. Overall, mitochondrial compromise plays a potent role in class I ferroptosis, which is dependent on GSH exhaustion, but not in class II ferroptosis, which is dependent on genetic ablation or pharmacological inhibition of GPX4. Opposite conclusions proposed from different studies may be due to the different cell lines or different methodologies utilized. Specifically, the cell viability assays based on mitochondrial respiration maybe not suitable to evaluate the role of mitochondria in ferroptosis, as manipulation of mitochondrial function may impact the outcome of the cell viability assays [116].
4. Mitochondria Manipulating Ferroptosis
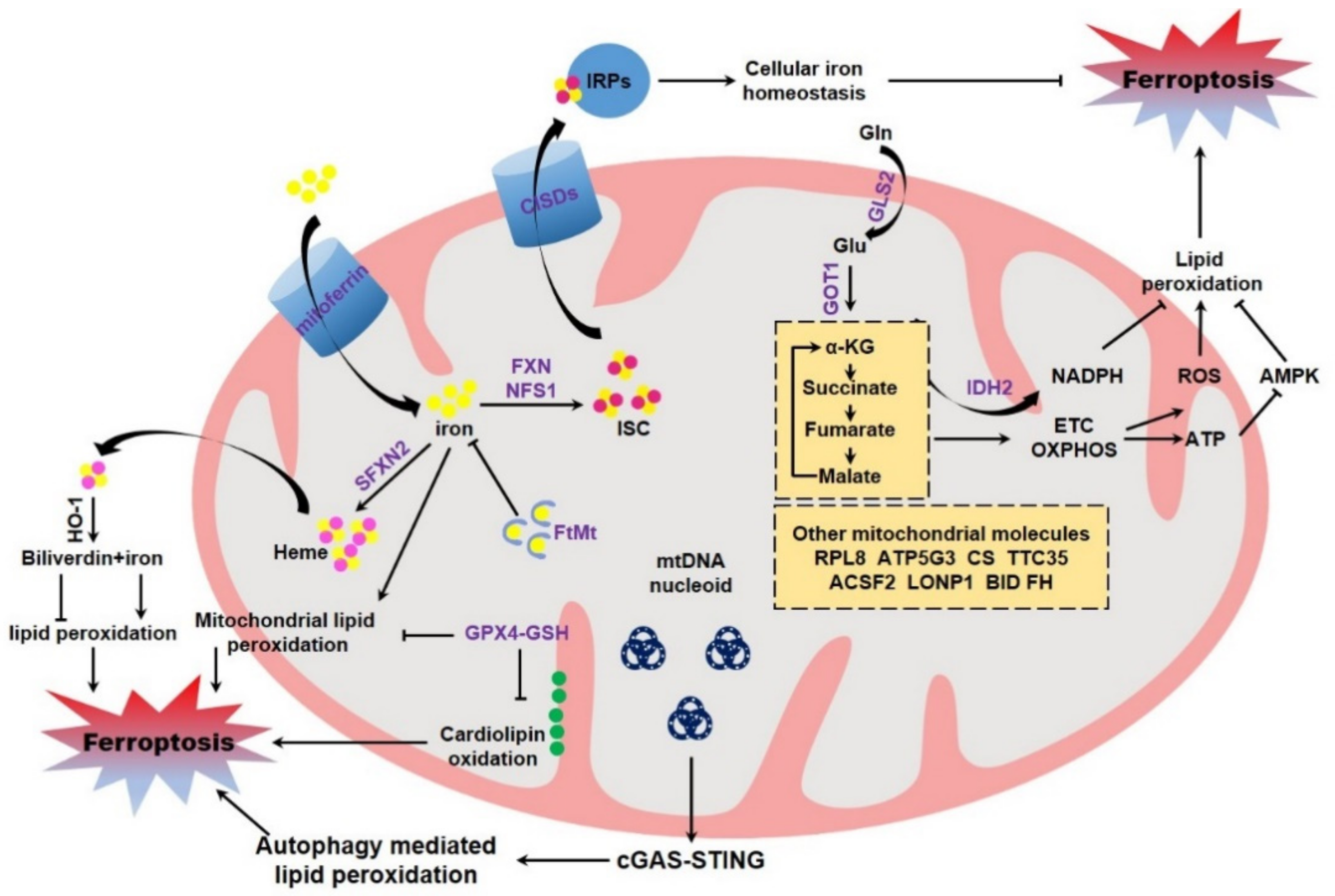
The direct evidence to evaluate the potential role of mitochondria in modulating ferroptosis originates from the studies using mitochondria deficient cells. Gao et al. constructed the Parkin over-expressing HT-1080, and then CCCP was supplemented to remove mitochondria by continuous activation of PINK-Parkin dependent mitophagy [121,122]. By using these cells, the authors found that cells deficient in mitochondria are resistant to cystine deprivation or erastin exposure induced lipid peroxidation and ferroptosis, but remain sensitive to RSL3 induced lipid peroxidation and ferroptosis [116]. However, an opposite conclusion was drawn by Gaschler and colleagues. These authors reported that mitochondria are dispensable for ferroptosis execution by utilizing the identical methodology [123]. Mitochondria contain their own DNA (mtDNA) within the mitochondrial matrix. mtDNA is circular, double stranded, maternally inherited, and composed of 16,569 bp to encode 13 polypeptides of electron transport chain required for ATP production, as well as 22 tRNAs (transfer RNAs) and 2 rRNAs (ribosomal RNAs) [124,125]. It was reported that the mtDNA-depleted cells (termed ρ0 cells) generated from a long-term culture with ethidium bromide display an equivalent sensitivity to ferroptosis when compared to the parental cells [3]. Other study found that ρ0 cells are more sensitive to hydrogen peroxide-induced cell death through accelerating lipid peroxidation [126]. However, it is still unknown whether ferroptosis is involved in this cell death. Recently, Li and colleagues reported that zalcitabine, an antiviral drug for human immunodeficiency virus infection, triggers TFAM degradation dependent ferroptotic cell death of pancreatic cancer cells. Specifically, the degradation of TFAM triggers mtDNA stress, mtDNA release to the cytosol, and ultimate activation of the cGAS-STING pathway, the central cellular cytosolic double-stranded DNA sensor, allowing innate immune to respond to infections, inflammation, and cancer. STING mediated autophagy contributes to zalcitabine-induced ferroptosis by mediating lipid peroxidation [127] (Figure 3). However, it is unclear whether the typical ferroptosis inducers, including erastin, RSL3, GPX4 knockout and cystine deprivation, could trigger the similar mtDNA stress.
4.1. Mitochondrial GPX4 Modulating Ferroptosis
Ferroptosis is dictated by multiple proteins and cellular metabolisms, of which some are tightly associated with mitochondria (Figure 3). GSH reduction and GPX4 inactivation are canonical hallmarks of ferroptosis. Mitochondria possess abundant GSH, with about 10–15% of total GSH [128], although GSH is not de novo synthesized inside of mitochondria [129]. Multiple mitochondria associated carrier proteins have been identified to mediate GSH importing into mitochondria for the maintenance of mitochondrial redox homeostasis. These include 2-oxoglutarate carrier (or SLC25A11), dicarboxylate carrier (or SLC25A10) and tricarboxylate carrier (or SLC25A1) [130,131]. Mitochondrial GSH modulates apoptotic cell death through remodeling mitochondrial membrane and restraining cardiolipin oxidation, which is postulated to facilitate cytochrome c release [132]. GPX4 is partially localized in the intermembrane space of mitochondria [133,134]. GPX4 knockout is embryonic lethal, while GPX4+/− heterozygous cells and the heterozygous mice are markedly sensitive to oxidative stress, as compared to the wild-type cells and the control littermates, respectively [135]. By using GPX4 transgenic mice as the model, it was evidenced that GPX4 could preserve mitochondrial ATP production and maintain mitochondrial membrane potential under oxidative stress [136]. Additionally, independent studies evidenced that specific expression of mitochondrial GPX4 mitigates lipid peroxidation and counteracts cell death. More specifically, mitochondrial GPX4 protects cells from oxidized low-density lipoprotein and cholesterol hydroperoxide [137,138], which may potently trigger ferroptotic cell death. The direct evidence originates from recent studies. The chemotherapeutic agent doxorubicin is commonly used to treat breast cancer, leukemia, and many other types of malignancies [139]. However, the clinical application is limited due to its cardiotoxicities, including irreversible degenerative cardiomyopathy and congestive heart failure [140]. It was reported that doxorubicin triggers a ferroptotic cardiomyopathy [64]. Over-expression of both cytosolic GPX4 and the mitochondrial GPX4 could equally suppress doxorubicin induced accumulation of lipid peroxidation, and thus protect against doxorubicin-induced ferroptotic cardiomyopathy [141]. This study thus shed light on a critical importance of mitochondrial GPX4 in antagonizing ferroptosis (Figure 3).
4.2. Mitochondrial Iron Modulating Ferroptosis
Mitochondria function as the center for cellular iron metabolism. Receptor-mediated endocytosis is the major mechanism for cellular iron uptake. Specifically, extracellular transferrin-bound iron is absorbed by the membrane associated transferrin receptor [142]. Alternatively, cells could also absorb iron directly via DMT1 (divalent metal transporter 1) with the assistance of cell surface ferrireductases DCYTB (duodenal cytochrome b) [143,144]. A large portion of cellular iron is safely sequestered by ferritin, while other is imported into mitochondria by the mitochondrial iron transporter mitoferrin1 in erythroblasts [145] and mitoferrin2 in non-erythroid cells [146]. Mitochondrial iron is stored by FtMt (mitochondrial ferritin) inside the mitochondrial matrix [147] or utilized for the biosynthesis of heme and ISC, two pivotal iron containing molecules essential for numerous heme- and ISC-containing proteins, respectively [148,149]. The excess cytosolic iron could be exported by ferrous iron exporter FPN1 (ferroportin 1), which is precisely controlled by the peptide hormone hepcidin [150]. The cellular iron homeostasis and metabolism is under strict control, mainly by the IRPs (iron regulatory proteins)/IREs (iron-responsive elements) system. During iron deficiency, IRPs (including IRP1 and IRP2) are activated to bind to the conserved stem-loop IREs in UTRs (untranslated regions) of target mRNAs, which mostly encode proteins associated with iron metabolism. The IRPs binding thus determines the fates of target mRNAs and leads to increased iron absorption, as well as decreased iron export, iron utilization and iron storage, thereby maintaining appropriate intracellular iron concentrations [151]. It was not unexpectedly that IRPs are crucial in determining the ferroptotic sensitivity. Independent studies using shRNA screening assay suggested that knockdown of both IRPs leads to an overt resistance to ferroptosis [3,60], which is possibly due to the reduced iron level in IRPs depleted cells. Iron overload, as mentioned above, could facilitate ferroptosis both in vivo and in vitro by catalyzing lipid peroxidation [152]. Immuno-depleted transferrin in serum or knockdown TFRC (transferrin receptor) protects cultured cells from ferroptotic cell death [115]. Additionally, mitochondria are iron overloaded in multiple ferroptosis models [64,153,154], although the underlying mechanism is unclear. It is reasonable that iron accumulation in mitochondria would lead to mitochondrial dysfunction, including disrupted mitochondrial morphology and compromised mitochondrial functionalities as mentioned above. Additionally, mitochondrial iron deposition also catalyzes lipid peroxidation in mitochondrial membrane and triggers ferroptosis. Therefore, it is not surprised that expression of FtMt significantly protects cells from ferroptosis by safely sequestering iron within mitochondria. Moreover, transgenic expression of FtMt protects the survival of fruit fly drosophila feeding an erastin containing diet [155,156], highlighting a fundamental role of mitochondrial iron in determining the ferroptotic sensitivity (Figure 3).
4.3. Mitochondrial ISC Assembly Modulating Ferroptosis
ISC constitutes the reactive center and is functionally important for numerous proteins associated with fundamental cellular processes, including the TCA cycle, electron transport chain, DNA damage/repair and oxygen metabolism, due to the chemical and structural versatility [157]. ISC is mainly assembled in the mitochondrial matrix by the conserved mitochondrially localized ISC assembly machinery, which is mainly composed of the dedicated scaffold protein ISCU, the iron donor molecule FXN (Frataxin), the cysteine desulfurase NFS1, and the small partner protein ISD11 [158]. It has been reported that FXN, the major causative gene of Friedreich ataxia, is possibly involved in ferroptosis. Friedreich ataxia is a progressive neuro- and cardio-degenerative disorder characterized by ataxia, sensory loss, and hypertrophic cardiomyopathy. Friedreich ataxia patient-derived fibroblasts, as well as the FXN mutated murine fibroblasts, are more sensitive to erastin than the normal control cells [159]. Silencing FXN in HT-1080 cells represses cell proliferation and induces mitochondrial damage with elevated iron deposition, which leads to the higher ferroptotic sensitivity to erastin. Conversely, ectopic expression of FXN maintains mitochondrial integrity and thus mitigates ferroptosis [160]. Similarly, FXN deficiency also increases ferroptosis susceptibility in cultured adipocytes. RNAseq analysis indicated ferroptosis as one of the enriched pathways during FXN ablation, further arguing the potent role of FXN in ferroptosis [161]. These studies combined with the previous identification that Friedreich ataxia is pathologically manifested by mitochondrial dysfunction, lipid peroxidation, elevated ROS generation, GSH depletion, and increased iron availability, indicate the possibility of pathological engagement of ferroptosis in cell degeneration during the pathogenesis of Friedreich ataxia [159,162]. It is supposed that defective expression of FXN (experimental knockout or inherited mutation leading to FXN loss in Friedreich ataxia) leading to a disturbed ISC assembly and thus accumulation of mitochondrial iron would facilitate mitochondrial lipid peroxidation and aggravate ferroptosis. Additionally, defective ISC assembly due to FXN deficiency results in loss of ISC binding of IRP1 and robustly activates the cytosolic iron-starvation response. The subsequent upregulation of TFRC and decrease of ferritin thus leads to elevated labile free iron, which further catalyzes lipid peroxidation and navigates ferroptosis (Figure 3). This is verified by the other study reporting that suppression of NFS1, the cysteine desulfurase abstracting the sulfur atom from cysteine for ISC assembly, predisposes cancer cells to ferroptosis and slows tumor growth [163]. Cystine/cysteine deprivation could trigger ferroptosis in diverse cells. However, it is still elusive whether disturbed ISC biosynthesis due to the cystine/cysteine deprivation is responsible for this ferroptosis initiation [164]. Moreover, CISDs (CDGSH iron sulfur domain, also known as mitoNEET including CISD1, CISD2 and CISD3) are mitochondria associated iron-containing proteins responsible for the export of ISC [165]. Knockdown of CISD1 and CISD2 renders ferroptosis [153,154], further shedding light on the pivotal role of mitochondrial ISC metabolism in modulating ferroptosis. Alternatively, ISC could be also synthesized in cytosol by the CIA (cytosolic iron–sulfur protein assembly) machinery [166]. However, it is still elusive whether CIA is coupled with the ferroptotic sensitivity or not.
4.4. Mitochondrial Heme Biosynthesis Modulating Ferroptosis
Heme is other ubiquitous iron-containing molecules important for numerous metabolic enzymes. De novo heme biosynthesis depends on the inter-metabolites shuttling across the mitochondria. Whether heme biosynthesis itself is associated with ferroptosis or not is still unclear. Similar to ISC assembly, it is possible that disturbed heme biosynthesis leads to deposition of mitochondrial iron, which catalyzes mitochondrial lipid peroxidation and initiates ferroptosis. Mitochondrial SFXN2 (sideroflexin 2) is involved in heme biosynthesis with an unclear mechanism. It was reported that SFXN2 deficient cells possess higher mitochondrial iron accompanied with decreased heme and reduced heme-dependent enzyme activities, and these mutant cells are more sensitive to erastin [167]. After ICH (intracerebral hemorrhage), hemoglobin is released from lysed erythrocytes, then absorbed by microglia and metabolized to heme and free iron, which induce lethal ROS accumulation and are thus neurotoxic [168]. Several types of cell death have been identified for neuronal death during ICH in human patients and experimental animals, including apoptosis and necrosis. It was reported that ferroptosis is also involved in this ICH injury. Ferrostatin-1 significantly reduces hemoglobin induced lipid ROS accumulation, GPX activity deficit and neuronal death in organotypic hippocampal slice cultures in vitro. Moreover, ferrostatin-1 also alleviates neuronal degeneration, iron deposition, injury volume, and neurologic deficit after ICH in vivo, further indicating an involvement of hemoglobin/heme in ferroptosis [169]. Additionally, heme produced from hemoglobin degradation also induces ferroptosis in anucleate platelets [170] and in HT-1080 fibrosarcoma cells [62]. It is still unclear whether heme itself or its catabolite iron is responsible for this ferroptosis regulation. HO-1 catabolically degrades heme and generates carbon monoxide, biliverdin, and iron [171]. It has been reported that the expression of HO-1 aggravates ferroptosis, which is possibly due to the iron release via heme catabolism. Pharmacological inhibition or genetic ablation of HO-1 overtly ameliorates ferroptosis [62,64,172]. However, the opposite conclusion was drawn by independent studies. Depletion of HO-1 increases the sensitivity of renal proximal tubule cells to erastin induced ferroptosis [173]. Similarly, Sun and colleagues showed that knockdown of HO-1 by RNAi facilitates ferroptosis induced by erastin and the system XC- inhibitor sorafenib in hepatocellular carcinoma cells [65]. This is possibly due to the catabolite biliverdin, which possesses reducing properties and is recognized as potent antioxidants [174]. Altogether, mitochondria act as the center for cellular iron metabolism and dominate both mitochondrial and the whole cellular iron status, thus determining the ferroptotic sensitivity.
4.5. Mitochondrial Glutaminolysis-TCA-ETC Axis Modulating Ferroptosis
Glutaminolysis-TCA cycle with the coupled ETC-ATP production in mitochondria was characterized as one of the most important metabolisms involved in the regulation of ferroptosis. Glutamine is the most abundant amino acid and serves as the nitrogen source for the anabolism of nucleotides, amino acids, and hexamine, as well as acts as the carbon source to fuel TCA cycle through glutaminolysis [175]. Multiple cancer cells are addicted to high concentration of glutamine for the growth and proliferation in culture [176,177]. An earlier study reported that transporter SLC38A1 essential for glutamine uptake is a positive regulator of ferroptosis by using a pooled shRNA lentivirus library screening assay [60]. Moreover, miR-137 suppresses ferroptosis by targeting glutamine importer SLC1A5. Similarly, chemical inhibition of SLC1A5/SLC38A1 by the inhibitor GPNA (L-g-glutamyl-p-nitroanilide) sharply reduces cytotoxicity of erastin in melanoma cells [178]. Furthermore, mitochondrial glutaminase GLS including GLS1 and GLS2 are responsible for the glutaminolysis to catabolize glutamine and yield glutamate. Knockdown of GLS2, but not GLS1, or chemical inhibition of GLS by Compound 968, significantly decelerates ferroptotic cell death. Glutamic-oxaloacetic transaminase GOT1 is subsequently responsible for the conversion of glutamate into α-KG (α-ketoglutarate), which further fuels TCA cycle. GOT1 RNAi or pharmacological inhibition of the transaminases activity by AOA (amino-oxyacetate) inhibits the execution of ferroptosis, while this ferroptosis could be restored by supplementation of α-KG [115,178,179,180]. Similarly, miR-9 could reduce erastin and RSL3 induced ferroptosis by targeting GOT1 [181]. It was also elaborated that glutaminolysis product α-KG and its downstream TCA metabolites including succinate, fumarate, and malate are all intrinsic metabolites which could modulate cysteine-deprivation induced ferroptosis [116]. It is supposed that glutaminolysis-TCA cycle supported electron transport may facilitate lipid peroxidation through generating ROS, which is produced as the inevitable by-product of mitochondrial respiration. Alternatively, the glutaminolysis-TCA cycle and the coupled ETC-ATP production could modulate AMPK activity and thus determine ferroptotic sensitivity [182]. More specifically, AMPK activation phosphorylates and thus inhibits acetyl-CoA carboxylase 1 and 2 (ACC1/2), leading to a reduced conversion of acetyl-CoA to malonyl-CoA required for the biosynthesis PUFAs [183], which in turn protects cells from lipid peroxidation and ferroptosis [183,184]. Moreover, AMPK activation leads to downregulation of SREBP (sterol regulatory element-binding protein 1) and its downstream SCD1 (stearoyl-coenzyme A (CoA) desaturase-1), the primary enzyme for the biosynthesis of anti-ferroptotic MUFAs [185]. Altogether, these studies thus highlight a critical role of mitochondrial metabolism in regulation of lipid peroxidation and ferroptosis via the metabolic process including glutaminolysis-TCA cycle-ETC-ATP production (Figure 3).
4.6. Mitochondrial NADPH Modulating Ferroptosis
NADPH is always used as an essential electron donor in several redox reaction catalyzed by some enzymes. It is recognized that NADPH functions importantly in fine-tuning ferroptosis. Tonnus and Linkermann hypothesized that NADPH gradient defines the ferroptotic risk and directs the progression of synchronized regulated necrosis of renal tubules cells in ischemia-reperfusion injury [186,187]. By using a compound screening with high cell-line-selective lethality, NADPH was identified as a biomarker for ferroptotic sensitivity across multiple cell lines [188]. Mechanistically, it seems complicated for NADPH to antagonize ferroptosis. NADPH could support GSH reductase to convert GSSG and yield reduced GSH [189,190]. Furthermore, NADPH assists ferroptosis regulator FSP1 for the reduction of CoQ10 to trap lipophilic radical and halts the propagation of lipid peroxides and suppresses ferroptosis [34,35]. Although the pentose phosphate pathway, a metabolic branch from glycolysis, is the major metabolic process responsible to produce NADPH [191], mitochondria are one of the major compartments for NADPH metabolism. NADPH could be generated by the oxidation of malate to pyruvate by malic enzyme inside mitochondria [192]. Alternatively, mitochondrial IDH2 (isocitrate dehydrogenase 2) could catalyze the oxidative decarboxylation of isocitrate to α-KG by reducing NADP+ to NADPH [193]. NADPH could also be generated by cytosolic NADP+-dependent dehydrogenases (including isocitrate dehydrogenase 1 and malic enzyme) utilizing mitochondrial TCA intermediates isocitrate and malate as metabolic precursors [194]. Knockdown of IDH2 substantially increases the susceptibility of ferroptosis in cultured tumor cells and in allografted Hepa1–6 tumor in nude mice [195]. Although the extra-mitochondrial NADPH (mainly plasma membrane) was evidenced as the major lipophilic radical-trapping antioxidant to prevent lipid damage and counteract ferroptosis, these studies put forward the possibility that mitochondria may modulate ferroptosis through NADPH metabolism.
4.7. Other Mitochondrial Components and Ferroptosis
Besides, there are other research elaborating additional mitochondria associated molecules in determining ferroptosis (Figure 3). The shRNA screening by using the library targeting mitochondrial genes identified that RPL8 (ribosomal protein L8), ATP5G3 (ATP synthase F0 complex subunit C3), CS (citrate synthase), TTC35 (tetratricopeptide repeat domain 35), and ACSF2 (acyl-CoA synthetase family member 2) are required for erastin induced ferroptosis. Among them, ACSF2 and CS are implicated in mitochondrial fatty acid metabolism, indicating that lipid synthesis may supply a specific lipid precursor required for ferroptosis [3]. Mutation of mitochondrial TCA cycle enzyme FH (fumarate hydratase) leads to ferroptotic resistance, further suggesting an involvement of mitochondrial TCA cycle in ferroptosis modulation, as discussed above [116]. Furthermore, the mitochondrial protease LONP1 (Lon peptidase 1) [196] and the pro-apoptotic BID [110] are also regulators for ferroptosis under the corresponding contexts, although the underlying mechanism is unclear. Many canonical ferroptosis proteins including ASCL4 and P53 possess partially mitochondrial localization. However, it is still elusive whether this portion has any potential role in the regulation of ferroptosis.
5. Conclusive Remarks and Perspective
Ferroptosis is a newly characterized form of RCD, which is typically manifested by the GSH exhaustion, GPX4 inactivation, and lethal accumulation of lipid peroxidation. It has been widely recognized that dysregulated ferroptosis accounts, at least partially, for the cell degeneration and tissue damage during the pathogenesis of diverse human diseases, especially the neurodegenerative diseases and organ injury related diseases (Table 1). Targeted induction of ferroptosis is considered as a highly potential therapeutic approach for the clinical intervention of other therapy-resistant cancers.
As the powerhouse for ATP production, mitochondria also provide the proper compartment for series of cellular metabolisms. Besides, mitochondria act as a signaling organelle for diverse signaling pathways in response to intracellular and extracellular clues. It has been extensively evidenced that mitochondria determine several types of RCDs. Although some studies proposed that other organelles are involved in ferroptosis, including endoplasmic reticulum [22], Golgi [197] and lysosome [198], increasing studies as mentioned above have put forward the possibility of multifaceted regulation of ferroptosis by mitochondria. Mitochondria are compromised as evidenced by the abnormal morphology, burst of mitochondrial ROS, alteration of mitochondrial membrane potential, elevated mitochondrial lipid peroxidation, as well as accumulation of mitochondrial iron during ferroptosis in different models. More importantly, emerging evidence has been presented to elaborate a multifaceted regulation of ferroptosis by mitochondria associated metabolisms and molecules, although some controversies still exist. It is worth noting that both mitochondrial compromise and typical ferroptosis characteristics are manifested in diseased tissues of patients with neurodegenerative diseases or organ injury related diseases. For example, mitochondrial dysfunction has been widely recognized as an early event during the pathogenesis of Alzheimer’s disease, including elevated mitochondrial oxidative damage and abnormal mitochondrial morphology due to the imbalanced mitochondrial fission and fusion [199]. The typical hallmarks of ferroptosis including iron deposition, depleted GSH and elevated lipid peroxidation, were also observed during the pathogenesis of Alzheimer’s disease [200,201,202]. Besides, both mitochondria-targeting therapeutic approaches and iron-targeting therapeutic strategies have been developed [203,204]. More extensive investigations are needed to further dissect the relevance of mitochondrial dysfunction and ferroptosis in the pathogenesis of Alzheimer’s disease and other related diseases. It is thus of potential treatment efficacy by combination of ferroptosis-targeting and mitochondria-targeting therapeutic approaches for the clinical treatment of these diseases.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Human diseases linked to ferroptosis.
| Disease | Model | Ferroptosis Related Index | Ferroptosis Related Reagents | References |
|---|---|---|---|---|
| Intracerebral hemorrhage | 1. Rat intracerebral injection of autologous whole blood 2. Murine models with striatum injection of collagenase | GPX4↓ GSH↓ ROS↑ | Iron chelator: VK-28 Inhibitor: Ferrostatin-1 | [205,206,207] |
| Cardiomyopathy | 1. Murine models of doxorubicin-induced cardiomyopathy 2. Murine models of ischemia/reperfusion-induced cardiomyopathy | GSH↓ GPX4↓ Ptgs2↑ 4-HNE↑ | Iron chelators: DFO, dexrazoxane Inhibitor: Ferrostatin-1 | [64,115] |
| Atherosclerosis | Iron-loaded ApoE-/-FPNwt/C326S mice | GPX4↓ GSH↓ ROS↑ MDA↑ HO-1↑ 4-HNE↑ | Iron chelator: DFO | [208,209] |
| Nonalcoholic fatty liver disease | Mice fed by the CDAA-based high-fat diet | GSH↓ MDA↑ | Iron chelator: DFP | [210,211] |
| Acute Kidney Injury | 1. Rats renal ischemia/reperfusion 2. Mice models with lipopolysaccharide injection 3. Mice models with BSO injection 4. GPX4 knockout mice | GPX4↓ GSH↓ HO-1 ↑ MDA↑ 4-HNE↑ ROS↑ | Inducer: RSL3 Inhibitor: Liproxstatin-1, Ferrostatin-1 Iron chelators: DFO | [212,213,214] |
| Chronic obstructive pulmonary disease | 1. Mice models exposed to whole body mainstream cigarette smoke 2. GPX4 knockout mice and transgenic mice | GPX4↓ GSH↓ iron↑ Transferrin↑ MDA↑ 4-HNE↑ | Iron chelator: DFO Inhibiter: Ferrostatin-1 | [215,216,217] |
Abbreviations: ApoE, apolipoprotein E; BSO, buthionine sulfoximine; CDAA, choline-deficient L-amino acid defined diet; DFO, deferoxamine; DFP, deferiprone; FPN, ferroportin; GPX4, glutathione peroxidase 4; GSH, glutathione; 4-HNE: 4-hydroxynonenal; HO-1: heme oxygenase 1; MDA: malondialdehyde; Ptgs2: prostaglandin-endoperoxide synthase 2; ROS, reactive oxygen species.
Author Contributions
Conceptualization, H.W.; writing—original draft preparation, H.W., F.W. and N.T.; writing—review and editing, H.W., F.W., N.T., T.Z. and W.G.; supervision, H.W.; funding acquisition, H.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (32070738), the Fundamental Research Funds for the Central Universities (Project NO. 2662020DKPY009), the State Key Laboratory of Medicinal Chemical Biology, Nankai University (2020015), and the Fundamental Research Funds for the Central Universities, Huazhong University of Science and Technology (Project NO. 2019kfyXJJS089).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Morrison, B.; Stockwell, B.R.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 1955, 122, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H. The specific amino acid requirements of a human carcinoma cell (Strain hela) in tissue culture. J. Exp. Med. 1955, 102, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Bannai, S.; Tsukeda, H.; Okumura, H. Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem. Biophys. Res. Commun. 1977, 74, 1582–1588. [Google Scholar] [CrossRef]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Tan, S.; Schubert, D.; Maher, P. Science Publisher David Schubert; P Bentham Science Publisher Pamela Maher Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 2001, 1, 497–506. [Google Scholar] [CrossRef]
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1998, 1, 366–373. [Google Scholar] [CrossRef]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Ates, G.; Methner, A.; Conrad, M.; Maher, P. Oxytosis/Ferroptosis—(Re-) Emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Neurosci. 2018, 12, 214. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-ras-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and cancer: Mitochondria meet the “Iron Maiden” cell death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef]
- Yagoda, N.; Von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef] [Green Version]
- Wolpaw, A.J.; Shimada, K.; Skouta, R.; Welsch, M.E.; Akavia, U.D.; Pe’Er, D.; Shaik, F.; Bulinski, J.C.; Stockwell, B.R. Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proc. Natl. Acad. Sci. USA 2011, 108, E771–E780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/Glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Brown, L.M.; Girotti, A.W.; Cornish, V.W.; Schreiber, S.L.; Stockwell, B.R.; Welsch, M.E.; Shimada, K.; Skouta, R.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, S.; Yu, J.; Liao, W.; Shi, M.; He, W.; Huang, Q.; Zhao, Y.; Liang, B.; Zhang, S.; Wen, Z.; et al. Cysteine dioxygenase 1 mediates erastin-induced ferroptosis in human gastric cancer cells. Neoplasia 2017, 19, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Imai, H.; Hirao, F.; Sakamoto, T.; Sekine, K.; Mizukura, Y.; Saito, M.; Kitamoto, T.; Hayasaka, M.; Hanaoka, K.; Nakagawa, Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 2003, 305, 278–286. [Google Scholar] [CrossRef]
- Yoo, S.-E.; Chen, L.; Na, R.; Liu, Y.; Rios, C.; Van Remmen, H.; Richardson, A.; Ran, Q. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free. Radic. Biol. Med. 2012, 52, 1820–1827. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef] [Green Version]
- Seiler, A.; Schneider, M.; Bornkamm, G.W.; Schweizer, U.; Conrad, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and aif-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Carlson, B.A.; Tobe, R.; Yefremova, E.; Tsuji, P.A.; Hoffmann, V.J.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L.; Conrad, M. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016, 9, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Wortmann, M.; Schneider, M.; Bornkamm, G.W.; Conrad, M.; Beck, H.; Pircher, J.; Hellfritsch, J.; Aichler, M.; Vegi, N.; Kölle, P.; et al. Combined deficiency in glutathione peroxidase 4 and vitamin e causes multiorgan thrombus formation and early death in mice. Circ. Res. 2013, 113, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Ueta, T.; Inoue, T.; Furukawa, T.; Tamaki, Y.; Nakagawa, Y.; Imai, H.; Yanagi, Y. Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J. Biol. Chem. 2012, 287, 7675–7682. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Xu, L.-G.; Li, X.; Zhai, Z.; Shu, H.-B. Amid, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J. Biol. Chem. 2002, 277, 25617–25623. [Google Scholar] [CrossRef]
- Wu, M.; Xu, L.-G.; Su, T.; Tian, Y.; Zhai, Z.; Shu, H.-B. AMID is a p53-inducible gene downregulated in tumors. Oncogene 2004, 23, 6815–6819. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nat. Cell Biol. 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; Da Silva, M.C.; Ingold, I.; Grocin, A.G.; Da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nat. Cell Biol. 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Webb, S.M.; Zhang, B.; Shu, H.-B. The p53-inducible apoptotic protein AMID is not required for normal development and tumor suppression. Oncogene 2005, 25, 849–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, V.A.N.; Bezjian, C.T.; Brandner, S.; Daniels, J.D.; Schmitt-Kopplin, P.; Hauck, S.M.; Stockwell, B.R.; Hadian, K.; Schick, J.A.; Pfeiffer, S.; et al. Gtp cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent. Sci. 2019, 6, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skouta, R.; Dixon, S.J.; Stockwell, B.R.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, M.I.J.; Aichler, M.; Walch, M.A.A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.-J.; Olzmann, J.A.; Dixon, S.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 1–31. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Kenny, E.M.; Yang, Q.; Rosenbaum, J.C.; Sparvero, L.J.; Emlet, D.R.; Wen, X.; et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brütsch, S.H.; Wang, C.C.; Li, L.; Stender, H.; Neziroglu, N.; Richter, C.; Kuhn, H.; Borchert, A. Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the ALOX15 gene does not rescue such knock-in mice. Antioxid. Redox Signal. 2015, 22, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Central Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Masters, B.S.S. The journey from NADPH-cytochrome P450 oxidoreductase to nitric oxide synthases. Biochem. Biophys. Res. Commun. 2005, 338, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Dey, A.; Parmar, D.; Dhawan, A.; Dash, D.; Seth, P.K. Cytochrome P450 2E1 dependent catalytic activity and lipid peroxidation in rat blood lymphocytes. Life Sci. 2002, 71, 2509–2519. [Google Scholar] [CrossRef]
- Bast, A.; Brenninkmeijer, J.W.; Savenije-Chapel, E.; Noordhoek, J. Cytochrome P450 oxidase activity and its role in NADPH dependent lipid peroxidation. FEBS Lett. 1983, 151, 185–188. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases por and CYB5R1. Mol. Cell 2021, 81, 355–369.e10. [Google Scholar] [CrossRef]
- Andrews, N.C.; Schmidt, P.J. Iron homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef]
- Spangler, B.; Morgan, C.W.; Fontaine, B.S.C.W.M.S.D.; Wal, M.N.V.; Chang, M.N.V.W.C.J.; Wells, J.A.; Renslo, J.A.W.A.R. A reactivity-based probe of the intracellular labile ferrous iron pool. Nat. Chem. Biol. 2016, 12, 680–685. [Google Scholar] [CrossRef]
- Breuer, W.; Shvartsman, M.; Cabantchik, Z.I. Intracellular labile iron. Int. J. Biochem. Cell Biol. 2008, 40, 350–354. [Google Scholar] [CrossRef]
- De Domenico, I.; Vaughn, M.B.; Li, L.; Bagley, D.; Musci, G.; Ward, D.M.; Kaplan, J. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 2006, 25, 5396–5404. [Google Scholar] [CrossRef] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nat. Cell Biol. 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.-Y.; Park, E.; Lee, S.-J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [Green Version]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Haining, J.L.; Axelrod, B. Induction period in the lipoxidase-catalyzed oxidation of linoleic acid and its abolition by substrate peroxide. J. Biol. Chem. 1958, 232, 193–202. [Google Scholar] [CrossRef]
- Pandey, A.V.; Flück, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef]
- Kagan, E.V.; Tyurin, A.V.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, A.N.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Kang, R.; Zeng, L.; Wang, H.; Billiar, T.R.; Jiang, J.; Tang, D.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe 2018, 24, 97–108.e4. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.J.; Rathinam, V.A. Lipid peroxidation adds fuel to pyr(Optosis). Cell Host Microbe 2018, 24, 8–9. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Catalá, A.; Díaz, M. Editorial: Impact of lipid peroxidation on the physiology and pathophysiology of cell membranes. Front. Physiol. 2016, 7, 423. [Google Scholar] [CrossRef] [Green Version]
- Agmon, E.; Solon, J.; Bassereau, P.; Stockwell, B.R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Schölmerich, J.; Becher, M.-S.; Schmidt, K.; Schubert, R.; Kremer, B.; Feldhaus, S.; Gerok, W. Influence of hydroxylation and conjugation of bile salts on their membrane-damaging properties-studies on isolated hepatocytes and lipid membrane vesicles. Hepatology 1984, 4, 661–666. [Google Scholar] [CrossRef]
- Wong-Ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.-M.; Tieleman, D.P.; Monticelli, L. Effect of lipid peroxidation on the properties of lipid bilayers: A molecular dynamics study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Agarwal, S. Liposomes as membrane model for study of lipid peroxidation. Free. Radic. Biol. Med. 1988, 4, 51–72. [Google Scholar] [CrossRef]
- Catalá, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef]
- Zarkovic, N.; Cipak, A.; Jaganjac, M.; Borovic, S.; Zarkovic, K. Pathophysiological relevance of aldehydic protein modifications. J. Proteom. 2013, 92, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.; Angeli, J.P.F.; Conrad, M.; Wang, C.; Lan, T.; Qin, W.; Zhu, Y.; Qin, K.; Gao, J.; et al. Quantitative profiling of protein carbonylations in ferroptosis by an aniline-derived probe. J. Am. Chem. Soc. 2018, 140, 4712–4720. [Google Scholar] [CrossRef] [Green Version]
- McElhanon, K.E.; Bose, C.; Sharma, R.; Wu, L.; Awasthi, Y.C.; Singh, S.P. Gsta4 null mouse embryonic fibroblasts exhibit enhanced sensitivity to oxidants: Role of 4-hydroxynonenal in oxidant toxicity. Open J. Apoptosis 2013, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Butow, R.A. Mitochondrial retrograde signaling. Annu. Rev. Genet. 2006, 40, 159–185. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Evolution of mitochondria as signaling organelles. Cell Metab. 2015, 22, 204–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome C. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, X. Cytochromec-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.-D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Marshall, K.D.; Baines, C.P. Necroptosis: Is there a role for mitochondria? Front. Physiol. 2014, 5, 323. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor a gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Chen, Q. Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid. Redox Signal. 2015, 22, 1032–1046. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wei, H.; Sehgal, S.A.; Liu, L.; Chen, Q. Mitophagy receptors sense stress signals and couple mitochondrial dynamic machinery for mitochondrial quality control. Free Radic. Biol. Med. 2016, 100, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauckman, A.K.; Haller, E.; Flores, I.; Nanjundan, M. Iron modulates cell survival in a Ras- and MAPK-dependent manner in ovarian cells. Cell Death Dis. 2013, 4, e592. [Google Scholar] [CrossRef] [Green Version]
- Rouault, T.A. Mitochondrial iron overload: Causes and consequences. Curr. Opin. Genet. Dev. 2016, 38, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil Lee, D.; Park, J.; Lee, H.-S.; Lee, S.-R.; Lee, N.-S. Iron overload-induced calcium signals modulate mitochondrial fragmentation in HT-22 hippocampal neuron cells. Toxicology 2016, 365, 17–24. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhao, Y.; Guo, J.; Zhao, S.; Fei, C.; Xiao, C.; Wu, D.; Wu, L.; Li, X.; Chang, C. Iron overload promotes mitochondrial fragmentation in mesenchymal stromal cells from myelodysplastic syndrome patients through activation of the AMPK/MFF/Drp1 pathway. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volani, C.; Doerrier, C.; Demetz, E.; Haschka, D.; Paglia, G.; Lavdas, A.A.; Gnaiger, E.; Weiss, G. Dietary iron loading negatively affects liver mitochondrial function. Metallomics 2017, 9, 1634–1644. [Google Scholar] [CrossRef]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Campian, J.L.; Qian, M.; Sun, X.-F.; Eaton, J.W. Mitochondrial DNA damage in iron overload. J. Biol. Chem. 2009, 284, 4767–4775. [Google Scholar] [CrossRef] [Green Version]
- Asín, J.; Pérez-Martos, A.; Fernández-Silva, P.; Montoya, J.; Andreu, A.L. Iron(II) induces changes in the conformation of mammalian mitochondrial DNA resulting in a reduction of its transcriptional rate. FEBS Lett. 2000, 480, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Yaffee, M.; Walter, P.; Richter, C.; Muller, M. Direct observation of iron-induced conformational changes of mitochondrial DNA by high-resolution field-emission in-lens scanning electron microscopy. Proc. Natl. Acad. Sci. USA 1996, 93, 5341–5346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, D.-S.; Kim, B.; Park, S.-J.; Kim, J.-H.; Lee, S.-R.; Chang, K.-T.; Lee, H.-S. Iron overload triggers mitochondrial fragmentation via calcineurin-sensitive signals in HT-22 hippocampal neuron cells. Toxicology 2015, 337, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Silva-Gomes, S.; Santos, A.G.; Caldas, C.; Silva, C.M.; Neves, J.V.; Lopes, J.; Carneiro, F.; Rodrigues, P.N.; Duarte, T.L. Transcription factor NRF2 protects mice against dietary iron-induced liver injury by preventing hepatocytic cell death. J. Hepatol. 2014, 60, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Krainz, T.; Gaschler, M.M.; Lim, C.; Sacher, J.R.; Stockwell, B.R.; Wipf, P. A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Cent. Sci. 2016, 2, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, E.N.; Patnaik, J.; Mullins, M.R.; Lemasters, J.J. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010, 70, 10192–10201. [Google Scholar] [CrossRef] [Green Version]
- Dehart, D.N.; Fang, D.; Heslop, K.; Li, L.; Lemasters, J.J.; Maldonado, E.N. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem. Pharmacol. 2018, 148, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Holmuhamedov, E. Voltage-dependent anion channel (VDAC) as mitochondrial governator—Thinking outside the box. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2006, 1762, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, S.W.; Oberst, A.; Kidd, G.; Wakefield, R.; Frase, S.; Krautwald, S.; Linkermann, A.; Green, D.R.; Quarato, G.; Milasta, S.; et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013, 5, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Hu, F.; Feng, H.; Linkermann, A.; Min, W.; Stockwell, B.R. Determination of the subcellular localization anda mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem. Biol. 2018, 13, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genom. Hum. Genet. 2010, 11, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.J.; Butow, R.A. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 2005, 6, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Takashi, Y.; Ouchi, Y.; Kuwahara, Y.; Igarashi, K.; Nagasawa, T.; Nabika, H.; Kurimasa, A.; Fukumoto, M.; Nishitani, Y.; et al. Lipid peroxidation increases hydrogen peroxide permeability leading to cell death in cancer cell lines that lack mtDNA. Cancer Sci. 2019, 110, 2856–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zhang, Y.; Liu, J.; Kang, R.; Klionsky, D.J.; Tang, D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Meredith, M.J.; Reed, D.J. Status of the mitochondrial pool of glutathione in the isolated hepatocyte. J. Biol. Chem. 1982, 257, 3747–3753. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, V.; Garcã-a-Ruiz, C.; Fernã¡ndez-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; Colell, A.; Fernández–Checa, J.C.; Morales, A.; Caballero, F.; Moles, A.; Fernández, A.; Terrones, O.; Basañez, G.; Antonsson, B.; et al. Mechanism of mitochondrial glutathione-dependent hepatocellular susceptibility to TNF despite NF-κb activation. Gastroenterology 2008, 134, 1507–1520. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Ufer, C.; Kühn, H.; Borchert, A. Molecular biology of glutathione peroxidase 4: From genomic structure to developmental expression and neural function. Biol. Chem. 2007, 388, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, A.T. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Liang, H.; Van Remmen, H.; Frohlich, V.; Lechleiter, J.; Richardson, A.; Ran, Q. Gpx4 protects mitochondrial ATP generation against oxidative damage. Biochem. Biophys. Res. Commun. 2007, 356, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Hurst, R.; Korytowski, W.; Kriska, T.; Esworthy, R.; Chu, F.-F.; Girotti, A.W. Hyperresistance to cholesterol hydroperoxide-induced peroxidative injury and apoptotic death in a tumor cell line that overexpresses glutathione peroxidase isotype-4. Free. Radic. Biol. Med. 2001, 31, 1051–1065. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maurer, S.; Lötzer, K.; Böl, G.-F.; Kallionpää, H.; Lehtolainen, P.; Viita, H.; Ylä-Herttuala, S. Overexpression of PHGPx inhibits hydroperoxide-induced oxidation, NFκB activation and apoptosis and affects oxLDL-mediated proliferation of rabbit aortic smooth muscle cells. Atherosclerosis 2000, 152, 307–316. [Google Scholar] [CrossRef]
- Young, R.C.; Ozols, R.F.; Myers, C.E. The anthracycline antineoplastic drugs. N. Engl. J. Med. 1981, 305, 139–153. [Google Scholar] [CrossRef]
- Muggia, F.M.; Speyer, J.L. Doxorubicin-induced cardiomyopathy. N. Eng. J. Med. 1999, 340, 654–655. [Google Scholar]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.-I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, 5. [Google Scholar] [CrossRef]
- Lawen, A.; Lane, D. Mammalian iron homeostasis in health and disease: Uptake, storage, transport, and molecular mechanisms of action. Antioxid. Redox Signal. 2013, 18, 2473–2507. [Google Scholar] [CrossRef]
- Gunshin, H.; MacKenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nat. Cell Biol. 1997, 388, 482–488. [Google Scholar] [CrossRef]
- McKie, A.T.; Barrow, D.; Peters, T.J.; Raja, K.B.; Shirali, S.; Hediger, M.A.; Farzaneh, F.; Simpson, R.J.; Latunde-Dada, G.O.; Rolfs, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nat. Cell Biol. 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of Mitochondrial Iron Import through Differential Turnover of Mitoferrin 1 and Mitoferrin 2. Mol. Cell. Biol. 2008, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, T.A.; Tong, W.-H. Iron–sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat. Rev. Mol. Cell Biol. 2005, 6, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P. Tissue-specific regulation of iron metabolism and heme synthesis: Distinct control mechanisms in erythroid cells. Blood 1997, 89, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.J.R.; Merlot, A.M.; Huang, M.-H.; Bae, D.-H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; An, P.; Li, G.; Yang, L.; Liu, W.; Min, J.; Wang, F.; Xie, E.; Wu, Q.; Fang, X.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.-L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Chang, S.-Y.; Chang, Y.-Z.; Wu, Q.; Gou, Y.-J.; Jia, L.; Cui, Y.-M.; Yu, P.; Shi, Z.-H.; Wu, W.-S.; et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Gallardo, A.K.; Missirlis, F. Loss of ferritin in developing wing cells: Apoptosis and ferroptosis coincide. PLoS Genet. 2020, 16, e1008503. [Google Scholar] [CrossRef] [Green Version]
- Braymer, J.J.; Lill, R. Iron–sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, T.A. Biogenesis of iron-sulfur clusters in mammalian cells: New insights and relevance to human disease. Dis. Model. Mech. 2012, 5, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Cotticelli, M.G.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a novel therapeutic target for Friedreich’s ataxia. J. Pharmacol. Exp. Ther. 2019, 369, 47–54. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021, 38, 101791. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nat. Cell Biol. 2017, 551, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Novera, W.; Lee, Z.-W.; Wu, W.; Huang, R.Y.-J.; Deng, L.-W.; Nin, D.S.; Dai, M.Z.-Y.; Idres, S.B.; Wu, H.; Damen, J.M.A.; et al. Cysteine deprivation targets ovarian clear cell carcinoma via oxidative stress and iron−sulfur cluster biogenesis deficit. Antioxid. Redox Signal. 2020, 33, 1191–1208. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. Neet proteins: A new link between iron metabolism, reactive oxygen species, and cancer. Antioxid. Redox Signal. 2019, 30, 1083–1095. [Google Scholar] [CrossRef]
- Sharma, A.K.; Pallesen, L.J.; Spang, R.J.; Walden, W.E. Cytosolic iron-sulfur cluster assembly (CIA) System: Factors, mechanism, and relevance to cellular iron regulation. J. Biol. Chem. 2010, 285, 26745–26751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mon, E.E.; Wei, F.-Y.; Ahmad, R.N.R.; Yamamoto, T.; Moroishi, T.; Tomizawa, K. Regulation of mitochondrial iron homeostasis by sideroflexin 2. J. Physiol. Sci. 2018, 69, 359–373. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.-Y.; Wang, J.; Qian, Z.-M.; Yang, Q.-W. Iron and intracerebral hemorrhage: From mechanism to translation. Transl. Stroke Res. 2013, 5, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naveenkumar, S.K.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: Protection by Melatonin. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.-C.; Chiang, S.-K.; Chen, S.-E.; Yu, Y.-L.; Chou, R.-H.; Chang, W.-C. Heme oxygenase-1 mediates BAY 11–7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Adedoyin, O.; Boddu, R.; Traylor, A.M.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef] [Green Version]
- Ejansen, T.; Edaiber, A. Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Jiang, X. Metabolism and iron signaling in ferroptotic cell death. Oncotarget 2015, 6, 35145–35146. [Google Scholar] [CrossRef]
- Jennis, M.; Kung, C.-P.; Basu, S.; Budina-Kolomets, A.; Leu, J.I.-J.; Khaku, S.; Scott, J.P.; Cai, K.Q.; Campbell, M.R.; Porter, D.K.; et al. An African-specific polymorphism in theTP53gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016, 30, 918–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Wu, L.; Zhang, P.; Luo, M.; Du, J.; Gao, T.; O’Connell, D.; Wang, G.; Wang, H.; Yang, Y. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol. Carcinog. 2018, 57, 1566–1576. [Google Scholar] [CrossRef]
- Ma, Y.; Han, F.; Min, J.; Lin, W. Energy metabolism as a regulator of ferroptosis. Cell Cycle 2020, 19, 2960–2962. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Li, C.; Dong, X.; Du, W.; Shi, X.; Chen, K.; Zhang, W.; Gao, M. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct. Target. Ther. 2020, 5, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, M.; Yao, X.; Fei, Y.; Lin, Z.; Li, Z.; Cai, K.; Zhao, Y.; Luo, Z. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep. 2020, 33, 108487. [Google Scholar] [CrossRef]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.-S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnus, W.; Linkermann, A. “Death is my heir”—ferroptosis connects cancer pharmacogenomics and ischemia-reperfusion injury. Cell Chem. Biol. 2016, 23, 202–203. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanzok, S.M. Substitution of the thioredoxin system for glutathione reductase in drosophila melanogaster. Science 2001, 291, 643–646. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenkel, R. Regulation and physiological functions of malic enzymes. Curr. Topics Cell. Reg. 1975, 9, 157–181. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef] [Green Version]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, J.H.; Park, J.-W. Down-regulation of IDH2 sensitizes cancer cells to erastin-induced ferroptosis. Biochem. Biophys. Res. Commun. 2020, 525, 366–371. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Zhao, Y.; Zhang, W.; Xu, K.; Li, D.; Zhou, Y.; Li, H.; Xiao, G.; Lu, B.; et al. Inhibition of LONP1 protects against erastin-induced ferroptosis in Pancreatic ductal adenocarcinoma PANC1 cells. Biochem. Biophys. Res. Commun. 2020, 522, 1063–1068. [Google Scholar] [CrossRef]
- Alborzinia, H.; Ignashkova, T.I.; Dejure, F.R.; Gendarme, M.; Theobald, J.; Wölfl, S.; Lindemann, R.K.; Reiling, J.H. Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun. Biol. 2018, 1, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.J.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s disease: An update on emerging mechanisms. J. Alzheimers Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Sultana, R.; Scapagnini, G.; Guagliano, E.; Sapienza, M.; Bella, R.; Kanski, J.; Pennisi, G.; Mancuso, C.; Stella, A.M.G.; et al. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxidants Redox Signal. 2006, 8, 1975–1986. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.I.; Lynn, B.C.; Markesbery, W.R.; Lovell, M.A. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, D.R.C.; Kruck, T.; Kalow, W.; Andrews, D.; Dalton, A.; Bell, M.; Smith, W. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, X.; Zhou, Z.; Xiang, W.; Jiang, Y.; Tian, N.; Tang, X.; Chen, S.; Wen, J.; Chen, M.; Liu, K.; et al. Glutathione alleviates acute intracerebral hemorrhage injury via reversing mitochondrial dysfunction. Brain Res. 2020, 1727, 146514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, Y.; Yuan, S.; Zhang, P.; Zhang, J.; Li, H.; Li, X.; Shen, H.; Wang, Z.; Chen, G. Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 2018, 1701, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Ren, H.; Wang, J. Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc. Neurol. 2019, 4, 93–95. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, E.S.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Hear. J. 2020, 41, 2681–2695. [Google Scholar] [CrossRef]
- Guo, Z.; Ran, Q.; Roberts, L.J.; Zhou, L.; Richardson, A.; Sharan, C.; Wu, D.; Yang, H. Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein E-deficient mice. Free Radic. Biol. Med. 2008, 44, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Kok, C.Y.-Y.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bi, J.; Ren, Y.; Du, Z.; Li, T.; Wang, T.; Zhang, L.; Wang, M.; Wei, S.; Lv, Y.; et al. Involvement of GPX4 in irisin’s protection against ischemia reperfusion-induced acute kidney injury. J. Cell. Physiol. 2021, 236, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhang, H.; Yang, S.-K.; Wu, X.; He, D.; Cao, K.; Zhang, W. Emerging role of ferroptosis in acute kidney injury. Oxidative Med. Cell. Longev. 2019, 2019, 8010614. [Google Scholar] [CrossRef] [PubMed]
- Scindia, Y.; Leeds, J.; Swaminathan, S. Iron homeostasis in healthy kidney and its role in acute kidney injury. Semin. Nephrol. 2019, 39, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloonan, S.M.; Glass, K.; Laucho-Contreras, M.E.; Bhashyam, A.R.; Cervo, M.; Pabón, M.A.; Konrad, C.; Polverino, F.; Siempos, I.I.; Perez, E.; et al. Mitochondrial iron chelation ameliorates cigarette smoke–induced bronchitis and emphysema in mice. Nat. Med. 2016, 22, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Minagawa, S.; Araya, J.; Sakamoto, T.; Hara, H.; Tsubouchi, K.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Hammerschmidt, S.; Büchler, N.; Wahn, H. Tissue lipid peroxidation and reduced glutathione depletion in hypochlorite-induced lung injury. Chest 2002, 121, 573–581. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The main regulatory circuit of ferroptosis. System XC- mediated cystine uptake-GSH synthesis-GPX4, FSP1-CoQ-NADPH and GCH1-BH4/BH2 are major pathways for suppressing lipid peroxidation. Erastin, BSO and RSL3 are typically targeting to system XC-, GSH synthesis, and GPX4, respectively, to disrupt these endogenous lipophilic antioxidant systems and elevate lipid peroxidation. The LOXs and POR could enzymatically catalyze the formation of lipid peroxides, while cellular free iron catalyzes lipid peroxidation via Fenton rection or via the LOXs or POR. Autophagic ferritin degradation (ferritinophagy) could motivate cellular iron and facilitate lipid peroxidation. Iron chelators, as well as the autophagy inhibitors including BafA1 and chloroquine, could suppress lipid peroxidation and counteract ferroptosis by decreasing cellular free iron. Furthermore, the lipophilic antioxidants could directly trap lipophilic radical to halt the propagation of lipid peroxidation and suppress ferroptosis.The lethal outburst of lipid peroxides ultimately drives ferroptosis. Abbreviations: ATGs, autophagy genes; BafA1, bafilomycin A1; BH4/BH2, tetrahydrobiopterin/dihydrobiopterin; BSO, buthionine sulfoximine; FSP1, ferroptosis suppressor protein 1; GCH1, GTP cyclohydrolase 1; Glu: glutamate; GPX4, glutathione peroxidase 4; GSH, glutathione; LOXs, lipoxygenases; NADPH, β-nicotinamide adenine dinucleotide 2′-phosphate, reduced; NCOA4, nuclear receptor coactivator 4; POR, cytochrome P450 oxidoreductase; PUFAs, polyunsaturated fatty acids.
Figure 1.
The main regulatory circuit of ferroptosis. System XC- mediated cystine uptake-GSH synthesis-GPX4, FSP1-CoQ-NADPH and GCH1-BH4/BH2 are major pathways for suppressing lipid peroxidation. Erastin, BSO and RSL3 are typically targeting to system XC-, GSH synthesis, and GPX4, respectively, to disrupt these endogenous lipophilic antioxidant systems and elevate lipid peroxidation. The LOXs and POR could enzymatically catalyze the formation of lipid peroxides, while cellular free iron catalyzes lipid peroxidation via Fenton rection or via the LOXs or POR. Autophagic ferritin degradation (ferritinophagy) could motivate cellular iron and facilitate lipid peroxidation. Iron chelators, as well as the autophagy inhibitors including BafA1 and chloroquine, could suppress lipid peroxidation and counteract ferroptosis by decreasing cellular free iron. Furthermore, the lipophilic antioxidants could directly trap lipophilic radical to halt the propagation of lipid peroxidation and suppress ferroptosis.The lethal outburst of lipid peroxides ultimately drives ferroptosis. Abbreviations: ATGs, autophagy genes; BafA1, bafilomycin A1; BH4/BH2, tetrahydrobiopterin/dihydrobiopterin; BSO, buthionine sulfoximine; FSP1, ferroptosis suppressor protein 1; GCH1, GTP cyclohydrolase 1; Glu: glutamate; GPX4, glutathione peroxidase 4; GSH, glutathione; LOXs, lipoxygenases; NADPH, β-nicotinamide adenine dinucleotide 2′-phosphate, reduced; NCOA4, nuclear receptor coactivator 4; POR, cytochrome P450 oxidoreductase; PUFAs, polyunsaturated fatty acids.

Figure 2.
Lipid Peroxidation drives ferroptotic cell death. ACSL4 and LPCAT3 motivate and esterify the PUFA (AA and AdA) into PE for the next peroxidation, which is enzymatically catalyzed by LOXs or POR, or non-enzymatically mediated by cellular free iron via Fenton reaction. Lipid peroxides could be antagonized by lipophilic antioxidant system (including GPX4-GSH, FSP1-CoQ-NADPH and GCH1-BH4/BH2 axises). The lethal accumulation of lipid peroxides could drive ferroptotic cell death directly through perturbing the integrity of bio-membrane, or indirectly via the peroxidation-derived aldehydes. Abbreviations: AA, arachidonic acid; ACSL4, acyl-CoA synthetase long-chain family member 4; AdA, adrenaline; LOXs, lipoxygenases; LPCAT3, lysophosphatidylcholine acyltransferase 3; PE, phosphatidylethanolamine; POR, cytochrome P450 oxidoreductase; PUFAs, polyunsaturated fatty acids.
Figure 2.
Lipid Peroxidation drives ferroptotic cell death. ACSL4 and LPCAT3 motivate and esterify the PUFA (AA and AdA) into PE for the next peroxidation, which is enzymatically catalyzed by LOXs or POR, or non-enzymatically mediated by cellular free iron via Fenton reaction. Lipid peroxides could be antagonized by lipophilic antioxidant system (including GPX4-GSH, FSP1-CoQ-NADPH and GCH1-BH4/BH2 axises). The lethal accumulation of lipid peroxides could drive ferroptotic cell death directly through perturbing the integrity of bio-membrane, or indirectly via the peroxidation-derived aldehydes. Abbreviations: AA, arachidonic acid; ACSL4, acyl-CoA synthetase long-chain family member 4; AdA, adrenaline; LOXs, lipoxygenases; LPCAT3, lysophosphatidylcholine acyltransferase 3; PE, phosphatidylethanolamine; POR, cytochrome P450 oxidoreductase; PUFAs, polyunsaturated fatty acids.

Figure 3.
Mitochondrial regulation of ferroptosis. Mitochondria modulate ferroptotic cell death through multifaceted mechanisms. As the major compartment for cellular iron metabolism, mitochondria determine the ISC assembly and heme bio-synthesis, and thus dominate the enzymatic activities of numerous proteins containing ISC or heme. Mitochondria also govern cytosolic iron via ISC/IRPs or heme/HO-1 axis. The related mitochondrial proteins including mitoferrin, FXN, NFS1, SFXN2, FtMt, and CISDs thus modulate cellular iron metabolism and ferroptotic sensitivity. The mitochondrial glutaminolysis-TCA-ETC axis is other major pathway to determine ferroptotic cell death, during which the production of ROS, NADPH, and ATP regulates ferroptosis through distinct mechanisms. Mitochondrial GPX4/GSH also determines ferroptosis, which may be partially via modulating cardiolipin oxidation. mtDNA stress could activate the cGAS-STING pathway to facilitate autophagy mediated lipid peroxidation. Moreover, other mitochondria associate molecules also participate in ferroptosis regulation. Abbreviations: ACSF2, acyl-CoA synthetase family member 2; AMPK, adenosine 5′-monophosphate (AMP)-activated protein kinase; ATP, adenosine triphosphate; ATP5G3, ATP synthase F0 complex subunit C3; BID, BH3-interacting domain death agonist; cGAS, cyclic GMP-AMP synthase; CISDs, CDGSH iron sulfur domain; CS, citrate synthase; ETC, electron transport chain; FH, fumarate hydratase; FtMt, mitochondrial ferritin; FXN, Frataxin; Gln, glutamine; GLS2, glutaminase 2; Glu, glutamate; GOT1, glutamic-oxaloacetic transaminase 1; GPX4, glutathione peroxidase 4; GSH, glutathione; HO-1, heme oxygenase-1; IDH2, isocitrate dehydrogenase 2; IRPs, iron regulatory protein; ISC, iron-sulfur cluster; α-KG, α-ketoglutarate; LONP1, Lon peptidase 1; mtDNA, mitochondrial DNA; NADPH, β-nicotinamide adenine dinucleotide 2′-phosphate, reduced; NFS1, NFS1 cysteine desulfurase; ROS, reactive oxygen species; RPL8, ribosomal protein L8; SFXN2, sideroflexin 2; STING, stimulator of interferon genes; TTC35, tetratricopeptide repeat domain 35.
Figure 3.
Mitochondrial regulation of ferroptosis. Mitochondria modulate ferroptotic cell death through multifaceted mechanisms. As the major compartment for cellular iron metabolism, mitochondria determine the ISC assembly and heme bio-synthesis, and thus dominate the enzymatic activities of numerous proteins containing ISC or heme. Mitochondria also govern cytosolic iron via ISC/IRPs or heme/HO-1 axis. The related mitochondrial proteins including mitoferrin, FXN, NFS1, SFXN2, FtMt, and CISDs thus modulate cellular iron metabolism and ferroptotic sensitivity. The mitochondrial glutaminolysis-TCA-ETC axis is other major pathway to determine ferroptotic cell death, during which the production of ROS, NADPH, and ATP regulates ferroptosis through distinct mechanisms. Mitochondrial GPX4/GSH also determines ferroptosis, which may be partially via modulating cardiolipin oxidation. mtDNA stress could activate the cGAS-STING pathway to facilitate autophagy mediated lipid peroxidation. Moreover, other mitochondria associate molecules also participate in ferroptosis regulation. Abbreviations: ACSF2, acyl-CoA synthetase family member 2; AMPK, adenosine 5′-monophosphate (AMP)-activated protein kinase; ATP, adenosine triphosphate; ATP5G3, ATP synthase F0 complex subunit C3; BID, BH3-interacting domain death agonist; cGAS, cyclic GMP-AMP synthase; CISDs, CDGSH iron sulfur domain; CS, citrate synthase; ETC, electron transport chain; FH, fumarate hydratase; FtMt, mitochondrial ferritin; FXN, Frataxin; Gln, glutamine; GLS2, glutaminase 2; Glu, glutamate; GOT1, glutamic-oxaloacetic transaminase 1; GPX4, glutathione peroxidase 4; GSH, glutathione; HO-1, heme oxygenase-1; IDH2, isocitrate dehydrogenase 2; IRPs, iron regulatory protein; ISC, iron-sulfur cluster; α-KG, α-ketoglutarate; LONP1, Lon peptidase 1; mtDNA, mitochondrial DNA; NADPH, β-nicotinamide adenine dinucleotide 2′-phosphate, reduced; NFS1, NFS1 cysteine desulfurase; ROS, reactive oxygen species; RPL8, ribosomal protein L8; SFXN2, sideroflexin 2; STING, stimulator of interferon genes; TTC35, tetratricopeptide repeat domain 35.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wu, H.; Wang, F.; Ta, N.; Zhang, T.; Gao, W. The Multifaceted Regulation of Mitochondria in Ferroptosis. Life 2021, 11, 222. https://doi.org/10.3390/life11030222
AMA Style
Wu H, Wang F, Ta N, Zhang T, Gao W. The Multifaceted Regulation of Mitochondria in Ferroptosis. Life. 2021; 11(3):222. https://doi.org/10.3390/life11030222
Chicago/Turabian StyleWu, Hao, Fengli Wang, Na Ta, Ting Zhang, and Weihua Gao. 2021. "The Multifaceted Regulation of Mitochondria in Ferroptosis" Life 11, no. 3: 222. https://doi.org/10.3390/life11030222
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.