Profiling and Identification of Omeprazole Metabolites in Mouse Brain and Plasma by Isotope Ratio-Monitoring Liquid Chromatography-Mass Spectrometric Method


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Animals
2.3. LC–QTOF–MS Condition
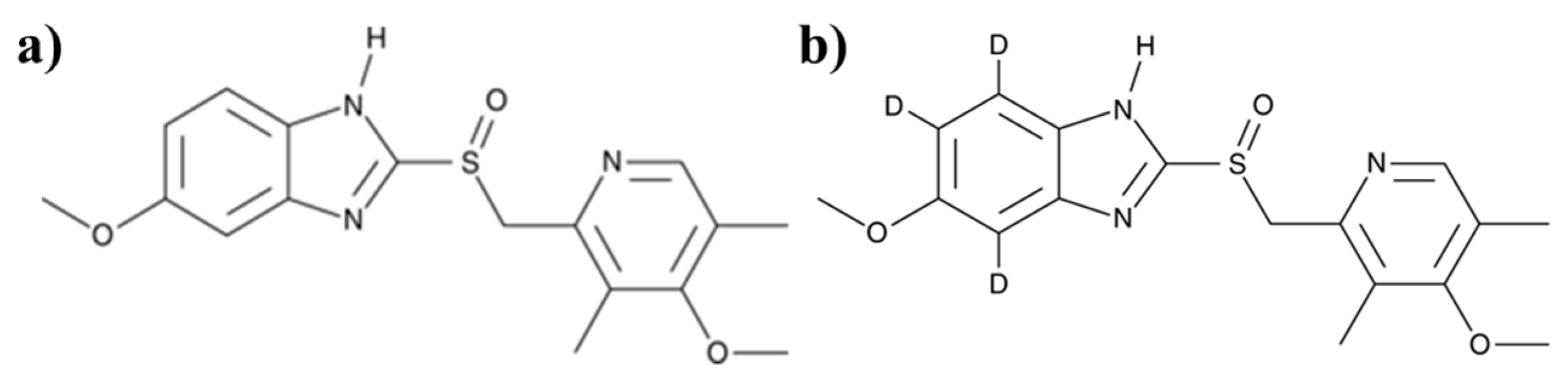
2.4. Pharmacokinetics and Metabolite Identification
2.5. Sample Preparation—In Vivo PK Samples
- Plasma samples: Ten microliters of the mouse plasma samples were placed in cluster tubes. As a make-up solution, 4 μL of DMSO and 10 μL of blank brain homogenate were added. Hundred microliters of ACN containing internal standard (ISTD, verapamil) was added to each sample for extraction. The mixture was capped, gently shaken for approximately 1 min and then was centrifuged for 5 min at 10,000 rpm (4 °C). Following the centrifugation, 50 μL supernatant was transferred to a clean test tube and was diluted with 100 μL of distilled water. The resulting mixture was then transferred to an LC–vial and 10 μL was injected to the LC–QTOF–MS.
- Brain samples: Ten microliters of the mouse brain homogenate were placed in cluster tubes. Four microliters of DMSO and 10 μL of blank plasma were added as a make-up solution. Hundred microliters of ACN containing internal standard (ISTD, verapamil) was added to each sample for extraction. The mixture was capped, gently shaken for approximately 1 min and then centrifuged for 5 min at 10,000 rpm (4 °C). Following the centrifugation, 50 μL supernatant was transferred to a clean test tube and was diluted with 100 μL of distilled water. The resulting mixture was then transferred to an LC-vial, and 10 μL was injected to the LC–QTOF–MS.
- Standard (STD) and quality control (QC) samples: Ten microliters of the blank mouse plasma samples were placed in cluster tubes, and 10 μL of blank brain homogenate were added. Four microliters of STD samples (final concentrations of 3.02, 9.05, 27.2, 81.5, 244, 733, 2200 and 6670 ng/mL, respectively) and QC samples (low QC [15.0 ng/mL], medium QC [165 ng/mL] and high QC [1820 ng/mL], as final concentrations) were added to each cluster tube. The mixture was capped, gently shaken for approximately 1 min and then centrifuged for 5 min at 10,000 rpm (4 °C). Following the centrifugation, 50 μL supernatant was transferred to a clean test tube and was diluted with 100 μL of distilled water. The resulting mixture was then transferred to an LC–vial, and 10 μL was injected to the LC–QTOF–MS.
2.6. Sample Preparation—Metabolite Identification
2.7. Software
3. Results
3.1. Method Development and Qualification
3.2. Application
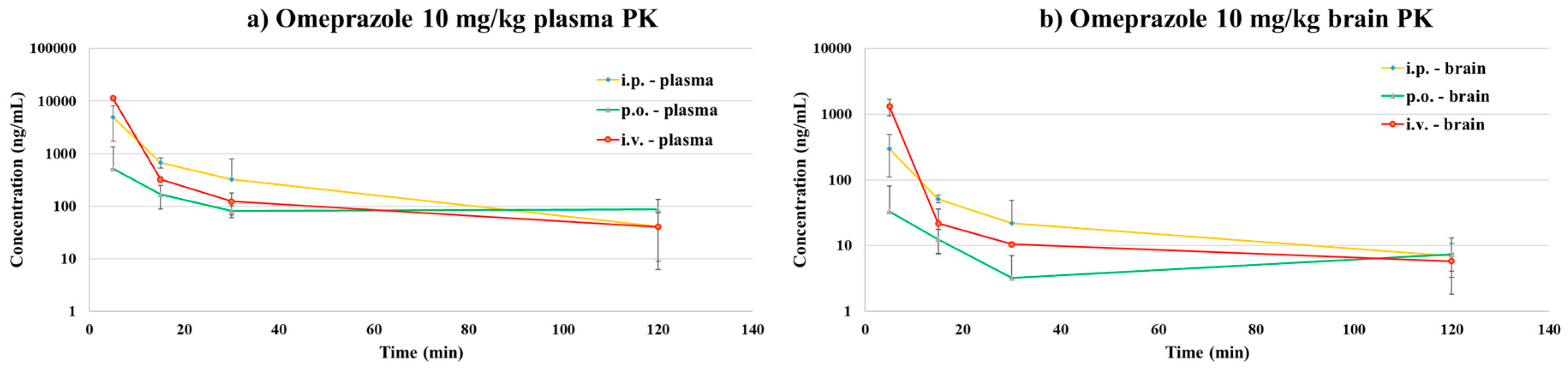
3.2.1. Pharmacokinetic Study
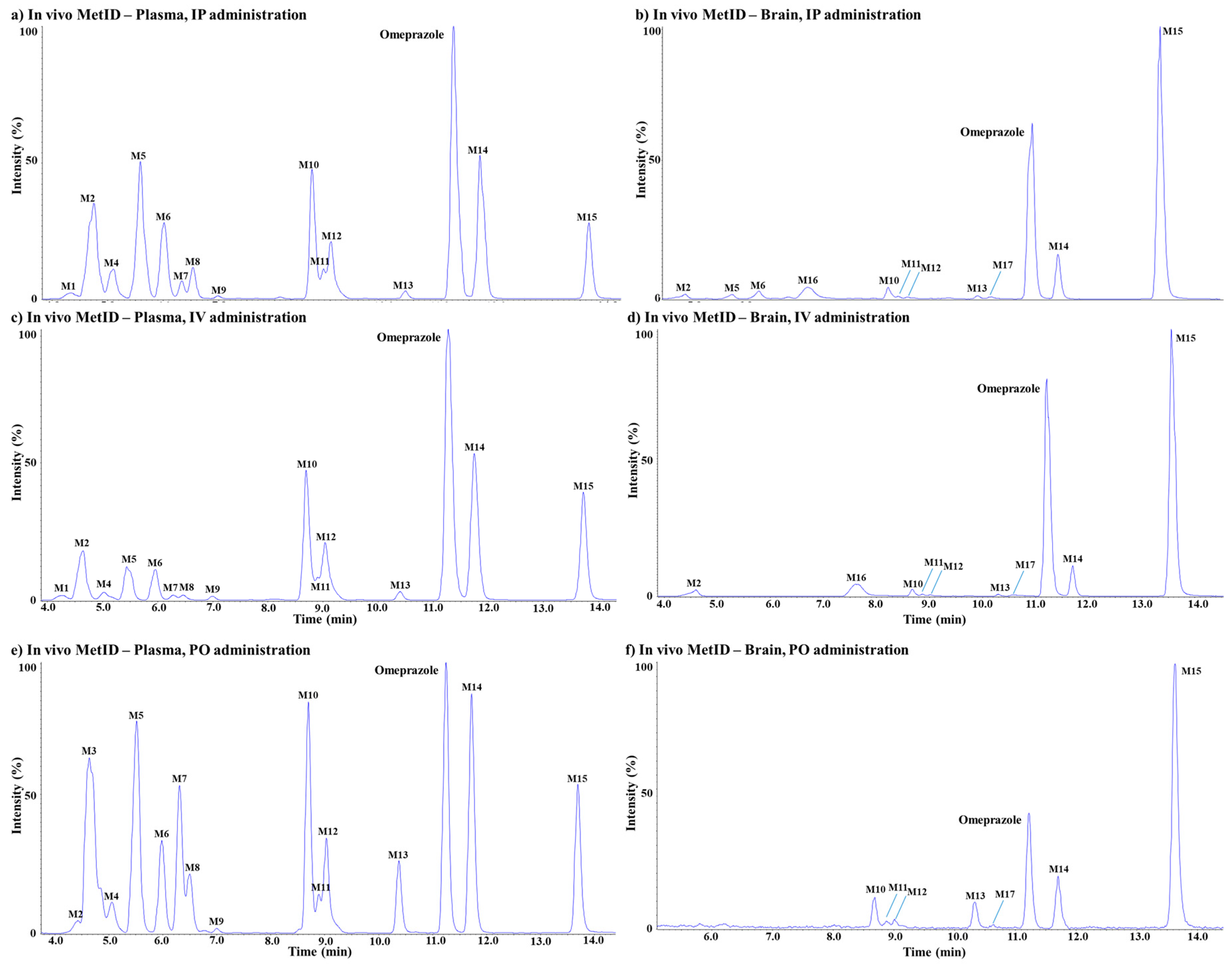
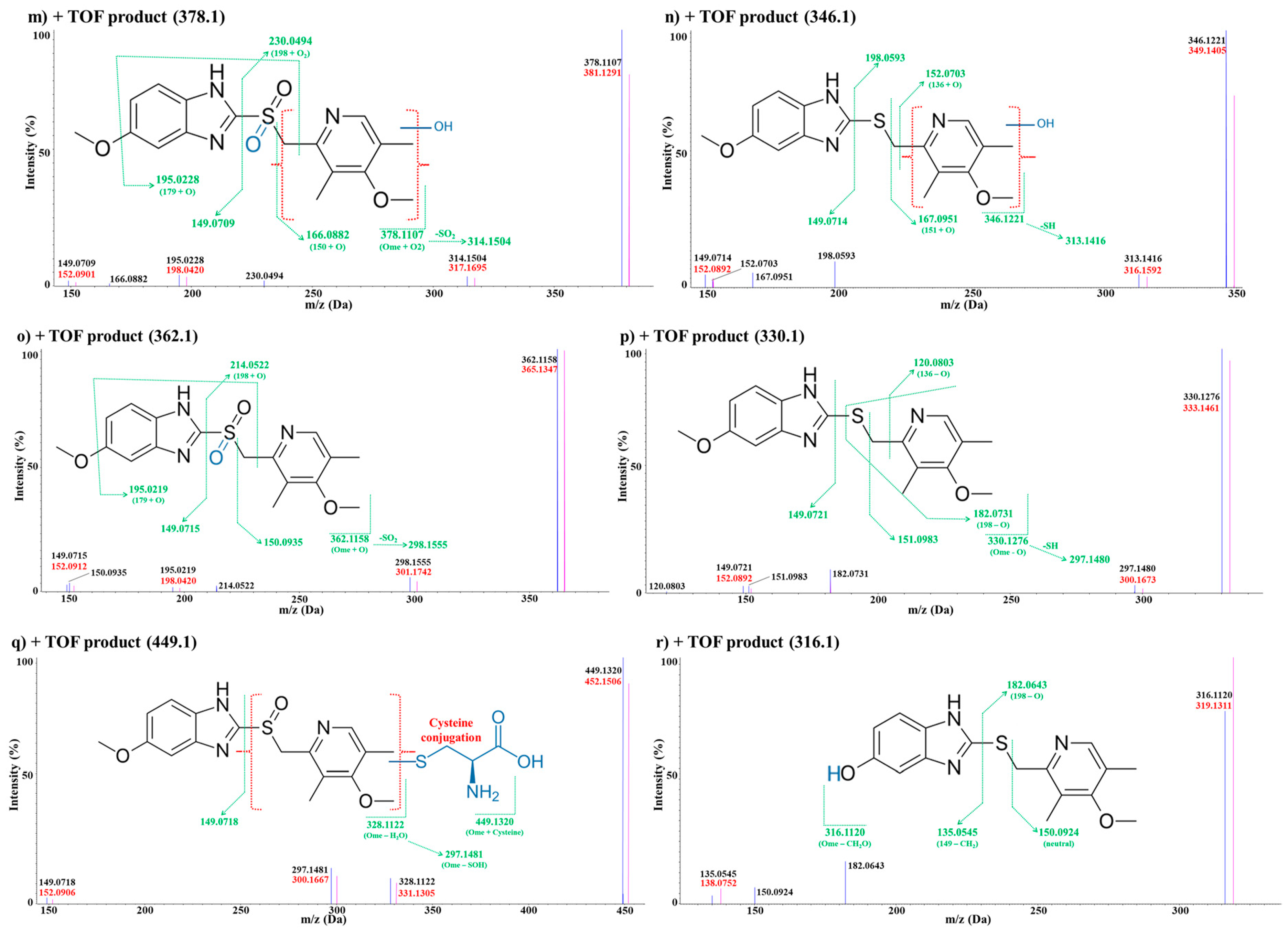
3.2.2. In Vivo Metabolite Identification
Omeprazole
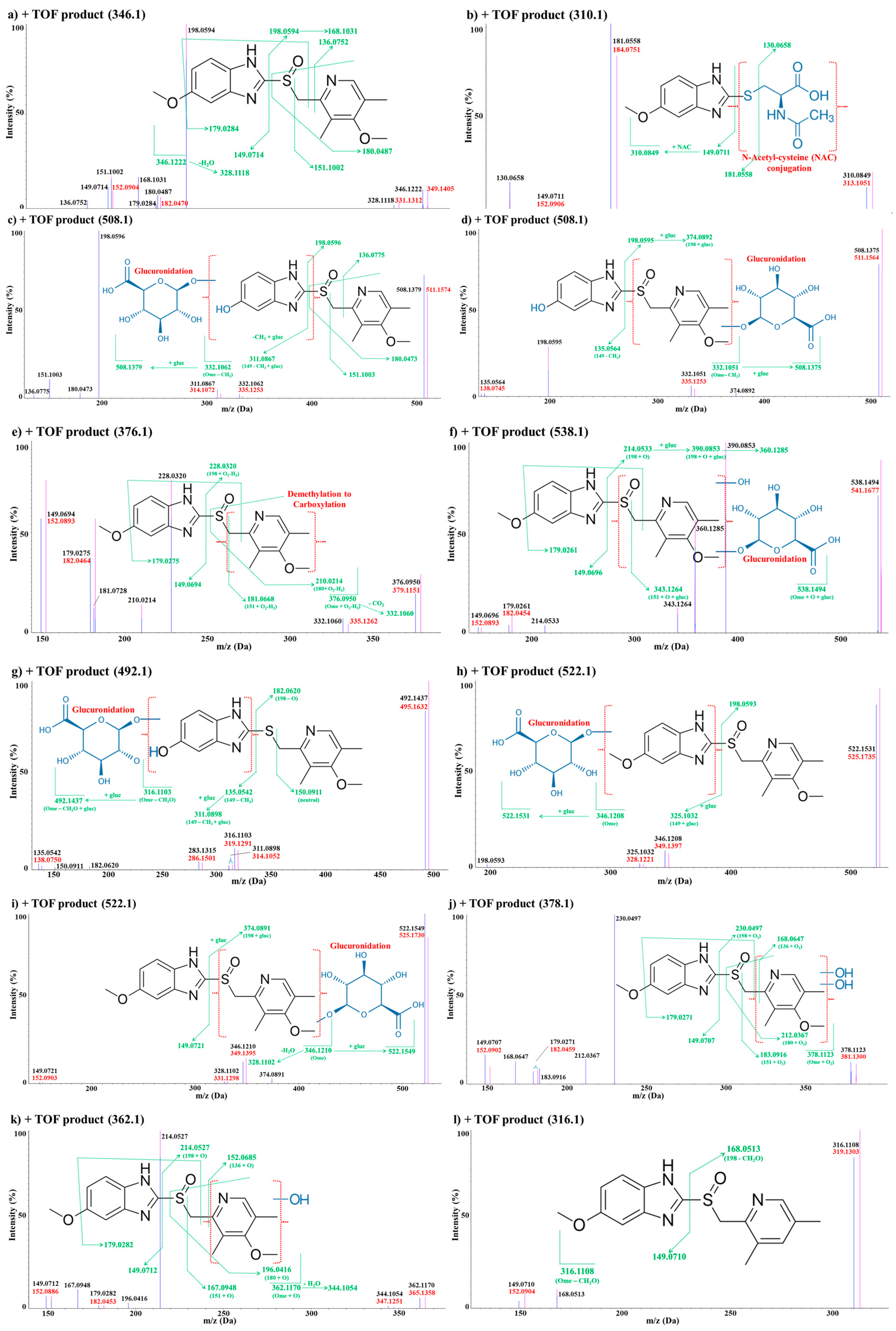
Metabolite M1
Metabolite M2
Metabolite M3
Metabolites M4
Metabolite M5
Metabolite M6
Metabolites M7
Metabolites M8
Metabolites M9
Metabolite M10
Metabolite M11
Metabolites M12
Metabolite M13
Metabolite M14
Metabolite M15
Metabolite M16
Metabolite M17
4. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lo, E.H. Degeneration and repair in central nervous system disease. Nat. Med. 2010, 16, 1205–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.J.; Sjogren, M.; Winblad, B. Neurofibrillary degeneration in Alzheimer’s disease: From molecular mechanisms to identification of drug targets. Curr. Opin. Psychiatr. 2008, 21, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Suh, H.-S. Alzheimer’s Disease: Clinical Trials and Future Perspectives. Korean J. Psychopharmacol. 2012, 23, 131–135. [Google Scholar]
- Rich, J. Degenerative central nervous system (CNS) disease. Pediatr. Rev. 2001, 22, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Theuns, J.; Van Broeckhoven, C. Locus–specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 2012, 33, 1340–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Agarwal, S.; Singh, S. Age related neurodegenerative Alzheimer’s disease: Usage of traditional herbs in therapeutics. Neurosci. Lett. 2020, 717, 134679. [Google Scholar] [CrossRef]
- Ascaso, F.J.; Mateo, J.; Cabezón, L.; Casas, P.; Grzybowski, A. Other Neurological Disorders: Migraine, Neurosarcoidosis, Schizophrenia, Obstructive Sleep Apnea–Hypopnea Syndrome and Bipolar Disorder. In OCT and Imaging in Central Nervous System Diseases; Springer: New York, NY, USA, 2020; pp. 317–342. [Google Scholar]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Tu, D.; Yang, R.; Chu, C.H.; Hong, J.S.; Gao, H.M. Through Reducing ROS Production, IL–10 Suppresses Caspase–1–Dependent IL–1beta Maturation, thereby Preventing Chronic Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 465. [Google Scholar] [CrossRef] [Green Version]
- Abu–Rumeileh, S.; Vacchiano, V.; Zenesini, C.; Polischi, B.; de Pasqua, S.; Fileccia, E.; Mammana, A.; Di Stasi, V.; Capellari, S.; Salvi, F.; et al. Diagnostic–prognostic value and electrophysiological correlates of CSF biomarkers of neurodegeneration and neuroinflammation in amyotrophic lateral sclerosis. J. Neurol. 2020, 267, 1699–1708. [Google Scholar] [CrossRef]
- Richter, J.E.; Kahrilas, P.J.; Johanson, J.; Maton, P.; Breiter, J.R.; Hwang, C.; Marino, V.; Hamelin, B.; Levine, J.G. Efficacy and safety of esomeprazole compared with omeprazole in GERD patients with erosive esophagitis: A randomized controlled trial. Am. J. Gastroenterol. 2001, 96, 656–665. [Google Scholar] [CrossRef]
- Olbe, L.; Carlsson, E.; Lindberg, P. A proton–pump inhibitor expedition: The case histories of omeprazole and esomeprazole. Nat. Rev. Drug Discov. 2003, 2, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Yeomans, N.D.; Tulassay, Z.; Juhasz, L.; Grp, A.S. Omeprazole was more effective for healing and preventing relapse of ulcers associated with long term NSAID use. Gut 1998, 43, 745. [Google Scholar]
- Chanchal, S.K.; Mahajan, U.B.; Siddharth, S.; Reddy, N.; Goyal, S.N.; Patil, P.H.; Bommanahalli, B.P.; Kundu, C.N.; Patil, C.R.; Ojha, S. In vivo and in vitro protective effects of omeprazole against neuropathic pain. Sci. Rep. 2016, 6, 3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashioka, S.; Klegeris, A.; McGeer, P.L. Proton Pump Inhibitors Reduce Interferon–gamma–Induced Neurotoxicity and STAT3 Phosphorylation of Human Astrocytes. Glia 2011, 59, 833–840. [Google Scholar] [CrossRef]
- Balza, E.; Piccioli, P.; Carta, S.; Lavieri, R.; Gattorno, M.; Semino, C.; Castellani, P.; Rubartelli, A. Proton pump inhibitors protect mice from acute systemic inflammation and induce long–term cross–tolerance. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef]
- Huskey, S.E.W.; Forseth, R.R.; Li, H.M.; Jian, Z.G.; Catoire, A.; Zhang, J.; Ray, T.; He, H.; Flarakos, J.; Mangold, J.B. Utilization of Stable Isotope Labeling to Facilitate the Identification of Polar Metabolites of KAF156, an Antimalarial Agent. Drug Metab. Dispos. 2016, 44, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.G.; Chowdhury, S.K.; Alton, K.B. Application of mass spectrometry for metabolite identification. Curr. Drug Metab. 2006, 7, 503–523. [Google Scholar] [CrossRef]
- Ando, S.; Tanaka, Y. Mass spectrometric studies on brain metabolism, using stable isotopes. Mass Spectrom. Rev. 2005, 24, 865–886. [Google Scholar] [CrossRef]
- Dawson, G. Measuring brain lipids. Bba–Mol. Cell Biol. L 2015, 1851, 1026–1039. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, R.A.; Garnett, W.R.; Kline, B.J. Determination of mean valproic acid serum level by assay of a single pooled sample. Clin. Pharmacol. Ther. 1981, 29, 408–413. [Google Scholar] [CrossRef]
- Sane, R.; Agarwal, S.; Elmquist, W.F. Brain Distribution and Bioavailability of Elacridar after Different Routes of Administration in the Mouse. Drug Metab. Dispos. 2012, 40, 1612–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gampa, G.; Kim, M.; Cook-Rostie, N.; Laramy, J.K.; Sarkaria, J.N.; Paradiso, L.; DePalatis, L.; Elmquist, W.F. Brain Distribution of a Novel MEK Inhibitor E6201: Implications in the Treatment of Melanoma Brain Metastases. Drug Metab. Dispos. 2018, 46, 658–666. [Google Scholar] [CrossRef]
- Radwan, M.F.; Dalby, K.N.; Kaoud, T.S. Propyphenazone-Based Analogues as Prodrugs and Selective Cyclooxygenase-2 Inhibitors. Acs Med. Chem. Lett. 2014, 5, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Redasani, V.K.; Shinde, A.B.; Surana, S.J.J.U. Anti–inflammatory and gastroprotective evaluation of prodrugs of piroxicam. Ulcers 2014. [Google Scholar] [CrossRef] [Green Version]
- Jeremy, J.Y.; Thompson, C.S.; Mikhailidis, D.P.; Dandona, P. Effect of the antiinflammatory prodrug, nabumetone and its principal active metabolite on rat gastric mucosal, aortic and platelet eicosanoid synthesis, in vitro and ex vivo. Prostaglandins Leukot Essent Fat. Acids 1990, 41, 195–199. [Google Scholar] [CrossRef]
- Maione, F.; Minosi, P.; Di Giannuario, A.; Raucci, F.; Chini, M.G.; De Vita, S.; Bifulco, G.; Mascolo, N.; Pieretti, S. Long–Lasting Anti-Inflammatory and Antinociceptive Effects of Acute Ammonium Glycyrrhizinate Administration: Pharmacological, Biochemical, and Docking Studies. Molecules 2019, 24, 2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El–Shenawy, S.M.; Abdel–Salam, O.M.; Baiuomy, A.R.; El–Batran, S.; Arbid, M.S. Studies on the anti-inflammatory and anti–nociceptive effects of melatonin in the rat. Pharmacol. Res. 2002, 46, 235–243. [Google Scholar] [CrossRef]
- de Lange, E.C.; Danhof, M.; de Boer, A.G.; Breimer, D.D. Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on drug transport across the blood-brain barrier. Brain Res. Brain Res. Rev. 1997, 25, 27–49. [Google Scholar] [CrossRef]
- Cheng, F.C.; Ho, Y.F.; Hung, L.C.; Chen, C.F.; Tsai, T.H. Determination and pharmacokinetic profile of omeprazole in rat blood, brain and bile by microdialysis and high–performance liquid chromatography. J. Chromatogr. A 2002, 949, 35–42. [Google Scholar] [CrossRef]
- Liu, X.R.; Chen, C.P.; Smith, B.J. Progress in brain penetration evaluation in drug discovery and development. J. Pharmacol. Exp. Ther. 2008, 325, 349–356. [Google Scholar] [CrossRef]
- Ball, K.; Bouzom, F.; Scherrmann, J.M.; Walther, B.; Decleves, X. Development of a physiologically based pharmacokinetic model for the rat central nervous system and determination of an in vitro–in vivo scaling methodology for the blood–brain barrier permeability of two transporter substrates, morphine and oxycodone. J. Pharm. Sci. 2012, 101, 4277–4292. [Google Scholar] [CrossRef]
- Shaffer, C.L. Defining Neuropharmacokinetic Parameters in CNS Drug Discovery to Determine Cross–Species Pharnnacologic Exposure-Response Relationships. Annu. Rep. Med. Chem. 2010, 45, 55–70. [Google Scholar]
- Benet, L.Z. Effect of route of administration and distribution on drug action. J. Pharmacokinet. Biopharm. 1978, 6, 559–585. [Google Scholar] [CrossRef] [PubMed]
- Gibaldi, M.; Feldman, S. Route of administration and drug metabolism. Eur. J. Pharmacol. 1972, 19, 323–329. [Google Scholar] [CrossRef]
- Qaisi, A.M.; Tutunji, M.F.; Tutunji, L.F. Acid decomposition of omeprazole in the absence of thiol: A differential pulse polarographic study at the static mercury drop electrode (SMDE). J. Pharm. Sci. 2006, 95, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Gul, W.; Sajid, S.; Hamid, F.; Bhatti, S.J.T.P.I. Effect of acidic Ph. and heat on the degradation of omeprazole and Esomeprazole. Pharma. Innov. 2015, 4, 19. [Google Scholar]
- Choi, M.S.; Lee, Y.H.; Shim, C.K. Bioavailabilities of Omeprazole Administered to Rats through Various Routes. Arch. Pharm. Res. 1995, 18, 141–145. [Google Scholar] [CrossRef]
- Kanazawa, H.; Okada, A.; Matsushima, Y.; Yokota, H.; Okubo, S.; Mashige, F.; Nakahara, K. Determination of omeprazole and its metabolites in human plasma by liquid chromatography-mass spectrometry. J. Chromatogr. A 2002, 949, 1–9. [Google Scholar] [CrossRef]
- Tassaneeyakul, W.; Tassaneeyakul, W.; Vannaprasaht, S.; Yamazoe, Y. Formation of omeprazole sulphone but not 5–hydroxyomeprazole is inhibited by grapefruit juice. Brit. J. Clin. Pharmacol. 2000, 49, 139–144. [Google Scholar] [CrossRef]
- Weidolf, L.; Karlsson, K.E.; Nilsson, I. A Metabolic Route of Omeprazole Involving Conjugation with Glutathione Identified in the Rat. Drug Metab. Dispos. 1992, 20, 262–267. [Google Scholar]
- Andersson, T. Pharmacokinetics, metabolism and interactions of acid pump inhibitors: Focus on omeprazole, lansoprazole and pantoprazole. Clin. Pharmacokinet. 1996, 31, 274. [Google Scholar] [CrossRef] [Green Version]
- Boix, C.; Ibanez, M.; Zamora, T.; Sancho, J.V.; Niessen, W.M.A.; Hernandez, F. Identification of new omeprazole metabolites in wastewaters and surface waters. Sci. Total Environ. 2014, 468, 706–714. [Google Scholar] [CrossRef]
- Martignoni, M.; Groothuis, G.M.; de Kanter, R.J.E. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.H.; Jin, H.J.; Liu, H.; Yue, X.Y.; Perlmutter, J.; Parsons, S.; Tu, Z.D. Exploration of new sulfur–containing analogues and in vivo evaluation of a lead F-18 PET tracer for imaging VAChT in rodent and nonhuman primate. J. Nucl. Med. 2016, 57, 160. [Google Scholar]
- Kumbar, S.S.; Hosamani, K.M.; Shettar, A.K. Physicochemical properties, antioxidant and anti–inflammatory activities of coumarin–carbonodithioate hybrids. Asian Pac. J. Trop. Biomed. 2018, 8, 201–206. [Google Scholar]
- Singh, D.P.; Hashim, S.R.; Singhal, R.G.J.I.J.D.D.R. Anti–inflammatory activity of some new thioether derivatives of quinoxaline. Int. J. Drug Dev. Res. 2010, 2, 810–815. [Google Scholar]
- Rho, H.S.; Ahn, S.M.; Yoo, D.S.; Kim, M.K.; Cho, D.H.; Cho, J.Y. Kojyl thioether derivatives having both tyrosinase inhibitory and anti–inflammatory properties. Bioorg. Med. Chem. Lett. 2010, 20, 6569–6571. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.J.; Fullone, J.S.; Anders, M.W. Brain uptake of S–(1,2–dichlorovinyl)glutathione and S–(1,2–dichlorovinyl)–L–cysteine, the glutathione and cysteine S–conjugates of the neurotoxin dichloroacetylene. Brain Res. Mol. Brain Res. 1993, 17, 53–58. [Google Scholar] [CrossRef]
- Mizuno, K.; Takeuchi, K.; Umehara, K.; Nakajima, M. Identification of Novel Metabolites of Vildagliptin in Rats: Thiazoline–Containing Thiol Adducts Formed via Cysteine or Glutathione Conjugation. Drug Metab. Dispos. 2019, 47, 809–817. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scan Mode | TOF-MS Scan | Product Ion Scan (SRMHS) | |
|---|---|---|---|
| Omeprazole | Verapamil (ISTD) | ||
| Mass range | m/z 100–500 | m/z 155–250 | m/z 100–500 |
| Parent ion | - | m/z 346.1 | m/z 455.3 |
| Product ion | - | m/z 198.1 | m/z 165.1 |
| DP | 80 V | 80 V | 80 V |
| CE | 7 V | 15 V | 33 V |
| Accumulation time | 0.2 s | 0.1 s | 0.1 s |
| Scan Mode | TOF-MS Scan | Product Ion Scan (IDA) |
|---|---|---|
| Mass range | m/z 100–1000 | m/z 50–1000 |
| DP | 80 V | 80 V |
| CE | 7 V | 18 V |
| Accumulation time | 0.2 s | 0.08 s |
| Number of information dependent scans | - | 7 scans |
| Intra-Run Assay | |||||
| Run | Nominal Concentration (ng/mL) | Calculated Concentration (ng/mL) | Mean Accuracy (%) | Precision (% CV) | n |
| Run 1 | QC low (15.02) | 16.9 | 112.3 | 10.6% | 3 |
| QC medium (165.29) | 185.2 | 112.0 | 4.7% | ||
| QC high (1818.18) | 1972.4 | 108.5 | 10.9% | ||
| Run 2 | QC low (15.02) | 16.3 | 108.8 | 0.9% | 3 |
| QC medium (165.29) | 177.2 | 107.2 | 5.6% | ||
| QC high (1818.18) | 1830.9 | 100.7 | 3.1% | ||
| Run 3 | QC low (15.02) | 14.8 | 98.6 | 1.6% | 3 |
| QC medium (165.29) | 170.0 | 102.9 | 0.8% | ||
| QC high (1818.18) | 1790.7 | 98.5 | 6.9% | ||
| Inter-Run Assay (Run 1~3) | |||||
| Nominal QC Concentration (ng/mL) | Calculated Concentration (ng/mL) | Mean Accuracy (%) | Precision (% CV) | n | |
| QC low (15.02) | 14.1 | 94.0 | 10.3% | 9 | |
| QC medium (165.29) | 159.9 | 96.8 | 5.1% | ||
| QC high (1818.18) | 1710.4 | 94.1 | 3.1% | ||
| Repeat injection of LLOQ | |||||
| Nominal Concentration (ng/mL) | Calculated Concentration (ng/mL) | Mean Accuracy (%) | Precision (% CV) | n | |
| 3.02 | 2.9 | 95.4 | 13.3% | 6 | |
| PK Parameters of Omeprazole. | ||||||||
|---|---|---|---|---|---|---|---|---|
| PK Study | Dose (mg/kg) | T1/2 (min) | Cmax (ng/mL) | AUClast (min·ng/mL) | Vd (mL/kg) | CL (mL/min/kg) | Brain/Plasma Ratio (%) | BA (%) |
| i.p.-brain | 10 | 51.31 | 300.61 | 4365.73 | - | - | 6.81% | 24.06% |
| i.p.-plasma | 28.81 | 4885.82 | 64,115.37 | - | - | |||
| p.o.-brain | 10 | 44.58 | 37.81 | 909.27 | - | - | 6.43% | 5.31% |
| p.o.-plasma | 54.76 | 610.15 | 14,149.52 | - | - | |||
| i.v.-brain | 10 | 48.24 | 11,233.77 | 39,105.36 | 17,793.24 | 254.76 | 14.68% | - |
| i.v.-plasma | 27.39 | 67,631.18 | 266,451.92 | 1491.70 | 37.75 | |||
| In Vivo MetID Result of Omeprazole | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peak ID | Name | Formula (H3 and D3) | m/z | Error (ppm) | Nominal Mass Change (Da) | RT (min) | A | B | C | D | E | F |
| Parent | Omeprazole [M + H]+ | C17H19N3O3S | 346.1220 | 0.6 | - | 11.250 | O | O | O | O | O | O |
| D3C17H16N3O3S | 349.1408 | −0.9 | ||||||||||
| M1 | Loss of C9H11NO2S followed by N-acetylcysteine conjugation [M + H]+ | C13H15N3O4S | 310.0856 | −2.3 | −36 | 4.332 | O | O | - | - | - | - |
| D3C13H12N3O4S | 313.1044 | 2.1 | ||||||||||
| M2 | Demethylation followed by glucuronide conjugation [M + H]+ | C22H25N3O9S | 508.1384 | −1 | +162 | 4.420 | O | O | O | O | O | - |
| D3C22H22N3O9S | 511.1573 | 0.3 | ||||||||||
| M3 | Demethylation followed by glucuronide conjugation [M + H]+ | C22H25N3O9S | 508.1384 | −1.8 | +162 | 4.657 | - | - | O | - | - | - |
| D3C22H22N3O9S | 511.1573 | −1.7 | ||||||||||
| M4 | Demethylation to carboxylic acid [M + H]+ | C17H17N3O5S | 376.0962 | −3.1 | +30 | 5.086 | O | O | O | - | - | - |
| D3C17H14N3O5S | 379.1150 | 0.3 | ||||||||||
| M5 | Oxidation followed by glucuronide conjugation [M + H]+ | C23H27N3O10S | 538.1490 | 0.8 | +192 | 5.586 | O | O | O | O | - | - |
| D3C23H24N3O10S | 541.1678 | −0.2 | ||||||||||
| M6 | Loss of hydroxymethylene followed by glucuronide conjugation [M + H]+ | C22H25N3O8S | 492.1435 | 0.4 | +146 | 6.013 | O | O | O | O | - | - |
| D3C22H22N3O8S | 495.1623 | 1.7 | ||||||||||
| M7 | Glucuronide conjugation [M + H]+ | C23H27N3O9S | 522.1541 | −1.9 | +176 | 6.333 | O | O | O | - | - | - |
| D3C23H24N3O9S | 525.1729 | 1.1 | ||||||||||
| M8 | Glucuronide conjugation [M + H]+ | C23H27N3O9S | 522.1541 | 1.6 | +176 | 6.533 | O | O | O | - | - | - |
| D3C23H24N3O9S | 525.1729 | 0.2 | ||||||||||
| M9 | Di-oxidation [M + H]+ | C17H19N3O5S | 378.1118 | 1.3 | +32 | 6.986 | O | O | O | - | - | - |
| D3C17H16N3O5S | 381.1307 | −1.7 | ||||||||||
| M10 | Mono-oxidation [M + H]+ | C17H19N3O4S | 362.1169 | 0.3 | +16 | 8.679 | O | O | O | O | O | O |
| D3C17H16N3O4S | 365.1357 | 0.2 | ||||||||||
| M11 | Loss of hydroxymethylene [M + H]+ | C16H17N3O2S | 316.1114 | −2.0 | −30 | 8.878 | O | O | O | O | O | O |
| D3C16H14N3O2S | 319.1303 | 0.1 | ||||||||||
| M12 | Di-oxidation [M + H]+ | C17H19N3O5S | 378.1118 | −3.0 | +32 | 9.016 | O | O | O | O | O | O |
| D3C17H16N3O5S | 381.1307 | −2.5 | ||||||||||
| M13 | Oxidation and Deoxidation [M + H]+ | C17H19N3O3S | 346.1220 | 0.3 | - | 10.356 | O | O | O | O | O | O |
| D3C17H16N3O3S | 349.1408 | −0.9 | ||||||||||
| M14 | Mono-oxidation [M + H]+ | C17H19N3O4S | 362.1169 | −3.0 | +16 | 11.710 | O | O | O | O | O | O |
| D3C17H16N3O4S | 365.1357 | −2.8 | ||||||||||
| M15 | Sulfoxide to thioether [M + H]+ | C17H19N3O2S | 330.1271 | 1.6 | −16 | 13.656 | O | O | O | O | O | O |
| D3C17H16N3O2S | 333.1459 | 0.6 | ||||||||||
| M16 | Cysteine conjugation [M + H]+ | C20H24N4O4S2 | 449.1312 | 1.8 | +103 | 7.235 | - | - | - | O | O | - |
| D3C20H21N4O4S2 | 452.1500 | 1.3 | ||||||||||
| M17 | Loss of hydroxymethylene [M + H]+ | C16H17N3O2S | 316.1114 | 1.8 | −30 | 10.489 | - | - | - | O | O | - |
| D3C16H14N3O2S | 319.1303 | 2.6 | ||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, S.-H.; Park, Y.; Park, M.-H.; Byeon, J.-J.; Lee, B.i.; Choi, J.; Shin, Y.G. Profiling and Identification of Omeprazole Metabolites in Mouse Brain and Plasma by Isotope Ratio-Monitoring Liquid Chromatography-Mass Spectrometric Method. Life 2020, 10, 115. https://doi.org/10.3390/life10070115
Shin S-H, Park Y, Park M-H, Byeon J-J, Lee Bi, Choi J, Shin YG. Profiling and Identification of Omeprazole Metabolites in Mouse Brain and Plasma by Isotope Ratio-Monitoring Liquid Chromatography-Mass Spectrometric Method. Life. 2020; 10(7):115. https://doi.org/10.3390/life10070115
Chicago/Turabian StyleShin, Seok-Ho, Yuri Park, Min-Ho Park, Jin-Ju Byeon, Byeong ill Lee, Jangmi Choi, and Young G. Shin. 2020. "Profiling and Identification of Omeprazole Metabolites in Mouse Brain and Plasma by Isotope Ratio-Monitoring Liquid Chromatography-Mass Spectrometric Method" Life 10, no. 7: 115. https://doi.org/10.3390/life10070115