The Role of Oxidative Stress in Cardiovascular Aging and Cardiovascular Diseases

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Behind the Scenes of Cardiovascular Aging
2.1. Sources of Oxidative Stress and Oxidative Damage
- production of IL-1α leading to a proinflammatory state, which increases nuclear factor kappa-β (NFκβ) activity [40];
- induction of MMPs expression, which is associated with age-related and chronic diseases such as cancer, Alzheimer’s, atherosclerosis, osteoarthritis, and lung emphysema [40];
- inhibition of FOXO (Forkhead box) proteins activity, which is involved in insulin/insulin-like growth factor-1-mediated protection from oxidative stress [40];
- reduction of sarco/endoplasmic reticulum Ca2+-ATPase activity leading to cardiac senescence;
- inhibition of sirtuins activity leading to an increased production of ROS and RNS by SOD inhibition, a proinflammatory state by preventing their inhibition of tumor necrosis factor alpha (TNFα) and NFκB, and tumorigenic effect by preventing their inhibitory effect on c-Jun and c-Myc [42];
- regulation of p16INK4a/pRB and p53/p21 pathways leading to senescence [40].
2.2. Aging, Oxidative Stress and Cellular Dysfunction
2.3. Aging in Cardiac Structural and Patho-Physiological Changes
2.4. Endothelial Dysfunction
2.5. Aging and Autonomic Nervous System Adaptations
2.6. Genetic and Epigenetic Regulation on Cardiovascular Aging and Oxidative Stress
3. Aging, Oxidative Stress, and Cardiovascular Diseases
3.1. Atrial Fibrillation
3.2. Heart Failure
3.3. Valvular Heart Disease
4. Aging, Oxidative Stress, and Vascular Diseases
4.1. Aging in Vascular Structural and Patho-Physiological Changes
4.2. Hypertension
4.3. Atherosclerosis
4.4. Deep Vein Thrombosis
4.5. Stroke, TIA, and Cerebrovascular Dementia
5. Possible Antiaging and Antioxidant Interventions
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hayflick, L. Biological aging is no longer an unsolved problem. Ann. N. Y. Acad. Sci. 2007, 1100, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pepe, S.; Lakatta, E.G. Aging hearts and vessels: Masters of adaptation and survival. Cardiovasc. Res. 2005, 66, 190–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzo, C.; Carrizzo, A.; Alfano, A.; Virtuoso, N.; Capunzo, M.; Calabrese, M.; De Simone, E.; Sciarretta, S.; Frati, G.; Oliveti, M.; et al. The Impact of Aging on Cardio and Cerebrovascular Diseases. Int. J. Mol. Sci. 2018, 19, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidenreich, P.A.; Trogdon, J.G.; Khavjou, O.A.; Butler, J.; Dracup, K.; Ezekowitz, M.D.; Finkelstein, E.A.; Hong, Y.; Johnston, S.C.; Khera, A.; et al. Forecasting the future of cardiovascular disease in the United States: A policy statement from the American Heart Association. Circulation 2011, 123, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Tarride, J.E.; Lim, M.; DesMeules, M.; Luo, W.; Burke, N.; O’Reilly, D.; Bowen, J.; Goeree, R. A review of the cost of cardiovascular disease. Can. J. Cardiol. 2009, 25, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- Scuteri, A.; Lakatta, E.G. Bringing prevention in geriatrics: Evidences from cardiovascular medicine supporting the new challenge. Exp. Gerontol. 2013, 48, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, 2181–2190. [Google Scholar] [CrossRef] [Green Version]
- Tahhan, A.S.; Sandesara, P.B.; Hayek, S.S.; Alkhoder, A.; Chivukula, K.; Hammadah, M.; Mohamed-Kelli, H.; O’Neal, W.T.; Topel, M.; Ghasemzadeh, N.; et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm 2017, 14, 1849–1855. [Google Scholar] [CrossRef]
- Baradaran, A.; Nasri, H.; Rafieian-Kopaei, M. Oxidative stress and hypertension: Possibility of hypertension therapy with antioxidants. J. Res. Med. Sci. 2014, 19, 358–367. [Google Scholar]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, F.; Francia, P.; Camici, G.G.; Pelicci, P.G.; Luscher, T.F.; Volpe, M. Final common molecular pathways of aging and cardiovascular disease: Role of the p66Shc protein. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 622–628. [Google Scholar] [CrossRef] [Green Version]
- Sudheesh, N.P.; Ajith, T.A.; Ramnath, V.; Janardhanan, K.K. Therapeutic potential of Ganoderma lucidum (Fr.) P. Karst. against the declined antioxidant status in the mitochondria of post-mitotic tissues of aged mice. Clin. Nutr. 2010, 29, 406–412. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Lyu, S.Y.; Kwon, Y.J.; Joo, H.J.; Park, W.B. Preparation of alginate/chitosan microcapsules and enteric coated granules of mistletoe lectin. Arch. Pharm. Res. 2004, 27, 118–126. [Google Scholar] [CrossRef]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef] [Green Version]
- van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.X.; Gutterman, D.D. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, 2023–2031. [Google Scholar] [CrossRef]
- Krause, K.H. Aging: A revisited theory based on free radicals generated by NOX family NADPH oxidases. Exp. Gerontol. 2007, 42, 256–262. [Google Scholar] [CrossRef]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Manas, L.; El-Assar, M.; Vallejo, S.; Lopez-Doriga, P.; Solis, J.; Petidier, R.; Montes, M.; Nevado, J.; Castro, M.; Gomez-Guerrero, C.; et al. Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell 2009, 8, 226–238. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 2001, 276, 14533–14536. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Fleming, I.; Busse, R. Endothelial aging. Cardiovasc. Res. 2005, 66, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreiro, E. Role of Protein Carbonylation in Skeletal Muscle Mass Loss Associated with Chronic Conditions. Proteomes 2016, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [Green Version]
- Spickett, C.M. The lipid peroxidation product 4-hydroxy-2-nonenal: Advances in chemistry and analysis. Redox Biol. 2013, 1, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Demissie, S.; Levy, D.; Benjamin, E.J.; Cupples, L.A.; Gardner, J.P.; Herbert, A.; Kimura, M.; Larson, M.G.; Meigs, J.B.; Keaney, J.F.; et al. Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell 2006, 5, 325–330. [Google Scholar] [CrossRef]
- Fitzpatrick, A.L.; Kronmal, R.A.; Gardner, J.P.; Psaty, B.M.; Jenny, N.S.; Tracy, R.P.; Walston, J.; Kimura, M.; Aviv, A. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am. J. Epidemiol. 2007, 165, 14–21. [Google Scholar] [CrossRef]
- Liu, S.; Hui, K.S.; Hui, K.N. Flower-like Copper Cobaltite Nanosheets on Graphite Paper as High-Performance Supercapacitor Electrodes and Enzymeless Glucose Sensors. ACS Appl. Mater. Interfaces 2016, 8, 3258–3267. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Idelchik, M.; Melendez, J.A. Redox control of senescence and age-related disease. Redox Biol. 2017, 11, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, N.; Rinaldi, B.; Corbi, G.; Conti, V.; Stiuso, P.; Boccuti, S.; Rengo, G.; Rossi, F.; Filippelli, A. Exercise training promotes SIRT1 activity in aged rats. Rejuvenation Res. 2008, 11, 139–150. [Google Scholar] [CrossRef]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 2010, 88, 993–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Garinis, G.A.; van der Horst, G.T.; Vijg, J.; Hoeijmakers, J.H. DNA damage and ageing: New-age ideas for an age-old problem. Nat. Cell Biol. 2008, 10, 1241–1247. [Google Scholar] [CrossRef]
- Gredilla, R.; Bohr, V.A.; Stevnsner, T. Mitochondrial DNA repair and association with aging—An update. Exp. Gerontol. 2010, 45, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Kavli, B.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Uracil in DNA—General mutagen, but normal intermediate in acquired immunity. DNA Repair 2007, 6, 505–516. [Google Scholar] [CrossRef]
- Gredilla, R.; Garm, C.; Stevnsner, T. Nuclear and mitochondrial DNA repair in selected eukaryotic aging model systems. Oxidative Med. Cell. Longev. 2012, 2012, 282438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.F.; Rabinovitch, P.S. Cardiac aging in mice and humans: The role of mitochondrial oxidative stress. Trends Cardiovasc. Med. 2009, 19, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wilhelmsson, H.; Graff, C.; Li, H.; Oldfors, A.; Rustin, P.; Brüning, J.C.; Kahn, C.R.; Clayton, D.A.; Barsh, G.S.; et al. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat. Genet. 1999, 21, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Silva, J.P.; Gustafsson, C.M.; Rustin, P.; Larsson, N.G. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 4038–4043. [Google Scholar] [CrossRef] [Green Version]
- Vermulst, M.; Wanagat, J.; Kujoth, G.C.; Bielas, J.H.; Rabinovitch, P.S.; Prolla, T.A.; Loeb, L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008, 40, 392–394. [Google Scholar] [CrossRef]
- Edgar, D.; Shabalina, I.; Camara, Y.; Wredenberg, A.; Calvaruso, M.A.; Nijtmans, L.; Nedergaard, J.; Cannon, B.; Larsson, N.G.; Trifunovic, A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab. 2009, 10, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Baris, O.R.; Ederer, S.; Neuhaus, J.F.; von Kleist-Retzow, J.C.; Wunderlich, C.M.; Pal, M.; Wunderlich, F.T.; Peeva, V.; Zsurka, G.; Kunz, W.S.; et al. Mosaic Deficiency in Mitochondrial Oxidative Metabolism Promotes Cardiac Arrhythmia during Aging. Cell Metab. 2015, 21, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Kujoth, G.C.; Leeuwenburgh, C.; Prolla, T.A. Mitochondrial DNA mutations and apoptosis in mammalian aging. Cancer Res. 2006, 66, 7386–7389. [Google Scholar] [CrossRef] [Green Version]
- Tower, J. Programmed cell death in aging. Ageing Res. Rev. 2015, 23, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Modrego, J.; de las Heras, N.; Zamorano-León, J.J.; Mateos-Cáceres, P.J.; Martín-Fernández, B.; Valero-Muñoz, M.; Lahera, V.; López-Farré, A.J. Changes in cardiac energy metabolic pathways in overweighed rats fed a high-fat diet. Eur. J. Nutr. 2013, 52, 847–856. [Google Scholar] [CrossRef]
- Opie, L.H.; Knuuti, J. The adrenergic-fatty acid load in heart failure. J. Am. Coll. Cardiol. 2009, 54, 1637–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.F.; Hsieh, E.J.; Liu, Y.; Chen, T.; Beyer, R.P.; Chin, M.T.; MacCoss, M.J.; Rabinovitch, P.S. Mitochondrial proteome remodelling in pressure overload-induced heart failure: The role of mitochondrial oxidative stress. Cardiovasc. Res. 2012, 93, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyer, G.R.; De Keulenaer, G.W.; Martinet, W. Role of autophagy in heart failure associated with aging. Heart Fail. Rev. 2010, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [Green Version]
- Upadhya, B.; Taffet, G.E.; Cheng, C.P.; Kitzman, D.W. Heart failure with preserved ejection fraction in the elderly: Scope of the problem. J. Mol. Cell. Cardiol. 2015, 83, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.G.; Haynes, P.; Kelsey Snapp, W.; Nava, K.E.; Campbell, K.S. Altered ventricular torsion and transmural patterns of myocyte relaxation precede heart failure in aging F344 rats. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, 676–686. [Google Scholar] [CrossRef] [Green Version]
- Boluyt, M.O.; Converso, K.; Hwang, H.S.; Mikkor, A.; Russell, M.W. Echocardiographic assessment of age-associated changes in systolic and diastolic function of the female F344 rat heart. J. Appl. Physiol. 2004, 96, 822–828. [Google Scholar] [CrossRef]
- Maruyama, Y. Aging and arterial-cardiac interactions in the elderly. Int. J. Cardiol. 2012, 155, 14–19. [Google Scholar] [CrossRef]
- Juhaszova, M.; Rabuel, C.; Zorov, D.B.; Lakatta, E.G.; Sollott, S.J. Protection in the aged heart: Preventing the heart-break of old age? Cardiovasc. Res. 2005, 66, 233–244. [Google Scholar] [CrossRef]
- Mirza, M.; Strunets, A.; Shen, W.K.; Jahangir, A. Mechanisms of arrhythmias and conduction disorders in older adults. Clin. Geriatr. Med. 2012, 28, 555–573. [Google Scholar] [CrossRef] [Green Version]
- Strait, J.B.; Lakatta, E.G. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail. Clin. 2012, 8, 143–164. [Google Scholar] [CrossRef] [Green Version]
- Dhahbi, J.M.; Tsuchiya, T.; Kim, H.J.; Mote, P.L.; Spindler, S.R. Gene expression and physiologic responses of the heart to the initiation and withdrawal of caloric restriction. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 218–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakatta, E.G. Cardiovascular regulatory mechanisms in advanced age. Physiol. Rev. 1993, 73, 413–467. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Silbermann, M.; Gershon, D.; Reznick, A.Z. Giant mitochondria in the myocardium of aging and endurance-trained mice. Gerontology 1987, 33, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Muscari, C.; Finelli, C.; Stefanelli, C.; Flamigni, F.; Guarnieri, C.; Caldarera, C.M. Age-dependent differences of ATP breakdown and ATP-catabolite release in ischemic and reperfused hearts. Mech. Ageing Dev. 1993, 67, 1–11. [Google Scholar] [CrossRef]
- Muscari, C.; Caldarera, C.M.; Guarnieri, C. Age-dependent production of mitochondrial hydrogen peroxide, lipid peroxides and fluorescent pigments in the rat heart. Basic Res. Cardiol. 1990, 85, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Sohal, B.H. Hydrogen peroxide release by mitochondria increases during aging. Mech. Ageing Dev. 1991, 57, 187–202. [Google Scholar] [CrossRef]
- Liu, F.; Li, N.; Long, B.; Fan, Y.Y.; Liu, C.Y.; Zhou, Q.Y.; Murtaza, I.; Wang, K.; Li, P.F. Cardiac hypertrophy is negatively regulated by miR-541. Cell Death Dis. 2014, 5, e1171. [Google Scholar] [CrossRef] [Green Version]
- Fajemiroye, J.O.; da Cunha, L.C.; Saavedra-Rodriguez, R.; Rodrigues, K.L.; Naves, L.M.; Mourao, A.A.; da Silva, E.F.; Williams, N.E.E.; Martins, J.L.R.; Sousa, R.B.; et al. Aging-Induced Biological Changes and Cardiovascular Diseases. Biomed. Res. Int. 2018, 2018, 7156435. [Google Scholar] [CrossRef] [Green Version]
- Olivetti, G.; Melissari, M.; Capasso, J.M.; Anversa, P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ. Res. 1991, 68, 1560–1568. [Google Scholar] [CrossRef] [Green Version]
- Hees, P.S.; Fleg, J.L.; Lakatta, E.G.; Shapiro, E.P. Left ventricular remodeling with age in normal men versus women: Novel insights using three-dimensional magnetic resonance imaging. Am. J. Cardiol. 2002, 90, 1231–1236. [Google Scholar] [CrossRef]
- Olivetti, G.; Giordano, G.; Corradi, D.; Melissari, M.; Lagrasta, C.; Gambert, S.R.; Anversa, P. Gender differences and aging: Effects on the human heart. J. Am. Coll. Cardiol. 1995, 26, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Kajstura, J.; Cheng, W.; Sarangarajan, R.; Li, P.; Li, B.; Nitahara, J.A.; Chapnick, S.; Reiss, K.; Olivetti, G.; Anversa, P. Necrotic and apoptotic myocyte cell death in the aging heart of Fischer 344 rats. Am. J. Physiol. 1996, 271, 1215–1228. [Google Scholar] [CrossRef]
- Sheydina, A.; Riordon, D.R.; Boheler, K.R. Molecular mechanisms of cardiomyocyte aging. Clin. Sci. 2011, 121, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Chen, T.; Johnson, S.C.; Szeto, H.; Rabinovitch, P.S. Cardiac aging: From molecular mechanisms to significance in human health and disease. Antioxid. Redox Signal. 2012, 16, 1492–1526. [Google Scholar] [CrossRef] [Green Version]
- Leon, L.J.; Gustafsson, A.B. Staying young at heart: Autophagy and adaptation to cardiac aging. J. Mol. Cell. Cardiol. 2016, 95, 78–85. [Google Scholar] [CrossRef] [Green Version]
- AlGhatrif, M.; Lakatta, E.G. The conundrum of arterial stiffness, elevated blood pressure, and aging. Curr. Hypertens. Rep. 2015, 17, 12. [Google Scholar] [CrossRef] [Green Version]
- Gerstenblith, G.; Frederiksen, J.; Yin, F.C.; Fortuin, N.J.; Lakatta, E.G.; Weisfeldt, M.L. Echocardiographic assessment of a normal adult aging population. Circulation 1977, 56, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Schulman, S.P.; Lakatta, E.G.; Fleg, J.L.; Lakatta, L.; Becker, L.C.; Gerstenblith, G. Age-related decline in left ventricular filling at rest and exercise. Am. J. Physiol. 1992, 263, 1932–1938. [Google Scholar] [CrossRef]
- Miller, T.R.; Grossman, S.J.; Schectman, K.B.; Biello, D.R.; Ludbrook, P.A.; Ehsani, A.A. Left ventricular diastolic filling and its association with age. Am. J. Cardiol. 1986, 58, 531–535. [Google Scholar] [CrossRef]
- Howden, E.J.; Carrick-Ranson, G.; Sarma, S.; Hieda, M.; Fujimoto, N.; Levine, B.D. Effects of Sedentary Aging and Lifelong Exercise on Left Ventricular Systolic Function. Med. Sci. Sports Exerc. 2018, 50, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Peverill, R.E. Aging and the relationships between long-axis systolic and early diastolic excursion, isovolumic relaxation time and left ventricular length-Implications for the interpretation of aging effects on e. PLoS ONE 2019, 14, e0210277. [Google Scholar] [CrossRef]
- Fleg, J.L.; O’Connor, F.; Gerstenblith, G.; Becker, L.C.; Clulow, J.; Schulman, S.P.; Lakatta, E.G. Impact of age on the cardiovascular response to dynamic upright exercise in healthy men and women. J. Appl. Physiol. 1995, 78, 890–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silaghi, A.; Piercecchi-Marti, M.D.; Grino, M.; Leonetti, G.; Alessi, M.C.; Clement, K.; Dadoun, F.; Dutour, A. Epicardial adipose tissue extent: Relationship with age, body fat distribution, and coronaropathy. Obesity 2008, 16, 2424–2430. [Google Scholar] [CrossRef]
- Horn, M.A.; Trafford, A.W. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J. Mol. Cell. Cardiol. 2016, 93, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Biernacka, A.; Frangogiannis, N.G. Aging and Cardiac Fibrosis. Aging Dis. 2011, 2, 158–173. [Google Scholar]
- Jugdutt, B.I.; Jelani, A.; Palaniyappan, A.; Idikio, H.; Uweira, R.E.; Menon, V.; Jugdutt, C.E. Aging-related early changes in markers of ventricular and matrix remodeling after reperfused ST-segment elevation myocardial infarction in the canine model: Effect of early therapy with an angiotensin II type 1 receptor blocker. Circulation 2010, 122, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Mendes, A.B.; Ferro, M.; Rodrigues, B.; Souza, M.R.; Araujo, R.C.; Souza, R.R. Quantification of left ventricular myocardial collagen system in children, young adults, and the elderly. Medicina 2012, 72, 216–220. [Google Scholar]
- Burstein, B.; Nattel, S. Atrial fibrosis: Mechanisms and clinical relevance in atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 802–809. [Google Scholar] [CrossRef] [Green Version]
- Everett, T.H.T.; Li, H.; Mangrum, J.M.; McRury, I.D.; Mitchell, M.A.; Redick, J.A.; Haines, D.E. Electrical, morphological, and ultrastructural remodeling and reverse remodeling in a canine model of chronic atrial fibrillation. Circulation 2000, 102, 1454–1460. [Google Scholar] [CrossRef] [Green Version]
- Virdis, A.; Ghiadoni, L.; Giannarelli, C.; Taddei, S. Endothelial dysfunction and vascular disease in later life. Maturitas 2010, 67, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Toda, N. Age-related changes in endothelial function and blood flow regulation. Pharmacol. Ther. 2012, 133, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Matz, R.L.; Andriantsitohaina, R. Age-related endothelial dysfunction: Potential implications for pharmacotherapy. Drugs Aging 2003, 20, 527–550. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Luo, N.; Chi, Y. Aging-shifted prostaglandin profile in endothelium as a factor in cardiovascular disorders. J. Aging Res. 2012, 2012, 121390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrage, W.G.; Eisenach, J.H.; Joyner, M.J. Ageing reduces nitric-oxide- and prostaglandin-mediated vasodilatation in exercising humans. J. Physiol. 2007, 579, 227–236. [Google Scholar] [CrossRef]
- Nicholson, W.T.; Vaa, B.; Hesse, C.; Eisenach, J.H.; Joyner, M.J. Aging is associated with reduced prostacyclin-mediated dilation in the human forearm. Hypertension 2009, 53, 973–978. [Google Scholar] [CrossRef] [Green Version]
- Dere, E.; De Souza Silva, M.A.; Topic, B.; Fiorillo, C.; Li, J.S.; Sadile, A.G.; Frisch, C.; Huston, J.P. Aged endothelial nitric oxide synthase knockout mice exhibit higher mortality concomitant with impaired open-field habituation and alterations in forebrain neurotransmitter levels. Genes Brain Behav. 2002, 1, 204–213. [Google Scholar] [CrossRef]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Cau, S.B.; Carneiro, F.S.; Tostes, R.C. Differential modulation of nitric oxide synthases in aging: Therapeutic opportunities. Front. Physiol. 2012, 3, 218. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuteri, A.; Tesauro, M.; Rizza, S.; Iantorno, M.; Federici, M.; Lauro, D.; Campia, U.; Turriziani, M.; Fusco, A.; Cocciolillo, G.; et al. Endothelial function and arterial stiffness in normotensive normoglycemic first-degree relatives of diabetic patients are independent of the metabolic syndrome. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Avolio, A.P.; Mergner, W.J.; Robinowitz, M.; Herderick, E.E.; Cornhill, J.F.; Guo, S.Y.; Liu, T.H.; Ou, D.Y.; O’Rourke, M. Effect of aging on aortic morphology in populations with high and low prevalence of hypertension and atherosclerosis. Comparison between occidental and Chinese communities. Am. J. Pathol. 1991, 139, 1119–1129. [Google Scholar] [PubMed]
- Tao, J.; Wang, Y.; Yang, Z.; Tu, C.; Xu, M.G.; Wang, J.M. Circulating endothelial progenitor cell deficiency contributes to impaired arterial elasticity in persons of advancing age. J. Hum. Hypertens. 2006, 20, 490–495. [Google Scholar] [CrossRef]
- Edelberg, J.M.; Reed, M.J. Aging and angiogenesis. Front. Biosci. 2003, 8, 1199–1209. [Google Scholar] [CrossRef]
- Williamson, K.; Stringer, S.E.; Alexander, M.Y. Endothelial progenitor cells enter the aging arena. Front. Physiol. 2012, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part I: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, S.E. Ageing of the conduit arteries. J. Pathol. 2007, 211, 157–172. [Google Scholar] [CrossRef]
- Elliott, H.L.; Sumner, D.J.; McLean, K.; Reid, J.L. Effect of age on the responsiveness of vascular alpha-adrenoceptors in man. J. Cardiovasc. Pharmacol. 1982, 4, 388–392. [Google Scholar] [CrossRef]
- Vaitkevicius, P.V.; Fleg, J.L.; Engel, J.H.; O’Connor, F.C.; Wright, J.G.; Lakatta, L.E.; Yin, F.C.; Lakatta, E.G. Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation 1993, 88, 1456–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egashira, K.; Inou, T.; Hirooka, Y.; Kai, H.; Sugimachi, M.; Suzuki, S.; Kuga, T.; Urabe, Y.; Takeshita, A. Effects of age on endothelium-dependent vasodilation of resistance coronary artery by acetylcholine in humans. Circulation 1993, 88, 77–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, S.; Virdis, A.; Mattei, P.; Ghiadoni, L.; Gennari, A.; Fasolo, C.B.; Sudano, I.; Salvetti, A. Aging and endothelial function in normotensive subjects and patients with essential hypertension. Circulation 1995, 91, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Hotta, H.; Uchida, S. Aging of the autonomic nervous system and possible improvements in autonomic activity using somatic afferent stimulation. Geriatr. Gerontol. Int. 2010, 10, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.W.; Huang, J.C.; Chen, S.C.; Lin, H.Y.; Kuo, I.C.; Wu, P.Y.; Chiu, Y.W.; Chang, J.M. Association between Age and Changes in Heart Rate Variability after Hemodialysis in Patients with Diabetes. Front. Aging Neurosci. 2018, 10, 43. [Google Scholar] [CrossRef]
- Bonnemeier, H.; Richardt, G.; Potratz, J.; Wiegand, U.K.; Brandes, A.; Kluge, N.; Katus, H.A. Circadian profile of cardiac autonomic nervous modulation in healthy subjects: Differing effects of aging and gender on heart rate variability. J. Cardiovasc. Electrophysiol. 2003, 14, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Britton, A.; Shipley, M.; Malik, M.; Hnatkova, K.; Hemingway, H.; Marmot, M. Changes in heart rate and heart rate variability over time in middle-aged men and women in the general population (from the Whitehall II Cohort Study). Am. J. Cardiol. 2007, 100, 524–527. [Google Scholar] [CrossRef]
- Lipsitz, L.A.; Mietus, J.; Moody, G.B.; Goldberger, A.L. Spectral characteristics of heart rate variability before and during postural tilt. Relations to aging and risk of syncope. Circulation 1990, 81, 1803–1810. [Google Scholar] [CrossRef] [Green Version]
- Kanegusuku, H.; Queiroz, A.C.; Silva, V.J.; de Mello, M.T.; Ugrinowitsch, C.; Forjaz, C.L. High-Intensity Progressive Resistance Training Increases Strength With No Change in Cardiovascular Function and Autonomic Neural Regulation in Older Adults. J. Aging Phys. Act. 2015, 23, 339–345. [Google Scholar] [CrossRef]
- Masuki, S.; Morikawa, M.; Nose, H. Interval Walking Training Can Increase Physical Fitness in Middle-Aged and Older People. Exerc. Sport Sci. Rev. 2017, 45, 154–162. [Google Scholar] [CrossRef]
- Guedes, J.M.; Pieri, B.; Luciano, T.F.; Marques, S.O.; Guglielmo, L.G.A.; Souza, C.T. Muscular resistance, hypertrophy and strength training equally reduce adiposity, inflammation and insulin resistance in mice with diet-induced obesity. Einstein 2020, 18, eAO4784. [Google Scholar] [CrossRef] [Green Version]
- Masuda, M.; Yamada, T.; Mizuno, H.; Minamiguchi, H.; Konishi, S.; Ohtani, T.; Yamaguchi, O.; Okuyama, Y.; Uematsu, M.; Sakata, Y. Impact of atrial fibrillation ablation on cardiac sympathetic nervous system in patients with and without heart failure. Int. J. Cardiol. 2015, 199, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Tegene, E.; Tadesse, I.; Markos, Y.; Gobena, T. Prevalence and risk factors for atrial fibrillation and its anticoagulant requirement in adults aged >/=40 in Jimma Town, Southwest Ethiopia: A community based cross-sectional study. Int. J. Cardiol. Heart Vasc. 2019, 22, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Cheng, J. Dysfunction of the autonomic nervous system in atrial fibrillation. J. Thorac. Dis. 2015, 7, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Stavrakis, S.; Nakagawa, H.; Po, S.S.; Scherlag, B.J.; Lazzara, R.; Jackman, W.M. The role of the autonomic ganglia in atrial fibrillation. JACC Clin. Electrophysiol. 2015, 1, 1–13. [Google Scholar] [CrossRef]
- Newaz, M.A.; Yousefipour, Z.; Oyekan, A. Oxidative stress-associated vascular aging is xanthine oxidase-dependent but not NAD(P)H oxidase-dependent. J. Cardiovasc. Pharmacol. 2006, 48, 88–94. [Google Scholar] [CrossRef]
- Donato, A.J.; Eskurza, I.; Silver, A.E.; Levy, A.S.; Pierce, G.L.; Gates, P.E.; Seals, D.R. Direct evidence of endothelial oxidative stress with aging in humans: Relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ. Res. 2007, 100, 1659–1666. [Google Scholar] [CrossRef] [Green Version]
- Paolisso, G.; Tagliamonte, M.R.; Rizzo, M.R.; Manzella, D.; Gambardella, A.; Varricchio, M. Oxidative stress and advancing age: Results in healthy centenarians. J. Am. Geriatr. Soc. 1998, 46, 833–838. [Google Scholar] [CrossRef]
- Suzuki, M.; Willcox, D.C.; Rosenbaum, M.W.; Willcox, B.J. Oxidative stress and longevity in okinawa: An investigation of blood lipid peroxidation and tocopherol in okinawan centenarians. Curr. Gerontol. Geriatr. Res. 2010, 2010, 380460. [Google Scholar] [CrossRef] [Green Version]
- Soerensen, M.; Christensen, K.; Stevnsner, T.; Christiansen, L. The Mn-superoxide dismutase single nucleotide polymorphism rs4880 and the glutathione peroxidase 1 single nucleotide polymorphism rs1050450 are associated with aging and longevity in the oldest old. Mech. Ageing Dev. 2009, 130, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Schiattarella, G.G.; Hill, J.A. Metabolic control and oxidative stress in pathological cardiac remodelling. Eur. Heart J. 2017, 38, 1399–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laina, A.; Stellos, K.; Stamatelopoulos, K. Vascular ageing: Underlying mechanisms and clinical implications. Exp. Gerontol. 2018, 109, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, A.J.; Magerko, K.A.; Lawson, B.R.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 2011, 589, 4545–4554. [Google Scholar] [CrossRef] [PubMed]
- Zarzuelo, M.J.; Lopez-Sepulveda, R.; Sanchez, M.; Romero, M.; Gomez-Guzman, M.; Ungvary, Z.; Perez-Vizcaino, F.; Jimenez, R.; Duarte, J. SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: Implications for vascular aging. Biochem. Pharmacol. 2013, 85, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell. Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.M.; Wagner, R.; Rzucidlo, E.M. Age-related loss of SirT1 expression results in dysregulated human vascular smooth muscle cell function. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, 533–541. [Google Scholar] [CrossRef]
- TenNapel, M.J.; Lynch, C.F.; Burns, T.L.; Wallace, R.; Smith, B.J.; Button, A.; Domann, F.E. SIRT6 minor allele genotype is associated with >5-year decrease in lifespan in an aged cohort. PLoS ONE 2014, 9, e115616. [Google Scholar] [CrossRef]
- Stein, S.; Lohmann, C.; Schafer, N.; Hofmann, J.; Rohrer, L.; Besler, C.; Rothgiesser, K.M.; Becher, B.; Hottiger, M.O.; Boren, J.; et al. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur. Heart J. 2010, 31, 2301–2309. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Wang, Z.; Chen, H.Z.; Zhou, S.; Zheng, W.; Liu, G.; Wei, Y.S.; Cai, H.; Liu, D.P.; Liang, C.C. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc. Res. 2008, 80, 191–199. [Google Scholar] [CrossRef]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Yun, J.; Finkel, T. The emerging links between sirtuins and autophagy. Methods Mol. Biol 2013, 1077, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Dou, X.; Ning, H.; Song, Q.; Wei, W.; Zhang, X.; Shen, C.; Li, J.; Sun, C.; Song, Z. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology 2017, 66, 936–952. [Google Scholar] [CrossRef]
- Dai, S.H.; Chen, T.; Li, X.; Yue, K.Y.; Luo, P.; Yang, L.K.; Zhu, J.; Wang, Y.H.; Fei, Z.; Jiang, X.F. Sirt3 confers protection against neuronal ischemia by inducing autophagy: Involvement of the AMPK-mTOR pathway. Free Radic. Biol. Med. 2017, 108, 345–353. [Google Scholar] [CrossRef]
- He, J.; Zhang, G.; Pang, Q.; Yu, C.; Xiong, J.; Zhu, J.; Chen, F. SIRT6 reduces macrophage foam cell formation by inducing autophagy and cholesterol efflux under ox-LDL condition. FEBS J. 2017, 284, 1324–1337. [Google Scholar] [CrossRef] [Green Version]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef]
- Spescha, R.D.; Shi, Y.; Wegener, S.; Keller, S.; Weber, B.; Wyss, M.M.; Lauinger, N.; Tabatabai, G.; Paneni, F.; Cosentino, F.; et al. Deletion of the ageing gene p66(Shc) reduces early stroke size following ischaemia/reperfusion brain injury. Eur. Heart J. 2013, 34, 96–103. [Google Scholar] [CrossRef]
- Spescha, R.D.; Klohs, J.; Semerano, A.; Giacalone, G.; Derungs, R.S.; Reiner, M.F.; Rodriguez Gutierrez, D.; Mendez-Carmona, N.; Glanzmann, M.; Savarese, G.; et al. Post-ischaemic silencing of p66Shc reduces ischaemia/reperfusion brain injury and its expression correlates to clinical outcome in stroke. Eur. Heart J. 2015, 36, 1590–1600. [Google Scholar] [CrossRef] [Green Version]
- Camici, G.G.; Schiavoni, M.; Francia, P.; Bachschmid, M.; Martin-Padura, I.; Hersberger, M.; Tanner, F.C.; Pelicci, P.; Volpe, M.; Anversa, P.; et al. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc. Natl. Acad. Sci. USA 2007, 104, 5217–5222. [Google Scholar] [CrossRef] [Green Version]
- Paneni, F.; Osto, E.; Costantino, S.; Mateescu, B.; Briand, S.; Coppolino, G.; Perna, E.; Mocharla, P.; Akhmedov, A.; Kubant, R.; et al. Deletion of the activated protein-1 transcription factor JunD induces oxidative stress and accelerates age-related endothelial dysfunction. Circulation 2013, 127, 1229–1240. [Google Scholar] [CrossRef] [Green Version]
- Laurent, G.; Solari, F.; Mateescu, B.; Karaca, M.; Castel, J.; Bourachot, B.; Magnan, C.; Billaud, M.; Mechta-Grigoriou, F. Oxidative stress contributes to aging by enhancing pancreatic angiogenesis and insulin signaling. Cell Metab. 2008, 7, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Villa, F.; Carrizzo, A.; Spinelli, C.C.; Ferrario, A.; Malovini, A.; Maciag, A.; Damato, A.; Auricchio, A.; Spinetti, G.; Sangalli, E.; et al. Genetic Analysis Reveals a Longevity-Associated Protein Modulating Endothelial Function and Angiogenesis. Circ. Res. 2015, 117, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Villa, F.; Malovini, A.; Carrizzo, A.; Spinelli, C.C.; Ferrario, A.; Maciag, A.; Madonna, M.; Bellazzi, R.; Milanesi, L.; Vecchione, C.; et al. Serum BPIFB4 levels classify health status in long-living individuals. Immun. Ageing 2015, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Malavolta, M.; Dato, S.; Villa, F.; Rango, F.; Iannone, F.; Ferrario, A.; Maciag, A.; Ciaglia, E.; D’Amato, A.; Carrizzo, A.; et al. LAV-BPIFB4 associates with reduced frailty in humans and its transfer prevents frailty progression in old mice. Aging (Albany N. Y.) 2019, 11, 6555–6568. [Google Scholar] [CrossRef]
- Smith-Vikos, T.; Liu, Z.; Parsons, C.; Gorospe, M.; Ferrucci, L.; Gill, T.M.; Slack, F.J. A serum miRNA profile of human longevity: Findings from the Baltimore Longitudinal Study of Aging (BLSA). Aging (Albany N. Y.) 2016, 8, 2971–2987. [Google Scholar] [CrossRef] [Green Version]
- Quiat, D.; Olson, E.N. MicroRNAs in cardiovascular disease: From pathogenesis to prevention and treatment. J. Clin. Investig. 2013, 123, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Kinser, H.E.; Pincus, Z. MicroRNAs as modulators of longevity and the aging process. Hum. Genet. 2020, 139, 291–308. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Smith-Vikos, T.; Slack, F.J. MicroRNAs and their roles in aging. J. Cell Sci. 2012, 125, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Chen, X.; Inukai, S.; Zhao, H.; Slack, F.J. Age-associated changes in expression of small, noncoding RNAs, including microRNAs, in C. elegans. RNA 2011, 17, 1804–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, M.; Slack, F. A developmental timing microRNA and its target regulate life span in C. elegans. Science 2005, 310, 1954–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Almen, G.C.; Verhesen, W.; van Leeuwen, R.E.; van de Vrie, M.; Eurlings, C.; Schellings, M.W.; Swinnen, M.; Cleutjens, J.P.; van Zandvoort, M.A.; Heymans, S.; et al. MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell 2011, 10, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, X.; Wang, F.; Zhou, L.; Yin, Z.; Fan, J.; Nie, X.; Wang, P.; Fu, X.D.; Chen, C.; et al. MicroRNA-21 Lowers Blood Pressure in Spontaneous Hypertensive Rats by Upregulating Mitochondrial Translation. Circulation 2016, 134, 734–751. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, F.; Capri, M.; Bonafe, M.; Morsiani, C.; Jung, H.J.; Spazzafumo, L.; Vina, J.; Suh, Y. Circulating miRNAs and miRNA shuttles as biomarkers: Perspective trajectories of healthy and unhealthy aging. Mech. Ageing Dev. 2017, 165, 162–170. [Google Scholar] [CrossRef] [Green Version]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [Green Version]
- Morris, C.D.; Carson, S. Routine vitamin supplementation to prevent cardiovascular disease: A summary of the evidence for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2003, 139, 56–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, A.; Kuum, M.; Joubert, F.; Wilding, J.; Ventura-Clapier, R.; Veksler, V. Mitochondria as a source of mechanical signals in cardiomyocytes. Cardiovasc. Res. 2010, 87, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iribe, G.; Kaihara, K.; Yamaguchi, Y.; Nakaya, M.; Inoue, R.; Naruse, K. Mechano-sensitivity of mitochondrial function in mouse cardiac myocytes. Prog. Biophys. Mol. Biol. 2017, 130, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Sollott, S.J. Perspectives on mammalian cardiovascular aging: Humans to molecules. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2002, 132, 699–721. [Google Scholar] [CrossRef]
- Lakatta, E.G. Age-associated cardiovascular changes in health: Impact on cardiovascular disease in older persons. Heart Fail. Rev. 2002, 7, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Sollott, S.J.; Pepe, S. The old heart: Operating on the edge. In Novartis Foundation Symposium; Chichester: New York, NY, USA; John Wiley: Hoboken, NJ, USA, 1999; Volume 235, pp. 172–196. [Google Scholar]
- Sadoshima, J.; Xu, Y.; Slayter, H.S.; Izumo, S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 1993, 75, 977–984. [Google Scholar] [CrossRef]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 2006, 8, 691–728. [Google Scholar] [CrossRef] [Green Version]
- Papaconstantinou, J. The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells 2019, 8, 1383. [Google Scholar] [CrossRef] [Green Version]
- Aronow, W.S. Etiology, pathophysiology, and treatment of atrial fibrillation: Part 1. Cardiol. Rev. 2008, 16, 181–188. [Google Scholar] [CrossRef]
- Schueller, P.O.; Steiner, S.; Hennersdorf, M.G. Atrial fibrillation. Med. Mon. Pharm. 2009, 32, 204–210. [Google Scholar]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation: A translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef]
- Iwasaki, Y.K.; Nishida, K.; Kato, T.; Nattel, S. Atrial fibrillation pathophysiology: Implications for management. Circulation 2011, 124, 2264–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomstrom-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association of Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pan, N.H.; Tsao, H.M.; Chang, N.C.; Chen, Y.J.; Chen, S.A. Aging dilates atrium and pulmonary veins: Implications for the genesis of atrial fibrillation. Chest 2008, 133, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Ravassa, S.; Ballesteros, G.; Diez, J. Aging and atrial fibrillation: A matter of fibrosis. Aging (Albany N. Y.) 2019, 11, 9965–9966. [Google Scholar] [CrossRef]
- Ravassa, S.; Lopez, B.; Querejeta, R.; Echegaray, K.; San Jose, G.; Moreno, M.U.; Beaumont, F.J.; Gonzalez, A.; Diez, J. Phenotyping of myocardial fibrosis in hypertensive patients with heart failure. Influence on clinical outcome. J. Hypertens. 2017, 35, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Zhan, G.; Wenhua, G.; Jie, H.; Xiang, Q.; Lingzhi, C.; Xue, X.; Xing-Xing, C.; Qianli, Z.; Weijian, H.; Hao, Z. Potential roles of circulating matrix metalloproteinase-28 (MMP-28) in patients with atrial fibrillation. Life Sci. 2018, 204, 15–19. [Google Scholar] [CrossRef]
- Lewkowicz, J.; Knapp, M.; Tankiewicz-Kwedlo, A.; Sawicki, R.; Kaminska, M.; Waszkiewicz, E.; Musial, W.J. MMP-9 in atrial remodeling in patients with atrial fibrillation. In Annales de Cardiologie et D’angeiologie; Elsevier Masson: Paris, France, 2015; Volume 64, pp. 285–291. [Google Scholar] [CrossRef]
- Kamkin, A.; Kiseleva, I.; Isenberg, G.; Wagner, K.D.; Gunther, J.; Theres, H.; Scholz, H. Cardiac fibroblasts and the mechano-electric feedback mechanism in healthy and diseased hearts. Prog. Biophys. Mol. Biol. 2003, 82, 111–120. [Google Scholar] [CrossRef]
- Chang, S.L.; Chen, Y.C.; Chen, Y.J.; Wangcharoen, W.; Lee, S.H.; Lin, C.I.; Chen, S.A. Mechanoelectrical feedback regulates the arrhythmogenic activity of pulmonary veins. Heart 2007, 93, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Chen, Y.C.; Chen, Y.J.; Chang, S.L.; Tai, C.T.; Wongcharoen, W.; Yeh, H.I.; Lin, C.I.; Chen, S.A. Tumor necrosis factor-alpha alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci. 2007, 80, 1806–1815. [Google Scholar] [CrossRef]
- Lai, L.P.; Tsai, C.C.; Su, M.J.; Lin, J.L.; Chen, Y.S.; Tseng, Y.Z.; Huang, S.K. Atrial fibrillation is associated with accumulation of aging-related common type mitochondrial DNA deletion mutation in human atrial tissue. Chest 2003, 123, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, M.; Hisatome, I.; Morisaki, T.; Tanaka, M.; Tomikura, Y.; Takeda, S.; Shimoyama, M.; Ohtahara, A.; Ogino, K.; Igawa, O.; et al. Mitochondrial DNA deletion associated with the reduction of adenine nucleotides in human atrium and atrial fibrillation. Eur. J. Clin. Investig. 2001, 31, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.K.; Chen, Y.A.; Lee, T.I.; Chen, Y.C.; Chen, S.A.; Chen, Y.J. Aging Modulates the Substrate and Triggers Remodeling in Atrial Fibrillation. Circ. J. 2018, 82, 1237–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.J.; Pfeffer, M.A. Heart failure. Lancet 2005, 365, 1877–1889. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.; Adams, R.; Carnethon, M.; De Simone, G.; Ferguson, T.B.; Flegal, K.; Ford, E.; Furie, K.; Go, A.; Greenlund, K.; et al. Heart disease and stroke statistics—2009 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009, 119, 21–181. [Google Scholar] [CrossRef] [Green Version]
- Mosterd, A.; Hoes, A.W. Clinical epidemiology of heart failure. Heart 2007, 93, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Orso, F.; Fabbri, G.; Maggioni, A.P. Epidemiology of Heart Failure. Handb. Exp. Pharmacol. 2017, 243, 15–33. [Google Scholar] [CrossRef]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Kurmani, S.; Squire, I. Acute Heart Failure: Definition, Classification and Epidemiology. Curr. Heart Fail. Rep. 2017, 14, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S. The changing epidemiology of sudden death in heart failure. Curr. Heart Fail. Rep. 2004, 1, 93–97. [Google Scholar] [CrossRef]
- Penjaskovic, D.; Sakac, D.; Dejanovic, J.; Zec, R.; Zec Petkovic, N.; Stojsic Milosavljevic, A. Left ventricular diastolic dysfunction in patients with metabolic syndrome. Med. Pregl. 2012, 65, 18–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cil, H.; Bulur, S.; Turker, Y.; Kaya, A.; Alemdar, R.; Karabacak, A.; Aslantas, Y.; Ekinozu, I.; Albayrak, S.; Ozhan, H.; et al. Impact of body mass index on left ventricular diastolic dysfunction. Echocardiography 2012, 29, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D. Pathophysiology of Chronic Systolic Heart Failure. A View from the Periphery. Ann. Am. Thorac. Soc. 2018, 15, 38–41. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J. Clinical practice. Systolic heart failure. N. Engl. J. Med. 2010, 362, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K. Pathophysiology of systolic and diastolic heart failure. Med. Clin. 2012, 96, 891–899. [Google Scholar] [CrossRef]
- Henes, J.; Rosenberger, P. Systolic heart failure: Diagnosis and therapy. Curr. Opin. Anesthesiol. 2016, 29, 55–60. [Google Scholar] [CrossRef]
- Ennezat, P.V.; Le Jemtel, T.H.; Logeart, D.; Marechaux, S. Heart failure with preserved ejection fraction: A systemic disorder? Rev. Med. Interne 2012, 33, 370–380. [Google Scholar] [CrossRef]
- Wachter, R. Diastolic heart failure. Internist 2014, 55, 663–668. [Google Scholar] [CrossRef]
- Del Buono, M.G.; Buckley, L.; Abbate, A. Primary and Secondary Diastolic Dysfunction in Heart Failure with Preserved Ejection Fraction. Am. J. Cardiol. 2018, 122, 1578–1587. [Google Scholar] [CrossRef]
- Lodi, R.; Tonon, C.; Calabrese, V.; Schapira, A.H. Friedreich’s ataxia: From disease mechanisms to therapeutic interventions. Antioxid. Redox Signal. 2006, 8, 438–443. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Kawamori, D.; Miyatsuka, T.; Sakamoto, K.; Matsuoka, T.A.; Matsuhisa, M.; Yamasaki, Y. Involvement of oxidative stress in the pathogenesis of diabetes. Antioxid. Redox Signal. 2007, 9, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Panerai, A.E. Formalin injection in the tail facilitates hindpaw withdrawal reflexes induced by thermal stimulation in the rat: Effect of paracetamol. Neurosci. Lett. 1997, 237, 89–92. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohal, R.S.; Arnold, L.A.; Sohal, B.H. Age-related changes in antioxidant enzymes and prooxidant generation in tissues of the rat with special reference to parameters in two insect species. Free Radic. Biol. Med. 1990, 9, 495–500. [Google Scholar] [CrossRef]
- Yan, L.J.; Sohal, R.S. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc. Natl. Acad. Sci. USA 1998, 95, 12896–12901. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Woldgiorgis, G.; Elson, C.E.; Shrago, E. Age-related changes in respiration coupled to phosphorylation. I. Hepatic mitochondria. Mech. Ageing Dev. 1988, 46, 263–277. [Google Scholar] [CrossRef]
- Yokozawa, T.; Satoh, A.; Cho, E.J. Ginsenoside-Rd attenuates oxidative damage related to aging in senescence-accelerated mice. J. Pharm. Pharmacol. 2004, 56, 107–113. [Google Scholar] [CrossRef]
- Judge, S.; Jang, Y.M.; Smith, A.; Hagen, T.; Leeuwenburgh, C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: Implications for the mitochondrial theory of aging. FASEB J. 2005, 19, 419–421. [Google Scholar] [CrossRef]
- Mo, J.Q.; Hom, D.G.; Andersen, J.K. Decreases in protective enzymes correlates with increased oxidative damage in the aging mouse brain. Mech. Ageing Dev. 1995, 81, 73–82. [Google Scholar] [CrossRef]
- Hoffmann, B.; Stockl, A.; Schlame, M.; Beyer, K.; Klingenberg, M. The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. J. Biol. Chem. 1994, 269, 1940–1944. [Google Scholar] [CrossRef]
- Nehal, M.; Azam, M.; Baquer, N.Z. Changes in the levels of catecholamines, hexokinase and glucose 6-phosphate dehydrogenase in red cell aging. Biochem. Int. 1990, 22, 517–522. [Google Scholar] [PubMed]
- Karavidas, A.; Lazaros, G.; Tsiachris, D.; Pyrgakis, V. Aging and the cardiovascular system. Hell. J. Cardiol. 2010, 51, 421–427. [Google Scholar]
- Nassimiha, D.; Aronow, W.S.; Ahn, C.; Goldman, M.E. Association of coronary risk factors with progression of valvular aortic stenosis in older persons. Am. J. Cardiol. 2001, 87, 1313–1314. [Google Scholar] [CrossRef]
- Stewart, B.F.; Siscovick, D.; Lind, B.K.; Gardin, J.M.; Gottdiener, J.S.; Smith, V.E.; Kitzman, D.W.; Otto, C.M. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J. Am. Coll. Cardiol. 1997, 29, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Freeman, R.V.; Otto, C.M. Spectrum of calcific aortic valve disease: Pathogenesis, disease progression, and treatment strategies. Circulation 2005, 111, 3316–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, C.M.; Lind, B.K.; Kitzman, D.W.; Gersh, B.J.; Siscovick, D.S. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N. Engl. J. Med. 1999, 341, 142–147. [Google Scholar] [CrossRef]
- Olsen, M.H.; Wachtell, K.; Bella, J.N.; Gerdts, E.; Palmieri, V.; Nieminen, M.S.; Smith, G.; Ibsen, H.; Devereux, R.B.; Substudy, L. Aortic valve sclerosis relates to cardiovascular events in patients with hypertension (a LIFE substudy). Am. J. Cardiol. 2005, 95, 132–136. [Google Scholar] [CrossRef]
- Otto, C.M. Why is aortic sclerosis associated with adverse clinical outcomes? J. Am. Coll. Cardiol. 2004, 43, 176–178. [Google Scholar] [CrossRef] [Green Version]
- Faggiano, P.; Antonini-Canterin, F.; Erlicher, A.; Romeo, C.; Cervesato, E.; Pavan, D.; Piazza, R.; Huang, G.; Nicolosi, G.L. Progression of aortic valve sclerosis to aortic stenosis. Am. J. Cardiol. 2003, 91, 99–101. [Google Scholar] [CrossRef]
- Jeon, D.S.; Atar, S.; Brasch, A.V.; Luo, H.; Mirocha, J.; Naqvi, T.Z.; Kraus, R.; Berman, D.S.; Siegel, R.J. Association of mitral annulus calcification, aortic valve sclerosis and aortic root calcification with abnormal myocardial perfusion single photon emission tomography in subjects age < or =65 years old. J. Am. Coll. Cardiol. 2001, 38, 1988–1993. [Google Scholar] [CrossRef] [Green Version]
- Fulkerson, P.K.; Beaver, B.M.; Auseon, J.C.; Graber, H.L. Calcification of the mitral annulus: Etiology, clinical associations, complications and therapy. Am. J. Med. 1979, 66, 967–977. [Google Scholar] [CrossRef]
- Correia, L.C.; Lakatta, E.G.; O’Connor, F.C.; Becker, L.C.; Clulow, J.; Townsend, S.; Gerstenblith, G.; Fleg, J.L. Attenuated cardiovascular reserve during prolonged submaximal cycle exercise in healthy older subjects. J. Am. Coll. Cardiol. 2002, 40, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Akins, C.W.; Daggett, W.M.; Vlahakes, G.J.; Hilgenberg, A.D.; Torchiana, D.F.; Madsen, J.C.; Buckley, M.J. Cardiac operations in patients 80 years old and older. Ann. Thorac. Surg. 1997, 64, 606–614. [Google Scholar] [CrossRef]
- Kurz, D.J.; Kloeckener-Gruissem, B.; Akhmedov, A.; Eberli, F.R.; Buhler, I.; Berger, W.; Bertel, O.; Luscher, T.F. Degenerative aortic valve stenosis, but not coronary disease, is associated with shorter telomere length in the elderly. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 114–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ezquerro, J.D.; Rodriguez-Castaneda, A.; Ortiz-Ramirez, M.; Sanchez-Garcia, S.; Rosas-Vargas, H.; Sanchez-Arenas, R.; Garcia-de la Torre, P. Oxidative Stress, Telomere Length, and Frailty in an Old Age Population. bioRxiv 2019, 71, 414680. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.I.; Sakuma, I.; Sohn, I.S.; Jo, S.H.; Koh, K.K. Inflammatory and metabolic mechanisms underlying the calcific aortic valve disease. Atherosclerosis 2018, 277, 60–65. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 715–723. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, R.A.; Kolodgie, F.; Ravandi, A.; Leibundgut, G.; Hu, P.P.; Prasad, A.; Mahmud, E.; Dennis, E.; Curtiss, L.K.; Witztum, J.L.; et al. Differential expression of oxidation-specific epitopes and apolipoprotein(a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. J. Lipid Res. 2012, 53, 2773–2790. [Google Scholar] [CrossRef] [Green Version]
- Ravandi, A.; Leibundgut, G.; Hung, M.Y.; Patel, M.; Hutchins, P.M.; Murphy, R.C.; Prasad, A.; Mahmud, E.; Miller, Y.I.; Dennis, E.A.; et al. Release and capture of bioactive oxidized phospholipids and oxidized cholesteryl esters during percutaneous coronary and peripheral arterial interventions in humans. J. Am. Coll. Cardiol. 2014, 63, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Cote, C.; Pibarot, P.; Despres, J.P.; Mohty, D.; Cartier, A.; Arsenault, B.J.; Couture, C.; Mathieu, P. Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart 2008, 94, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Mohty, D.; Pibarot, P.; Despres, J.P.; Cote, C.; Arsenault, B.; Cartier, A.; Cosnay, P.; Couture, C.; Mathieu, P. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Ao, L.; Song, Y.; Babu, A.; Yang, X.; Wang, M.; Weyant, M.J.; Dinarello, C.A.; Cleveland, J.C., Jr.; Fullerton, D.A. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: Potential roles in aortic valve inflammation and stenosis. Am. J. Physiol. Cell Physiol. 2008, 294, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Fullerton, D.A.; Su, X.; Ao, L.; Cleveland, J.C., Jr.; Meng, X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J. Am. Coll. Cardiol. 2009, 53, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Yeang, C.; Wilkinson, M.J.; Tsimikas, S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr. Opin. Cardiol. 2016, 31, 440–450. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, G.; Sandhya Rani, M.R.; Gerber, C.E.; Mukhopadhyay, C.; Ransohoff, R.M.; Chisolm, G.M.; Kottke-Marchant, K. Lysophosphatidylcholine regulates human microvascular endothelial cell expression of chemokines. J. Mol. Cell. Cardiol. 2003, 35, 1375–1384. [Google Scholar] [CrossRef]
- Chang, M.C.; Lee, J.J.; Chen, Y.J.; Lin, S.I.; Lin, L.D.; Jein-Wen Liou, E.; Huang, W.L.; Chan, C.P.; Huang, C.C.; Jeng, J.H. Lysophosphatidylcholine induces cytotoxicity/apoptosis and IL-8 production of human endothelial cells: Related mechanisms. Oncotarget 2017, 8, 106177–106189. [Google Scholar] [CrossRef] [Green Version]
- Rolin, J.; Vego, H.; Maghazachi, A.A. Oxidized lipids and lysophosphatidylcholine induce the chemotaxis, up-regulate the expression of CCR9 and CXCR4 and abrogate the release of IL-6 in human monocytes. Toxins 2014, 6, 2840–2856. [Google Scholar] [CrossRef] [Green Version]
- Inoue, N.; Takeshita, S.; Gao, D.; Ishida, T.; Kawashima, S.; Akita, H.; Tawa, R.; Sakurai, H.; Yokoyama, M. Lysophosphatidylcholine increases the secretion of matrix metalloproteinase 2 through the activation of NADH/NADPH oxidase in cultured aortic endothelial cells. Atherosclerosis 2001, 155, 45–52. [Google Scholar] [CrossRef]
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Pena-Silva, R.; Heistad, D.D. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J. Am. Coll. Cardiol. 2008, 52, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Forstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, 13, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Milstien, S.; Katusic, Z. Oxidation of tetrahydrobiopterin by peroxynitrite: Implications for vascular endothelial function. Biochem. Biophys. Res. Commun. 1999, 263, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Rueckschloss, U.; Galle, J.; Holtz, J.; Zerkowski, H.R.; Morawietz, H. Induction of NAD(P)H oxidase by oxidized low-density lipoprotein in human endothelial cells: Antioxidative potential of hydroxymethylglutaryl coenzyme A reductase inhibitor therapy. Circulation 2001, 104, 1767–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liochev, S.I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 2013, 60, 1–4. [Google Scholar] [CrossRef]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Favero, G.; Paganelli, C.; Buffoli, B.; Rodella, L.F.; Rezzani, R. Endothelium and its alterations in cardiovascular diseases: Life style intervention. Biomed. Res. Int. 2014, 2014, 801896. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.R.; Seals, D.R.; Connell, M.L.; Russell, M.J.; Lawson, B.R.; Folian, B.J.; Donato, A.J.; Lesniewski, L.A. Voluntary wheel running restores endothelial function in conduit arteries of old mice: Direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down-regulation of NADPH oxidase. J. Physiol. 2009, 587, 3271–3285. [Google Scholar] [CrossRef]
- Pierce, G.L.; Lesniewski, L.A.; Lawson, B.R.; Beske, S.D.; Seals, D.R. Nuclear factor-κB activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation 2009, 119, 1284–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dichtl, W.; Dulak, J.; Frick, M.; Alber, H.F.; Schwarzacher, S.P.; Ares, M.P.; Nilsson, J.; Pachinger, O.; Weidinger, F. HMG-CoA reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, A.J.; Pierce, G.L.; Lesniewski, L.A.; Seals, D.R. Role of NFkappaB in age-related vascular endothelial dysfunction in humans. Aging (Albany N. Y.) 2009, 1, 678–680. [Google Scholar] [CrossRef]
- Anrather, J.; Racchumi, G.; Iadecola, C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 2006, 281, 5657–5667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seals, D.R.; Moreau, K.L.; Gates, P.E.; Eskurza, I. Modulatory influences on ageing of the vasculature in healthy humans. Exp. Gerontol. 2006, 41, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Froehlich, J.; Galis, Z.S.; Lakatta, E.G. Increased expression of matrix metalloproteinase-2 in the thickened intima of aged rats. Hypertension 1999, 33, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Denver, R.J.; Licht, P. Modulation of neuropeptide-stimulated pituitary hormone secretion in hatchling turtles. Gen. Comp. Endocrinol. 1990, 77, 107–115. [Google Scholar] [CrossRef]
- Chung, A.W.; Booth, A.D.; Rose, C.; Thompson, C.R.; Levin, A.; van Breemen, C. Increased matrix metalloproteinase 2 activity in the human internal mammary artery is associated with ageing, hypertension, diabetes and kidney dysfunction. J. Vasc. Res. 2008, 45, 357–362. [Google Scholar] [CrossRef]
- McNulty, M.; Mahmud, A.; Spiers, P.; Feely, J. Collagen type-I degradation is related to arterial stiffness in hypertensive and normotensive subjects. J. Hum. Hypertens. 2006, 20, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.H.; Li, J.; Su, C.; Yang, Z.; Chen, L.; Wu, F.; Zhang, Y.Y.; Yu, B.B.; Qiu, Y.X.; Wang, S.M.; et al. Physical exercise attenuates age-associated reduction in endothelium-reparative capacity of endothelial progenitor cells by increasing CXCR4/JAK-2 signaling in healthy men. Aging Cell 2012, 11, 111–119. [Google Scholar] [CrossRef]
- Thum, T.; Hoeber, S.; Froese, S.; Klink, I.; Stichtenoth, D.O.; Galuppo, P.; Jakob, M.; Tsikas, D.; Anker, S.D.; Poole-Wilson, P.A.; et al. Age-dependent impairment of endothelial progenitor cells is corrected by growth-hormone-mediated increase of insulin-like growth-factor-1. Circ. Res. 2007, 100, 434–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, J.; Vallejo, S.; El Assar, M.; Garcia-Septiem, J.; Sanchez-Ferrer, C.F.; Rodriguez-Manas, L. Age-related differences in the effects of alpha and gamma peroxisome proliferator-activated receptor subtype agonists on endothelial vasodilation in human microvessels. Exp. Gerontol. 2012, 47, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, M.; Shimokawa, H.; Otsuji, Y.; Ueta, Y.; Sasaguri, Y.; Yanagihara, N. Nitric oxide synthases and cardiovascular diseases: Insights from genetically modified mice. Circ. J. 2009, 73, 986–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Huang, A.; Yan, E.H.; Wu, Z.; Yan, C.; Kaminski, P.M.; Oury, T.D.; Wolin, M.S.; Kaley, G. Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, 2249–2256. [Google Scholar] [CrossRef] [Green Version]
- Challah, M.; Nadaud, S.; Philippe, M.; Battle, T.; Soubrier, F.; Corman, B.; Michel, J.B. Circulating and cellular markers of endothelial dysfunction with aging in rats. Am. J. Physiol. 1997, 273, 1941–1948. [Google Scholar] [CrossRef]
- Cernadas, M.R.; Sanchez de Miguel, L.; Garcia-Duran, M.; Gonzalez-Fernandez, F.; Millas, I.; Monton, M.; Rodrigo, J.; Rico, L.; Fernandez, P.; de Frutos, T.; et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ. Res. 1998, 83, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Versari, D.; Salvetti, A. Endothelium, aging, and hypertension. Curr. Hypertens. Rep. 2006, 8, 84–89. [Google Scholar] [CrossRef]
- Bakris, G.; Ali, W.; Parati, G. ACC/AHA Versus ESC/ESH on Hypertension Guidelines: JACC Guideline Comparison. J. Am. Coll. Cardiol. 2019, 73, 3018–3026. [Google Scholar] [CrossRef]
- Burnier, M.; Wuerzner, G. Hypertension. Rev. Med. Suisse 2017, 13, 61–65. [Google Scholar]
- Oparil, S.; Acelajado, M.C.; Bakris, G.L.; Berlowitz, D.R.; Cifkova, R.; Dominiczak, A.F.; Grassi, G.; Jordan, J.; Poulter, N.R.; Rodgers, A.; et al. Hypertension. Nat. Rev. Dis. Primers 2018, 4, 18014. [Google Scholar] [CrossRef] [Green Version]
- Pereira da Silva, A.; Matos, A.; Aguiar, L.; Ramos-Marques, N.; Ribeiro, R.; Gil, A.; Gorjao-Clara, J.; Bicho, M. Hypertension and longevity: Role of genetic polymorphisms in renin-angiotensin-aldosterone system and endothelial nitric oxide synthase. Mol. Cell Biochem. 2019, 455, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, J.A.; World Health Organization. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J. Hypertens. 2003, 21, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.B.; Baba, N.; Boesel, C.P. Cardiovascular abnormalities in progeria. Case report and review of the literature. Arch. Pathol. Lab. Med. 1981, 105, 384–386. [Google Scholar] [PubMed]
- Huveneers, S.; Daemen, M.J.; Hordijk, P.L. Between Rho(k) and a hard place: The relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ. Res. 2015, 116, 895–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, P.M.; Boutouyrie, P.; Cunha, P.; Kotsis, V.; Narkiewicz, K.; Parati, G.; Rietzschel, E.; Scuteri, A.; Laurent, S. Early vascular ageing in translation: From laboratory investigations to clinical applications in cardiovascular prevention. J. Hypertens. 2013, 31, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Van Bortel, L.M.; Laurent, S.; Boutouyrie, P.; Chowienczyk, P.; Cruickshank, J.K.; De Backer, T.; Filipovsky, J.; Huybrechts, S.; Mattace-Raso, F.U.; Protogerou, A.D.; et al. Expert consensus document on the measurement of aortic stiffness in daily practice using carotid-femoral pulse wave velocity. J. Hypertens. 2012, 30, 445–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsis, V.; Stabouli, S.; Karafillis, I.; Nilsson, P. Early vascular aging and the role of central blood pressure. J. Hypertens. 2011, 29, 1847–1853. [Google Scholar] [CrossRef]
- Iwaki, T.; Nagahashi, K.; Takano, K.; Suzuki-Inoue, K.; Kanayama, N.; Umemura, K.; Urano, T. Mutation in a highly conserved glycine residue in strand 5B of plasminogen activator inhibitor 1 causes polymerisation. Thromb. Haemost. 2017, 117, 860–869. [Google Scholar] [CrossRef]
- Guzik, T.J.; Mikolajczyk, T. In search of the T cell involved in hypertension and target organ damage. Hypertension 2014, 64, 224–226. [Google Scholar] [CrossRef] [Green Version]
- Mikolajczyk, T.P.; Nosalski, R.; Szczepaniak, P.; Budzyn, K.; Osmenda, G.; Skiba, D.; Sagan, A.; Wu, J.; Vinh, A.; Marvar, P.J.; et al. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J. 2016, 30, 1987–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheiermann, C.; Frenette, P.S.; Hidalgo, A. Regulation of leucocyte homeostasis in the circulation. Cardiovasc. Res. 2015, 107, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czesnikiewicz-Guzik, M.; Lee, W.W.; Cui, D.; Hiruma, Y.; Lamar, D.L.; Yang, Z.Z.; Ouslander, J.G.; Weyand, C.M.; Goronzy, J.J. T cell subset-specific susceptibility to aging. Clin. Immunol. 2008, 127, 107–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youn, J.C.; Yu, H.T.; Lim, B.J.; Koh, M.J.; Lee, J.; Chang, D.Y.; Choi, Y.S.; Lee, S.H.; Kang, S.M.; Jang, Y.; et al. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 2013, 62, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itani, H.A.; McMaster, W.G., Jr.; Saleh, M.A.; Nazarewicz, R.R.; Mikolajczyk, T.P.; Kaszuba, A.M.; Konior, A.; Prejbisz, A.; Januszewicz, A.; Norlander, A.E.; et al. Activation of Human T Cells in Hypertension: Studies of Humanized Mice and Hypertensive Humans. Hypertension 2016, 68, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yodoi, K.; Yamashita, T.; Sasaki, N.; Kasahara, K.; Emoto, T.; Matsumoto, T.; Kita, T.; Sasaki, Y.; Mizoguchi, T.; Sparwasser, T.; et al. Foxp3+ regulatory T cells play a protective role in angiotensin II-induced aortic aneurysm formation in mice. Hypertension 2015, 65, 889–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.; Yu, X.M.; Odorico, S.; Clark, M.; Jaskula-Sztul, R.; Schienebeck, C.M.; Kupcho, K.R.; Harrison, A.D.; Winston-McPherson, G.N.; Tang, W.; et al. Novel analogs targeting histone deacetylase suppress aggressive thyroid cancer cell growth and induce re-differentiation. Cancer Gene Ther. 2015, 22, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, G.R.; Vinh, A.; Guzik, T.J.; Sobey, C.G. Immune mechanisms of hypertension. Nat. Rev. Immunol. 2019, 19, 517–532. [Google Scholar] [CrossRef]
- Urbanski, K.; Ludew, D.; Filip, G.; Filip, M.; Sagan, A.; Szczepaniak, P.; Grudzien, G.; Sadowski, J.; Jasiewicz-Honkisz, B.; Sliwa, T.; et al. CD14(+)CD16(++) “nonclassical” monocytes are associated with endothelial dysfunction in patients with coronary artery disease. Thromb. Haemost. 2017, 117, 971–980. [Google Scholar] [CrossRef]
- Weber, C.; Shantsila, E.; Hristov, M.; Caligiuri, G.; Guzik, T.; Heine, G.H.; Hoefer, I.E.; Monaco, C.; Peter, K.; Rainger, E.; et al. Role and analysis of monocyte subsets in cardiovascular disease. Joint consensus document of the European Society of Cardiology (ESC) Working Groups “Atherosclerosis & Vascular Biology” and “Thrombosis”. Thromb. Haemost. 2016, 116, 626–637. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, T.; Ley, K. Monocyte trafficking across the vessel wall. Cardiovasc. Res. 2015, 107, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.P.; Vinh, A.; Tuck, K.L.; Sakkal, S.; Krishnan, S.M.; Chan, C.T.; Lieu, M.; Samuel, C.S.; Diep, H.; Kemp-Harper, B.K.; et al. M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Skiba, D.S.; Nosalski, R.; Mikolajczyk, T.P.; Siedlinski, M.; Rios, F.J.; Montezano, A.C.; Jawien, J.; Olszanecki, R.; Korbut, R.; Czesnikiewicz-Guzik, M.; et al. Anti-atherosclerotic effect of the angiotensin 1–7 mimetic AVE0991 is mediated by inhibition of perivascular and plaque inflammation in early atherosclerosis. Br. J. Pharmacol. 2017, 174, 4055–4069. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; Fernandez-Laso, V.; Sastre, C.; Llamas-Granda, P.; Egido, J.; Martin-Ventura, J.L.; Zalba, G.; Blanco-Colio, L.M. TWEAK/Fn14 interaction promotes oxidative stress through NADPH oxidase activation in macrophages. Cardiovasc. Res. 2015, 108, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.G.; Guzik, T.J. Macrophages come to mind as keys to cognitive decline. J. Clin. Investig. 2016, 126, 4393–4395. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes, D.; Poittevin, M.; Dere, E.; Broqueres-You, D.; Bonnin, P.; Benessiano, J.; Pocard, M.; Mariani, J.; Kubis, N.; Merkulova-Rainon, T.; et al. Hypertension accelerates the progression of Alzheimer-like pathology in a mouse model of the disease. Hypertension 2015, 65, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef]
- Hopps, E.; Lo Presti, R.; Caimi, G. Matrix Metalloproteases in Arterial Hypertension and their Trend after Antihypertensive Treatment. Kidney Blood Press. Res. 2017, 42, 347–357. [Google Scholar] [CrossRef]
- Bunbupha, S.; Pakdeechote, P.; Maneesai, P.; Prachaney, P.; Boonprom, P. Carthamus Tinctorius L. extract attenuates cardiac remodeling in L-NAME-induced hypertensive rats by inhibiting the NADPH oxidase-mediated TGF-beta1 and MMP-9 pathway. Ann. Anat. Anat. Anz. 2019, 222, 120–128. [Google Scholar] [CrossRef]
- Di Gregoli, K.; George, S.J.; Jackson, C.L.; Newby, A.C.; Johnson, J.L. Differential effects of tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2 on atherosclerosis and monocyte/macrophage invasion. Cardiovasc. Res. 2016, 109, 318–330. [Google Scholar] [CrossRef]
- Ma, Y.; Chiao, Y.A.; Clark, R.; Flynn, E.R.; Yabluchanskiy, A.; Ghasemi, O.; Zouein, F.; Lindsey, M.L.; Jin, Y.F. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc. Res. 2015, 106, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Stout, L.C.; Whorton, E.B., Jr.; Vaghela, M. Pathogenesis of diffuse intimal thickening (DIT) in non-human primate thoracic aortas. Atherosclerosis 1983, 47, 1–6. [Google Scholar] [CrossRef]
- Petersen-Jones, H.G.; Johnson, K.B.; Hitomi, K.; Tykocki, N.R.; Thompson, J.M.; Watts, S.W. Transglutaminase activity is decreased in large arteries from hypertensive rats compared with normotensive controls. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, 592–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhang, J.; Jiang, L.Q.; Spinetti, G.; Pintus, G.; Monticone, R.; Kolodgie, F.D.; Virmani, R.; Lakatta, E.G. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension 2007, 50, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Neves, K.B.; Nguyen Dinh Cat, A.; Lopes, R.A.; Rios, F.J.; Anagnostopoulou, A.; Lobato, N.S.; de Oliveira, A.M.; Tostes, R.C.; Montezano, A.C.; Touyz, R.M. Chemerin Regulates Crosstalk Between Adipocytes and Vascular Cells Through Nox. Hypertension 2015, 66, 657–666. [Google Scholar] [CrossRef]
- Krug, A.W.; Allenhofer, L.; Monticone, R.; Spinetti, G.; Gekle, M.; Wang, M.; Lakatta, E.G. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension 2010, 55, 1476–1483. [Google Scholar] [CrossRef] [Green Version]
- Spiers, J.P.; Kelso, E.J.; Siah, W.F.; Edge, G.; Song, G.; McDermott, B.J.; Hennessy, M. Alterations in vascular matrix metalloproteinase due to ageing and chronic hypertension: Effects of endothelin receptor blockade. J. Hypertens. 2005, 23, 1717–1724. [Google Scholar] [CrossRef]
- Park, J.B.; Schiffrin, E.L. Cardiac and vascular fibrosis and hypertrophy in aldosterone-infused rats: Role of endothelin-1. Am. J. Hypertens. 2002, 15, 164–169. [Google Scholar] [CrossRef] [Green Version]
- Weigert, C.; Brodbeck, K.; Klopfer, K.; Haring, H.U.; Schleicher, E.D. Angiotensin II induces human TGF-beta 1 promoter activation: Similarity to hyperglycaemia. Diabetologia 2002, 45, 890–898. [Google Scholar] [CrossRef] [Green Version]
- Carver, K.A.; Smith, T.L.; Gallagher, P.E.; Tallant, E.A. Angiotensin-(1-7) prevents angiotensin II-induced fibrosis in cremaster microvessels. Microcirculation 2015, 22, 19–27. [Google Scholar] [CrossRef]
- Ishidoya, S.; Morrissey, J.; McCracken, R.; Reyes, A.; Klahr, S. Angiotensin II receptor antagonist ameliorates renal tubulointerstitial fibrosis caused by unilateral ureteral obstruction. Kidney Int. 1995, 47, 1285–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ortega, M.; Gonzalez, S.; Seron, D.; Condom, E.; Bustos, C.; Largo, R.; Gonzalez, E.; Ortiz, A.; Egido, J. ACE inhibition reduces proteinuria, glomerular lesions and extracellular matrix production in a normotensive rat model of immune complex nephritis. Kidney Int. 1995, 48, 1778–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, M.R.; Frazier, K.S.; Abramson, S.; Williams, S.; Klapper, H.; Huang, X.; Grotendorst, G.R. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: Down-regulation by cAMP. FASEB J. 1999, 13, 1774–1786. [Google Scholar] [CrossRef] [Green Version]
- Sakurabayashi-Kitade, S.; Aoka, Y.; Nagashima, H.; Kasanuki, H.; Hagiwara, N.; Kawana, M. Aldosterone blockade by Spironolactone improves the hypertensive vascular hypertrophy and remodeling in angiotensin II overproducing transgenic mice. Atherosclerosis 2009, 206, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Callera, G.E.; Yogi, A.; Briones, A.M.; Montezano, A.C.; He, Y.; Tostes, R.C.; Schiffrin, E.L.; Touyz, R.M. Vascular proinflammatory responses by aldosterone are mediated via c-Src trafficking to cholesterol-rich microdomains: Role of PDGFR. Cardiovasc. Res. 2011, 91, 720–731. [Google Scholar] [CrossRef] [Green Version]
- Savoia, C.; Touyz, R.M.; Amiri, F.; Schiffrin, E.L. Selective mineralocorticoid receptor blocker eplerenone reduces resistance artery stiffness in hypertensive patients. Hypertension 2008, 51, 432–439. [Google Scholar] [CrossRef] [Green Version]
- Briones, A.M.; Nguyen Dinh Cat, A.; Callera, G.E.; Yogi, A.; Burger, D.; He, Y.; Correa, J.W.; Gagnon, A.M.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: Implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension 2012, 59, 1069–1078. [Google Scholar] [CrossRef]
- Horstmeyer, A.; Licht, C.; Scherr, G.; Eckes, B.; Krieg, T. Signalling and regulation of collagen I synthesis by ET-1 and TGF-beta1. FEBS J. 2005, 272, 6297–6309. [Google Scholar] [CrossRef]
- Hafizi, S.; Wharton, J.; Chester, A.H.; Yacoub, M.H. Profibrotic effects of endothelin-1 via the ETA receptor in cultured human cardiac fibroblasts. Cell Physiol. Biochem. 2004, 14, 285–292. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Sanada, F.; Taniyama, Y.; Muratsu, J.; Otsu, R.; Shimizu, H.; Rakugi, H.; Morishita, R. Gene-Therapeutic Strategies Targeting Angiogenesis in Peripheral Artery Disease. Medicines 2018, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Jakovljevic, D.G. Physical activity and cardiovascular aging: Physiological and molecular insights. Exp. Gerontol. 2018, 109, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.S.; Xanthakis, V.; Sullivan, L.M.; Lieb, W.; Aragam, J.; Redfield, M.M.; Mitchell, G.F.; Benjamin, E.J.; Vasan, R.S. Aortic root remodeling over the adult life course: Longitudinal data from the Framingham Heart Study. Circulation 2010, 122, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.W.; Markus, H.S.; Bots, M.L.; Rosvall, M.; Sitzer, M. Prediction of clinical cardiovascular events with carotid intima-media thickness: A systematic review and meta-analysis. Circulation 2007, 115, 459–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldschmidt-Clermont, P.J.; Dong, C.; Seo, D.M.; Velazquez, O.C. Atherosclerosis, inflammation, genetics, and stem cells: 2012 update. Curr. Atheroscler. Rep. 2012, 14, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Shen, H.; Schenten, D.; Shan, P.; Lee, P.J.; Goldstein, D.R. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Spagnoli, L.G.; Orlandi, A.; Mauriello, A.; Santeusanio, G.; de Angelis, C.; Lucreziotti, R.; Ramacci, M.T. Aging and atherosclerosis in the rabbit. 1. Distribution, prevalence and morphology of atherosclerotic lesions. Atherosclerosis 1991, 89, 11–24. [Google Scholar] [CrossRef]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puca, A.A.; Carrizzo, A.; Villa, F.; Ferrario, A.; Casaburo, M.; Maciag, A.; Vecchione, C. Vascular ageing: The role of oxidative stress. Int. J. Biochem. Cell. Biol. 2013, 45, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Kou, S.J.; Lin, W.T.; Liu, C.S. Regulatory role of mitochondria in oxidative stress and atherosclerosis. World J. Cardiol. 2010, 2, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Vinciguerra, M.; Longo, V.D. Growth factors, nutrient signaling, and cardiovascular aging. Circ. Res. 2012, 110, 1139–1150. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Ogura, Y.; Koya, D. The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging (Albany N. Y.) 2016, 8, 2290–2307. [Google Scholar] [CrossRef] [Green Version]
- Romano, A.D.; Serviddio, G.; de Matthaeis, A.; Bellanti, F.; Vendemiale, G. Oxidative stress and aging. J. Nephrol. 2010, 23, 29–36. [Google Scholar]
- Leon, A.S.; Rice, T.; Mandel, S.; Despres, J.P.; Bergeron, J.; Gagnon, J.; Rao, D.C.; Skinner, J.S.; Wilmore, J.H.; Bouchard, C. Blood lipid response to 20 weeks of supervised exercise in a large biracial population: The HERITAGE Family Study. Metabolism 2000, 49, 513–520. [Google Scholar] [CrossRef]
- Badireddy, M.; Mudipalli, V.R. Deep Venous Thrombosis Prophylaxis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Jimenez, D.; Bikdeli, B.; Quezada, A.; Muriel, A.; Lobo, J.L.; de Miguel-Diez, J.; Jara-Palomares, L.; Ruiz-Artacho, P.; Yusen, R.D.; Monreal, M.; et al. Hospital volume and outcomes for acute pulmonary embolism: Multinational population based cohort study. BMJ 2019, 366, l4416. [Google Scholar] [CrossRef] [Green Version]
- Vaqar, S.; Graber, M. Thromboembolic Event. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Price, C.P.; Fay, M.; Hopstaken, R.M. Point-of-Care Testing for D-Dimer in the Diagnosis of Venous Thromboembolism in Primary Care: A Narrative Review. Cardiol. Ther. 2020. [Google Scholar] [CrossRef]
- Stone, J.; Hangge, P.; Albadawi, H.; Wallace, A.; Shamoun, F.; Knuttien, M.G.; Naidu, S.; Oklu, R. Deep vein thrombosis: Pathogenesis, diagnosis, and medical management. Cardiovasc. Diagn. Ther. 2017, 7, 276–284. [Google Scholar] [CrossRef]
- Konstantinides, S.V.; Meyer, G.; Becattini, C.; Bueno, H.; Geersing, G.J.; Harjola, V.P.; Huisman, M.V.; Humbert, M.; Jennings, C.S.; Jimenez, D.; et al. 2019 ESC Guidelines for the diagnosis and management of acute pulmonary embolism developed in collaboration with the European Respiratory Society (ERS). Eur. Heart J. 2020, 41, 543–603. [Google Scholar] [CrossRef]
- Waheed, S.M.; Kudaravalli, P.; Hotwagner, D.T. Deep Vein Thrombosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Tsai, A.W.; Cushman, M.; Rosamond, W.D.; Heckbert, S.R.; Polak, J.F.; Folsom, A.R. Cardiovascular risk factors and venous thromboembolism incidence: The longitudinal investigation of thromboembolism etiology. Arch. Intern. Med. 2002, 162, 1182–1189. [Google Scholar] [CrossRef]
- Esmon, C.T. Basic mechanisms and pathogenesis of venous thrombosis. Blood Rev. 2009, 23, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Rumley, A.; Emberson, J.R.; Wannamethee, S.G.; Lennon, L.; Whincup, P.H.; Lowe, G.D. Effects of older age on fibrin D-dimer, C-reactive protein, and other hemostatic and inflammatory variables in men aged 60–79 years. J. Thromb. Haemost. 2006, 4, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, E.J.; Franchini, M.; Lippi, G. Aging hemostasis: Changes to laboratory markers of hemostasis as we age—A narrative review. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2014; Volume 40, pp. 621–633. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zennadi, R. Oxidative Stress and Thrombosis during Aging: The Roles of Oxidative Stress in RBCs in Venous Thrombosis. Int. J. Mol. Sci. 2020, 21, 4259. [Google Scholar] [CrossRef]
- Carrizzo, A.; Izzo, C.; Oliveti, M.; Alfano, A.; Virtuoso, N.; Capunzo, M.; Di Pietro, P.; Calabrese, M.; De Simone, E.; Sciarretta, S.; et al. The Main Determinants of Diabetes Mellitus Vascular Complications: Endothelial Dysfunction and Platelet Hyperaggregation. Int. J. Mol. Sci. 2018, 19, 2968. [Google Scholar] [CrossRef] [Green Version]
- Lopes, R.A.; Neves, K.B.; Tostes, R.C.; Montezano, A.C.; Touyz, R.M. Downregulation of Nuclear Factor Erythroid 2-Related Factor and Associated Antioxidant Genes Contributes to Redox-Sensitive Vascular Dysfunction in Hypertension. Hypertension 2015, 66, 1240–1250. [Google Scholar] [CrossRef] [Green Version]
- Csiszar, A.; Pacher, P.; Kaley, G.; Ungvari, Z. Role of oxidative and nitrosative stress, longevity genes and poly(ADP-ribose) polymerase in cardiovascular dysfunction associated with aging. Curr. Vasc. Pharmacol. 2005, 3, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaku, A.S.; Tadi, P. Cerebrovascular Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Writing Group, M.; Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, 38–360. [Google Scholar] [CrossRef]
- Panuganti, K.K.; Tadi, P.; Lui, F. Transient Ischemic Attack. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Di Carlo, A. Human and economic burden of stroke. Age Ageing 2009, 38, 4–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosamond, W.; Flegal, K.; Furie, K.; Go, A.; Greenlund, K.; Haase, N.; Hailpern, S.M.; Ho, M.; Howard, V.; Kissela, B.; et al. Heart disease and stroke statistics—2008 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008, 117, 25–146. [Google Scholar] [CrossRef] [Green Version]
- Navis, A.; Garcia-Santibanez, R.; Skliut, M. Epidemiology and Outcomes of Ischemic Stroke and Transient Ischemic Attack in the Adult and Geriatric Population. J. Stroke Cerebrovasc. Dis. 2019, 28, 84–89. [Google Scholar] [CrossRef]
- Tadi, P.; Lui, F. Acute Stroke. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Coull, A.J.; Silver, L.E.; Fairhead, J.F.; Giles, M.F.; Lovelock, C.E.; Redgrave, J.N.; Bull, L.M.; Welch, S.J.; Cuthbertson, F.C.; et al. Population-based study of event-rate, incidence, case fatality, and mortality for all acute vascular events in all arterial territories (Oxford Vascular Study). Lancet 2005, 366, 1773–1783. [Google Scholar] [CrossRef]
- Kandlur, A.; Satyamoorthy, K.; Gangadharan, G. Oxidative Stress in Cognitive and Epigenetic Aging: A Retrospective Glance. Front. Mol. Neurosci. 2020, 13, 41. [Google Scholar] [CrossRef] [Green Version]
- Baierle, M.; Nascimento, S.N.; Moro, A.M.; Brucker, N.; Freitas, F.; Gauer, B.; Durgante, J.; Bordignon, S.; Zibetti, M.; Trentini, C.M.; et al. Relationship between inflammation and oxidative stress and cognitive decline in the institutionalized elderly. Oxidative Med. Cell. Longev. 2015, 2015, 804198. [Google Scholar] [CrossRef] [Green Version]
- Ueno, M.; Tomimoto, H.; Akiguchi, I.; Wakita, H.; Sakamoto, H. Blood-brain barrier disruption in white matter lesions in a rat model of chronic cerebral hypoperfusion. J. Cereb. Blood Flow Metab. 2002, 22, 97–104. [Google Scholar] [CrossRef]
- Shao, A.; Lin, D.; Wang, L.; Tu, S.; Lenahan, C.; Zhang, J. Oxidative Stress at the Crossroads of Aging, Stroke and Depression. Aging Dis. 2020, 11, 1537–1566. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Eid, A.H. Determination of Vascular Reactivity of Middle Cerebral Arteries from Stroke and Spinal Cord Injury Animal Models Using Pressure Myography. Methods Mol. Biol. 2016, 1462, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Zitnanova, I.; Siarnik, P.; Kollar, B.; Chomova, M.; Pazderova, P.; Andrezalova, L.; Jezovicova, M.; Konarikova, K.; Laubertova, L.; Krivosikova, Z.; et al. Oxidative Stress Markers and Their Dynamic Changes in Patients after Acute Ischemic Stroke. Oxidative Med. Cell. Longev. 2016, 2016, 9761697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Cui, Y.; Meng, X.; Su, Z.; Cao, Y.; Yang, Y.; Liu, C.; Dai, L.; Wang, Y.; Wang, Y. The relationship between oxidized low-density lipoprotein and the NIHSS score among patients with acute ischemic stroke: The SOS-Stroke Study. Atherosclerosis 2018, 270, 21–25. [Google Scholar] [CrossRef]
- Wang, A.; Yang, Y.; Su, Z.; Yue, W.; Hao, H.; Ren, L.; Wang, Y.; Cao, Y.; Wang, Y. Association of Oxidized Low-Density Lipoprotein With Prognosis of Stroke and Stroke Subtypes. Stroke 2017, 48, 91–97. [Google Scholar] [CrossRef]
- Wang, A.; Liu, J.; Meng, X.; Li, J.; Wang, H.; Wang, Y.; Su, Z.; Zhang, N.; Dai, L.; Wang, Y.; et al. Association between oxidized low-density lipoprotein and cognitive impairment in patients with ischemic stroke. Eur. J. Neurol. 2018, 25, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, A.R.; Larrick, J.W. The NAD+/PARP1/SIRT1 Axis in Aging. Rejuvenation Res. 2017, 20, 244–247. [Google Scholar] [CrossRef]
- Ham, P.B., 3rd; Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol. 2017, 157, 92–116. [Google Scholar] [CrossRef]
- Rodrigo, R.; Fernandez-Gajardo, R.; Gutierrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeutic opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714. [Google Scholar] [CrossRef]
- Tang, Z.; Li, M.; Zhang, X.; Hou, W. Dietary flavonoid intake and the risk of stroke: A dose-response meta-analysis of prospective cohort studies. BMJ Open 2016, 6, e008680. [Google Scholar] [CrossRef]
- Targum, S.D.; Wedel, P.C.; Fava, M. Changes in cognitive symptoms after a buspirone-melatonin combination treatment for Major Depressive Disorder. J. Psychiatr. Res. 2015, 68, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Su, X.T.; Wang, L.; Ma, S.M.; Cao, Y.; Yang, N.N.; Lin, L.L.; Fisher, M.; Yang, J.W.; Liu, C.Z. Mechanisms of Acupuncture in the Regulation of Oxidative Stress in Treating Ischemic Stroke. Oxidative Med. Cell. Longev. 2020, 2020, 7875396. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruzen, C.; Colman, R.J. Effects of caloric restriction on cardiovascular aging in non-human primates and humans. Clin. Geriatr. Med. 2009, 25, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.G.; Chiasson, K.B.; Leslie, T.H.; Thomas, D.; Beasley, T.M.; Kemnitz, J.W.; Weindruch, R. Auditory function in rhesus monkeys: Effects of aging and caloric restriction in the Wisconsin monkeys five years later. Hear. Res. 2010, 261, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Kastman, E.K.; Willette, A.A.; Coe, C.L.; Bendlin, B.B.; Kosmatka, K.J.; McLaren, D.G.; Xu, G.; Canu, E.; Field, A.S.; Alexander, A.L.; et al. A calorie-restricted diet decreases brain iron accumulation and preserves motor performance in old rhesus monkeys. J. Neurosci. 2010, 30, 7940–7947. [Google Scholar] [CrossRef] [Green Version]
- McKiernan, S.H.; Colman, R.J.; Lopez, M.; Beasley, T.M.; Aiken, J.M.; Anderson, R.M.; Weindruch, R. Caloric restriction delays aging-induced cellular phenotypes in rhesus monkey skeletal muscle. Exp. Gerontol. 2011, 46, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Niemann, B.; Chen, Y.; Issa, H.; Silber, R.E.; Rohrbach, S. Caloric restriction delays cardiac ageing in rats: Role of mitochondria. Cardiovasc. Res. 2010, 88, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Shinmura, K.; Tamaki, K.; Sano, M.; Murata, M.; Yamakawa, H.; Ishida, H.; Fukuda, K. Impact of long-term caloric restriction on cardiac senescence: Caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. J. Mol. Cell. Cardiol. 2011, 50, 117–127. [Google Scholar] [CrossRef]
- Varady, K.A.; Hellerstein, M.K. Alternate-day fasting and chronic disease prevention: A review of human and animal trials. Am. J. Clin. Nutr. 2007, 86, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Palacios, O.M.; Carmona, J.J.; Michan, S.; Chen, K.Y.; Manabe, Y.; Ward, J.L., 3rd; Goodyear, L.J.; Tong, Q. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging (Albany N. Y.) 2009, 1, 771–783. [Google Scholar] [CrossRef]
- Goswami, S.K.; Das, D.K. Autophagy in the myocardium: Dying for survival? Exp. Clin. Cardiol. 2006, 11, 183–188. [Google Scholar] [PubMed]
- Wohlgemuth, S.E.; Julian, D.; Akin, D.E.; Fried, J.; Toscano, K.; Leeuwenburgh, C.; Dunn, W.A., Jr. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007, 10, 281–292. [Google Scholar] [CrossRef]
- Saifutdinov, R.G.; Trifonova, E.V. Investigation on mice of the common toxicity of methyl-tert-butyl ether, substance for contact chemical litholys of cholesterol stones in a gallbladder. Exp. Clin. Gastroenterol. 2011, 4, 70–73. [Google Scholar]
- Shinmura, K.; Tamaki, K.; Bolli, R. Impact of 6-mo caloric restriction on myocardial ischemic tolerance: Possible involvement of nitric oxide-dependent increase in nuclear Sirt1. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, 2348–2355. [Google Scholar] [CrossRef] [Green Version]
- Hepple, R.T.; Baker, D.J.; McConkey, M.; Murynka, T.; Norris, R. Caloric restriction protects mitochondrial function with aging in skeletal and cardiac muscles. Rejuvenation Res. 2006, 9, 219–222. [Google Scholar] [CrossRef]
- Rohrbach, S.; Niemann, B.; Abushouk, A.M.; Holtz, J. Caloric restriction and mitochondrial function in the ageing myocardium. Exp. Gerontol. 2006, 41, 525–531. [Google Scholar] [CrossRef]
- Kris-Etherton, P.M.; Lichtenstein, A.H.; Howard, B.V.; Steinberg, D.; Witztum, J.L.; Nutrition Committee of the American Heart Association Council on Nutrition, Physical Activity, and Metabolism. Antioxidant vitamin supplements and cardiovascular disease. Circulation 2004, 110, 637–641. [Google Scholar] [CrossRef]
- Martinez Gonzalez, J.M.; Grana Gomez, J.L.; Trujillo Mendoza, H. Longitudinal study on quality of life, craving and psychological adjustment in alcohol-dependent patients: Variations depending on the personality disorders. Adicciones 2011, 23, 227–235. [Google Scholar]
- Lennox, S.D.; Mulkerrin, E.; Penney, M.; Woodhouse, K. Plasma atrial natriuretic polypeptide in the elderly: Age or hypertension? Arch. Gerontol. Geriatr. 1994, 19, 1–5. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Steinhubl, S.R. Why have antioxidants failed in clinical trials? Am. J. Cardiol. 2008, 101, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Scott, E.; Brown, V.A.; Gescher, A.J.; Steward, W.P.; Brown, K. Clinical trials of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Miranda, A.; Vergara, L. Modulation of endogenous antioxidant system by wine polyphenols in human disease. Clin. Chim. Acta 2011, 412, 410–424. [Google Scholar] [CrossRef]
- van Empel, V.P.; Bertrand, A.T.; van Oort, R.J.; van der Nagel, R.; Engelen, M.; van Rijen, H.V.; Doevendans, P.A.; Crijns, H.J.; Ackerman, S.L.; Sluiter, W.; et al. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J. Am. Coll. Cardiol. 2006, 48, 824–832. [Google Scholar] [CrossRef] [Green Version]
- Medina-Remon, A.; Zamora-Ros, R.; Rotches-Ribalta, M.; Andres-Lacueva, C.; Martinez-Gonzalez, M.A.; Covas, M.I.; Corella, D.; Salas-Salvado, J.; Gomez-Gracia, E.; Ruiz-Gutierrez, V.; et al. Total polyphenol excretion and blood pressure in subjects at high cardiovascular risk. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 323–331. [Google Scholar] [CrossRef]
- Bakker, G.C.; van Erk, M.J.; Pellis, L.; Wopereis, S.; Rubingh, C.M.; Cnubben, N.H.; Kooistra, T.; van Ommen, B.; Hendriks, H.F. An antiinflammatory dietary mix modulates inflammation and oxidative and metabolic stress in overweight men: A nutrigenomics approach. Am. J. Clin. Nutr. 2010, 91, 1044–1059. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, V.; Roy, A.; Bharadvaja, N. Current Prospects of Nutraceuticals: A Review. Curr. Pharm. Biotechnol. 2020, 21, 884–896. [Google Scholar] [CrossRef]
- Carrizzo, A.; Moltedo, O.; Damato, A.; Martinello, K.; Di Pietro, P.; Oliveti, M.; Acernese, F.; Giugliano, G.; Izzo, R.; Sommella, E.; et al. New Nutraceutical Combination Reduces Blood Pressure and Improves Exercise Capacity in Hypertensive Patients Via a Nitric Oxide-Dependent Mechanism. J. Am. Heart Assoc. 2020, 9, e014923. [Google Scholar] [CrossRef]
- Santini, A.; Cammarata, S.M.; Capone, G.; Ianaro, A.; Tenore, G.C.; Pani, L.; Novellino, E. Nutraceuticals: Opening the debate for a regulatory framework. Br. J. Clin. Pharmacol. 2018, 84, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Helal, N.A.; Eassa, H.A.; Amer, A.M.; Eltokhy, M.A.; Edafiogho, I.; Nounou, M.I. Nutraceuticals’ Novel Formulations: The Good, the Bad, the Unknown and Patents Involved. Recent Pat. Drug Deliv. Formul. 2019, 13, 105–156. [Google Scholar] [CrossRef] [PubMed]
- Sauer, S.; Plauth, A. Health-beneficial nutraceuticals-myth or reality? Appl. Microbiol. Biotechnol. 2017, 101, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Biesalski, H.K.; Tinz, J. Multivitamin/mineral supplements: Rationale and safety—A systematic review. Nutrition 2017, 33, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Carrizzo, A.; Izzo, C.; Forte, M.; Sommella, E.; Di Pietro, P.; Venturini, E.; Ciccarelli, M.; Galasso, G.; Rubattu, S.; Campiglia, P.; et al. A Novel Promising Frontier for Human Health: The Beneficial Effects of Nutraceuticals in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 8706. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Boeing, H.; Stelmach-Mardas, M.; Gottschald, M.; Dietrich, S.; Hoffmann, G.; Chaimani, A. Dietary Supplements and Risk of Cause-Specific Death, Cardiovascular Disease, and Cancer: A Systematic Review and Meta-Analysis of Primary Prevention Trials. Adv. Nutr. 2017, 8, 27–39. [Google Scholar] [CrossRef]
- Carrizzo, A.; Forte, M.; Damato, A.; Trimarco, V.; Salzano, F.; Bartolo, M.; Maciag, A.; Puca, A.A.; Vecchione, C. Antioxidant effects of resveratrol in cardiovascular, cerebral and metabolic diseases. Food Chem. Toxicol. 2013, 61, 215–226. [Google Scholar] [CrossRef]
- Carrizzo, A.; Puca, A.; Damato, A.; Marino, M.; Franco, E.; Pompeo, F.; Traficante, A.; Civitillo, F.; Santini, L.; Trimarco, V.; et al. Resveratrol improves vascular function in patients with hypertension and dyslipidemia by modulating NO metabolism. Hypertension 2013, 62, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Barger, J.L.; Kayo, T.; Vann, J.M.; Arias, E.B.; Wang, J.; Hacker, T.A.; Wang, Y.; Raederstorff, D.; Morrow, J.D.; Leeuwenburgh, C.; et al. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE 2008, 3, e2264. [Google Scholar] [CrossRef]
- de Cavanagh, E.M.; Piotrkowski, B.; Fraga, C.G. Concerted action of the renin-angiotensin system, mitochondria, and antioxidant defenses in aging. Mol. Asp. Med. 2004, 25, 27–36. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Gaddam, R.R.; Chambers, S.; Bhatia, M. ACE and ACE2 in inflammation: A tale of two enzymes. Inflamm. Allergy Drug Targets 2014, 13, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Waugh, W.J. Factors to consider in selecting an angiotensin-converting-enzyme inhibitor. Am. J. Health Syst. Pharm. 2000, 57, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Simões, E.; Silva, A.C.; Teixeira, M.M. ACE inhibition, ACE2 and angiotensin-(1-7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol. Res. 2016, 107, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Williams, B. The renin angiotensin system and cardiovascular disease: Hope or hype? J. Renin Angiotensin Aldosterone Syst. 2000, 1, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Vanhoutte, P.M.; Boulanger, C.M.; Illiano, S.C.; Nagao, T.; Vidal, M.; Mombouli, J.V. Endothelium-dependent effects of converting-enzyme inhibitors. J. Cardiovasc. Pharmacol. 1993, 22, 10–16. [Google Scholar] [CrossRef]
- Xu, Y.; Clanachan, A.S.; Jugdutt, B.I. Enhanced expression of angiotensin II type 2 receptor, inositol 1,4, 5-trisphosphate receptor, and protein kinase cepsilon during cardioprotection induced by angiotensin II type 2 receptor blockade. Hypertension 2000, 36, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Kees, F.; Bucher, M.; Schweda, F.; Gschaidmeier, H.; Faerber, L.; Seifert, R. Neoimmun versus Neoral: A bioequivalence study in healthy volunteers and influence of a fat-rich meal on the bioavailability of Neoimmun. Naunyn Schmiedeberg’s Arch. Pharmacol. 2007, 375, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Rueckschloss, U.; Quinn, M.T.; Holtz, J.; Morawietz, H. Dose-dependent regulation of NAD(P)H oxidase expression by angiotensin II in human endothelial cells: Protective effect of angiotensin II type 1 receptor blockade in patients with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1845–1851. [Google Scholar] [CrossRef] [Green Version]
- Mira, M.L.; Silva, M.M.; Queiroz, M.J.; Manso, C.F. Angiotensin converting enzyme inhibitors as oxygen free radical scavengers. Free Radic. Res. Commun. 1993, 19, 173–181. [Google Scholar] [CrossRef]
- Chopra, M.; Beswick, H.; Clapperton, M.; Dargie, H.J.; Smith, W.E.; McMurray, J. Antioxidant effects of angiotensin-converting enzyme (ACE) inhibitors: Free radical and oxidant scavenging are sulfhydryl dependent, but lipid peroxidation is inhibited by both sulfhydryl- and nonsulfhydryl-containing ACE inhibitors. J. Cardiovasc. Pharmacol. 1992, 19, 330–340. [Google Scholar] [CrossRef]
- Bagchi, D.; Prasad, R.; Das, D.K. Direct scavenging of free radicals by captopril, an angiotensin converting enzyme inhibitor. Biochem. Biophys Res. Commun. 1989, 158, 52–57. [Google Scholar] [CrossRef]
- de Cavanagh, E.M.; Inserra, F.; Toblli, J.; Stella, I.; Fraga, C.G.; Ferder, L. Enalapril attenuates oxidative stress in diabetic rats. Hypertension 2001, 38, 1130–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Cavanagh, E.M.; Inserra, F.; Ferder, L.; Romano, L.; Ercole, L.; Fraga, C.G. Superoxide dismutase and glutathione peroxidase activities are increased by enalapril and captopril in mouse liver. FEBS Lett. 1995, 361, 22–24. [Google Scholar] [CrossRef] [Green Version]
- de Cavanagh, E.M.; Inserra, F.; Ferder, L.; Fraga, C.G. Enalapril and captopril enhance glutathione-dependent antioxidant defenses in mouse tissues. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, 572–577. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. Oxidative stress: Biomarkers and novel therapeutic pathways. Exp. Gerontol. 2010, 45, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. From rapalogs to anti-aging formula. Oncotarget 2017, 8, 35492–35507. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.K.; Kim, H.H. The functions of mTOR in ischemic diseases. BMB Rep. 2011, 44, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Humar, R.; Kiefer, F.N.; Berns, H.; Resink, T.J.; Battegay, E.J. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002, 16, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Chong, Z.Z.; Shang, Y.C.; Maiese, K. Cardiovascular disease and mTOR signaling. Trends Cardiovasc. Med. 2011, 21, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Chong, Z.Z.; Hou, J.; Shang, Y.C.; Wang, S.; Maiese, K. EPO relies upon novel signaling of Wnt1 that requires Akt1, FoxO3a, GSK-3β, and β-catenin to foster vascular integrity during experimental diabetes. Curr. Neurovasc. Res. 2011, 8, 103–120. [Google Scholar] [CrossRef]
- Dormond, O.; Madsen, J.C.; Briscoe, D.M. The effects of mTOR-Akt interactions on anti-apoptotic signaling in vascular endothelial cells. J. Biol. Chem. 2007, 282, 23679–23686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiese, K.; Li, F.; Chong, Z.Z.; Shang, Y.C. The Wnt signaling pathway: Aging gracefully as a protectionist? Pharmacol. Ther. 2008, 118, 58–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigneron, F.; Dos Santos, P.; Lemoine, S.; Bonnet, M.; Tariosse, L.; Couffinhal, T.; Duplaà, C.; Jaspard-Vinassa, B. GSK-3β at the crossroads in the signalling of heart preconditioning: Implication of mTOR and Wnt pathways. Cardiovasc. Res. 2011, 90, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Liu, Y.; Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2009, 2, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easley, C.A.; Ben-Yehudah, A.; Redinger, C.J.; Oliver, S.L.; Varum, S.T.; Eisinger, V.M.; Carlisle, D.L.; Donovan, P.J.; Schatten, G.P. mTOR-mediated activation of p70 S6K induces differentiation of pluripotent human embryonic stem cells. Cell Reprogram. 2010, 12, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Das, J.; Ghosh, J.; Manna, P.; Sil, P.C. Taurine suppresses doxorubicin-triggered oxidative stress and cardiac apoptosis in rat via up-regulation of PI3-K/Akt and inhibition of p53, p38-JNK. Biochem. Pharmacol. 2011, 81, 891–909. [Google Scholar] [CrossRef]
- Gangloff, Y.G.; Mueller, M.; Dann, S.G.; Svoboda, P.; Sticker, M.; Spetz, J.F.; Um, S.H.; Brown, E.J.; Cereghini, S.; Thomas, G.; et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell Biol. 2004, 24, 9508–9516. [Google Scholar] [CrossRef] [Green Version]
- Földes, G.; Mioulane, M.; Wright, J.S.; Liu, A.Q.; Novak, P.; Merkely, B.; Gorelik, J.; Schneider, M.D.; Ali, N.N.; Harding, S.E. Modulation of human embryonic stem cell-derived cardiomyocyte growth: A testbed for studying human cardiac hypertrophy? J. Mol. Cell. Cardiol. 2011, 50, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Hernández, G.; Lal, H.; Fidalgo, M.; Guerrero, A.; Zalvide, J.; Force, T.; Pombo, C.M. A novel cardioprotective p38-MAPK/mTOR pathway. Exp. Cell Res. 2011, 317, 2938–2949. [Google Scholar] [CrossRef] [Green Version]
- Jaber, N.; Dou, Z.; Chen, J.S.; Catanzaro, J.; Jiang, Y.P.; Ballou, L.M.; Selinger, E.; Ouyang, X.; Lin, R.Z.; Zhang, J.; et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. USA 2012, 109, 2003–2008. [Google Scholar] [CrossRef] [Green Version]
- Lajoie, C.; El-Helou, V.; Proulx, C.; Clément, R.; Gosselin, H.; Calderone, A. Infarct size is increased in female post-MI rats treated with rapamycin. Can. J. Physiol. Pharmacol. 2009, 87, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Contu, R.; Latronico, M.V.; Zhang, J.; Zhang, J.L.; Rizzi, R.; Catalucci, D.; Miyamoto, S.; Huang, K.; Ceci, M.; et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Investig. 2010, 120, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Lemaître, V.; Dabo, A.J.; D’Armiento, J. Cigarette smoke components induce matrix metalloproteinase-1 in aortic endothelial cells through inhibition of mTOR signaling. Toxicol. Sci. 2011, 123, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Miriuka, S.G.; Rao, V.; Peterson, M.; Tumiati, L.; Delgado, D.H.; Mohan, R.; Ramzy, D.; Stewart, D.; Ross, H.J.; Waddell, T.K. mTOR inhibition induces endothelial progenitor cell death. Am. J. Transplant. 2006, 6, 2069–2079. [Google Scholar] [CrossRef]
- Leelahavanichkul, A.; Areepium, N.; Vadcharavivad, S.; Praditpornsilpa, K.; Avihingsanon, Y.; Karnjanabuchmd, T.; Eiam-Ong, S.; Tungsanga, K. Pharmacokinetics of sirolimus in Thai healthy volunteers. J. Med. Assoc. Thail. 2005, 88, 157–162. [Google Scholar]
- Singh, M.; Jensen, M.D.; Lerman, A.; Kushwaha, S.; Rihal, C.S.; Gersh, B.J.; Behfar, A.; Tchkonia, T.; Thomas, R.J.; Lennon, R.J.; et al. Effect of Low-Dose Rapamycin on Senescence Markers and Physical Functioning in Older Adults with Coronary Artery Disease: Results of a Pilot Study. J. Frailty Aging 2016, 5, 204–207. [Google Scholar] [CrossRef]
- Krebs, M.; Brunmair, B.; Brehm, A.; Artwohl, M.; Szendroedi, J.; Nowotny, P.; Roth, E.; Fürnsinn, C.; Promintzer, M.; Anderwald, C.; et al. The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes 2007, 56, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Stower, H. Senolytic CAR T cells. Nat. Med. 2020, 26, 1009. [Google Scholar] [CrossRef]
- Hajdu, K.L.; Bonamino, M.H. Senolytic chimeric antigen receptor (CAR) T cell: Driving the immune system to fight cell senescence. Immunol. Cell Biol. 2020, 98, 709–711. [Google Scholar] [CrossRef]
- Ravichandra, A.; Filliol, A.; Schwabe, R.F. CAR-T cells as senolytic and antifibrotic therapy. Hepatology 2020. [Google Scholar] [CrossRef]
- Li, J.H.; Chen, Y.Y. A Fresh Approach to Targeting Aging Cells: CAR-T Cells Enhance Senolytic Specificity. Cell Stem Cell 2020, 27, 192–194. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Senolytic CAR T Cells Targeting uPAR Treat Lung Cancer in Mice. Cancer Discov. 2020, 10, 1093. [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the Global Burden of Cardiovascular Disease, Part 1: The Epidemiology and Risk Factors. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef]
- Paneni, F.; Diaz Canestro, C.; Libby, P.; Luscher, T.F.; Camici, G.G. The Aging Cardiovascular System: Understanding It at the Cellular and Clinical Levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef]



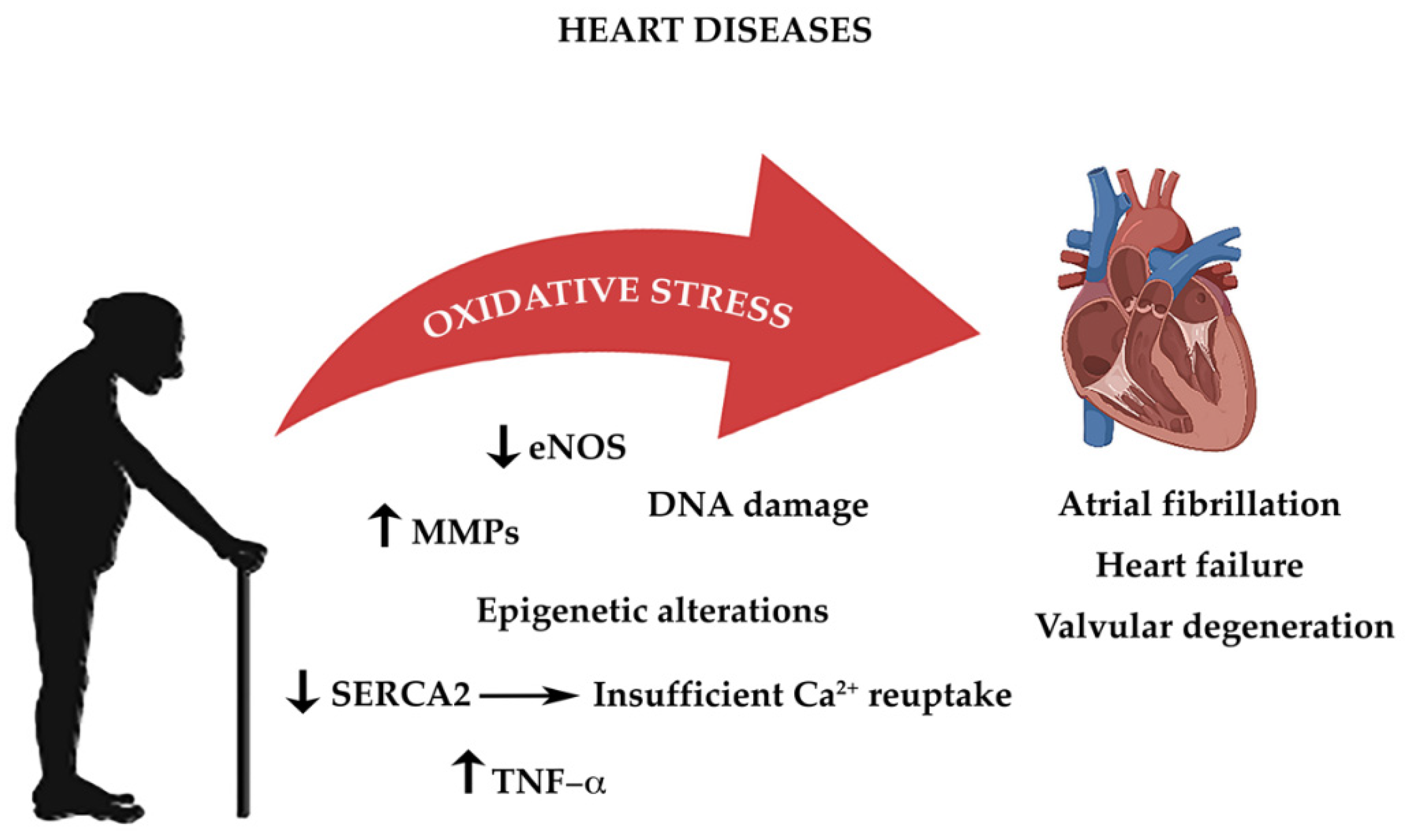{kind=link}
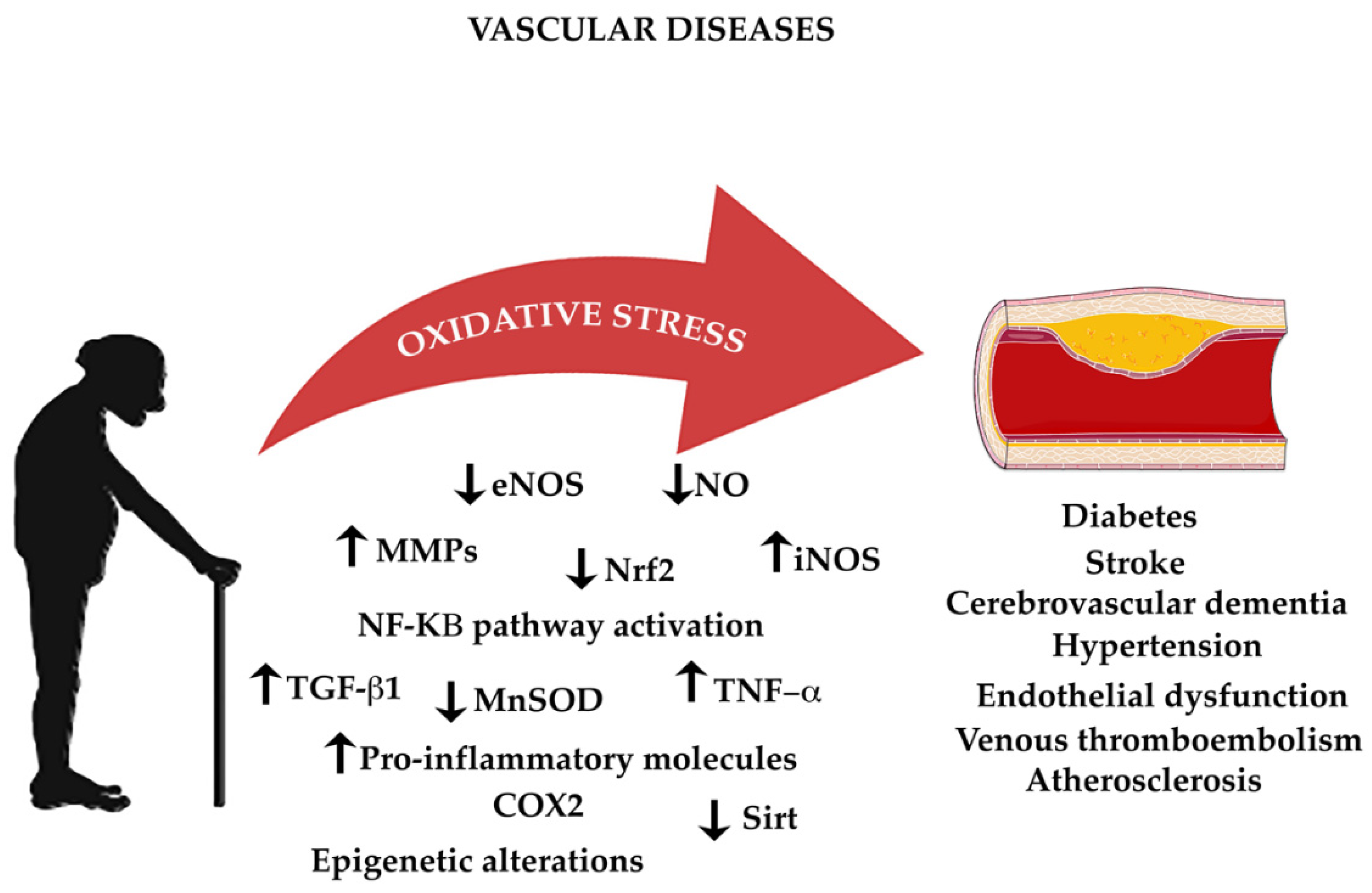{kind=link}
| Species | Intervention | Results | Reference |
|---|---|---|---|
| Mice | Caloric restriction | Protection of mitochondria in aging heart | Shinmura et al. [414]; Hepple et al. [415] |
| Mice | Resveratrol or caloric restriction | Prevention of age-related cardiac dysfunction | Barger et al. [437] |
| Mice | Nutraceutical combination | Reduction of blood pressure and oxidative stress, improvement of endothelial function and increase of nitric oxide release | Carrizzo et al. [428] |
| Mice | Enalapril and captopril | Enhancement of endogenous antioxidant defenses in mouse tissues | de Cavanagh et al. [452] |
| Mice | CAR-T cell | Elimination of senescent cells and extended survival in lung adenocarcinoma | Amor et al. [481]; [482] |
| Rats | Enalapril | Reduction of oxidative stress in diabetic rats | de Cavanagh et al. [451] |
| Rats | Caloric restriction | Improvement of cardiac aging and diastolic dysfunction in the senescent myocardium | Shinmura et al. [408]; Niemann et al. [407] |
| Rhesus monkeys | Caloric restriction | Delay in age-associated pathologies incidence (diabetes, brain atrophy, cancer, cardiovascular disease) and mortality | Colman et al. [402]; Fowler et al. [404]; McKiernan et al. [406] |
| Humans | Nutraceutical combination | Reduction of blood pressure, increase of nitric oxide release, and improvement of exercise capacity in hypertensive patients | Carrizzo et al. [428] |
| Humans | Caloric restriction or alternate-day fasting | Cardiovascular aging and chronic disease protection | Cruzen et al. [403]; Varady et al. [409] |
| Humans | Polyphenols | Reduction of systolic and diastolic blood pressure in high cardiovascular risk population | Medina-Remón et al. [425] |
| Humans | Dietary supplements, fish oil, green tea extract, resveratrol, vitamin E, vitamin C, and tomato extract | Improvement of endothelial function, reduction of inflammation and oxidative stress | Bakker et al. [426]; Schwingshackl et al. [434] |
| Humans | Resveratrol | Reduction of oxidative stress and endothelial dysfunction in hypertensive patients | Carrizzo et al. [436]; |
| Humans | Rapamycin | Reduction of senescent markers in patients with coronary artery disease | Singh et al. [475]; Krebs et al. [476] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izzo, C.; Vitillo, P.; Di Pietro, P.; Visco, V.; Strianese, A.; Virtuoso, N.; Ciccarelli, M.; Galasso, G.; Carrizzo, A.; Vecchione, C. The Role of Oxidative Stress in Cardiovascular Aging and Cardiovascular Diseases. Life 2021, 11, 60. https://doi.org/10.3390/life11010060
Izzo C, Vitillo P, Di Pietro P, Visco V, Strianese A, Virtuoso N, Ciccarelli M, Galasso G, Carrizzo A, Vecchione C. The Role of Oxidative Stress in Cardiovascular Aging and Cardiovascular Diseases. Life. 2021; 11(1):60. https://doi.org/10.3390/life11010060
Chicago/Turabian StyleIzzo, Carmine, Paolo Vitillo, Paola Di Pietro, Valeria Visco, Andrea Strianese, Nicola Virtuoso, Michele Ciccarelli, Gennaro Galasso, Albino Carrizzo, and Carmine Vecchione. 2021. "The Role of Oxidative Stress in Cardiovascular Aging and Cardiovascular Diseases" Life 11, no. 1: 60. https://doi.org/10.3390/life11010060