Embelin as Lead Compound for New Neuroserpin Polymerization Inhibitors

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. NS Expression and Purification
2.2. Compounds Mix Preparation
2.3. Polymerization Assays
2.4. COS-7 cells Culture and DNA Transfection
2.5. SDS and Non-Denaturing PAGE and Western Blot Analysis of Cellular Samples
2.6. Non-Denaturing Polyacrylamide gel Electrophoresis
2.7. Size exclusion Chromatography
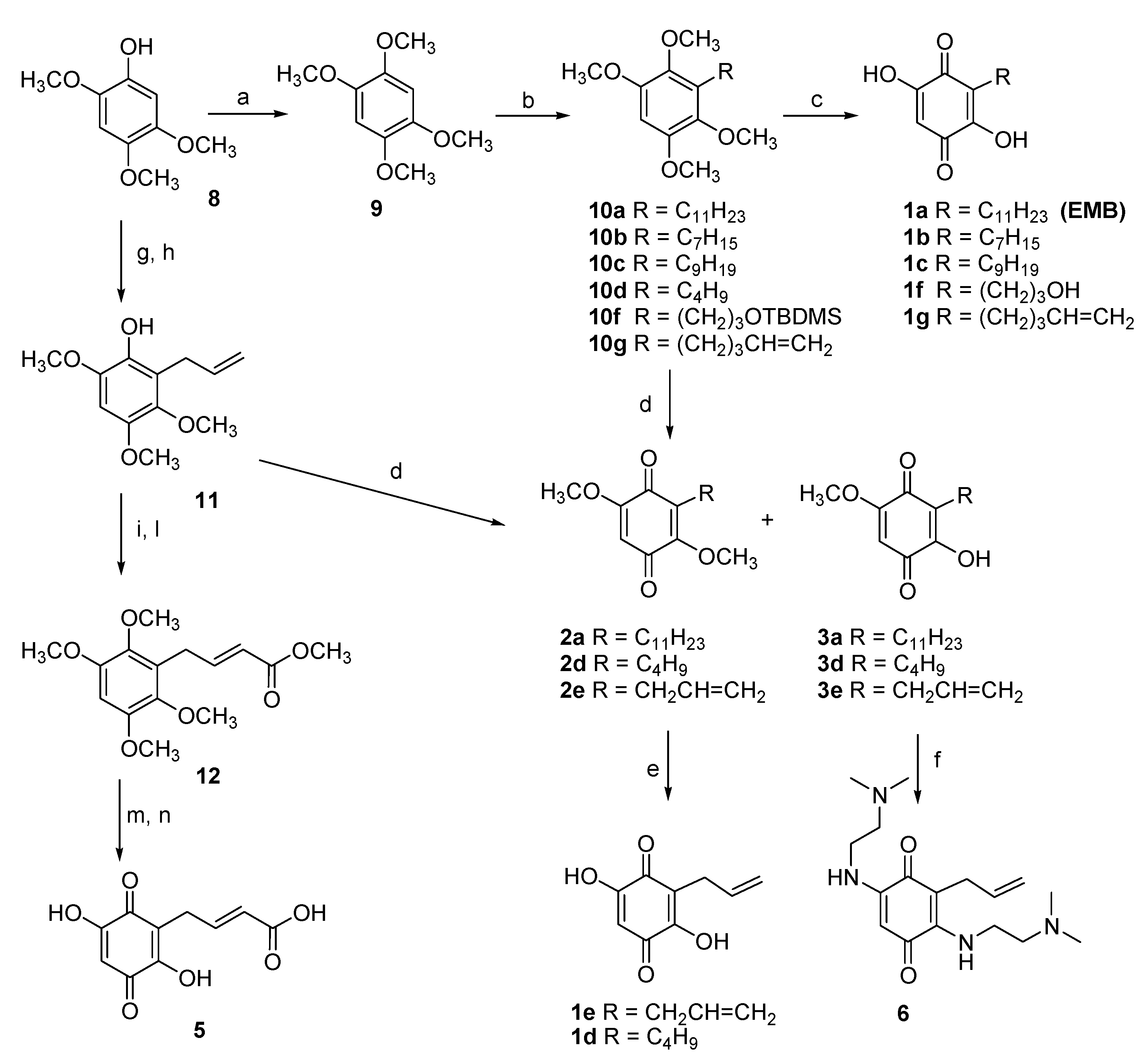
2.8. Synthesis of EMB Analogs
2.8.1. 3-allyl-2,5-dimethoxy-[1,4]benzoquinone (2e) and 3-allyl-2-hydroxy-5-methoxy-[1,4]benzoquinone (3e)
2.8.2. 3-allyl-2,5-dihdroxy-1,4-benzoquinone (1e)
2.8.3. 3-butyl-2,5-dimethoxy-[1,4] benzoquinone (2d) and 3-butyll-2-hydroxy-5-methoxy-[1,4] benzoquinone (3d)
2.8.4. 3-butyl-2,5-dihydroxy-1,4-benzoquinone (1d)
2.8.5. 3-heptyl-1,2,4,5-tetramethoxybenzene (10b)
2.8.6. 3-Heptyl-2,5-Dihydroxy-[1,4] Benzoquinone (1b)
2.8.7. 3-nonyl-1,2,4,5-tetramethoxybenzene (10c)
2.8.8. 3-nonyl-2,5-dihydroxy-[1,4] benzoquinone (1c)
2.8.9. Tert-butyldimethyl-[3-(2,3,5,6-tetramethoxyphenyl)-propoxy]-silane (10f)
2.8.10. 2,5-dihydroxy-3-(3-hydroxy-propyl)-[1,4] benzoquinone (1f)
2.8.11. (E)-methyl–4-(2,3,5,6-tetramethoxyphenyl)-but-2-enoate (12)
2.8.12. (2E)-4-(2,5-dihydroxy-3,6-dioxocyclohexa-1,4-dienyl)-but-2-enoic acid (5)
2.8.13. 1,2,4,5-tetramethoxy-3-(pent-4-en-1-yl)benzene (10g)
2.8.14. 2,5-dihydroxy-3-(pent-4-en-1-yl)cyclohexa-2,5-diene-1,4-dione (1g)
2.8.15. 3-allyl-2,5-bis-((2-(dimethylamino)ethyl)amino)-1,4-benzoquinone (6)
2.8.16. (E)-1,8-bis(2,3,5,6-tetramethoxyphenyl)oct-4-ene (13)
2.8.17. 1,8-bis(2,3,5,6-tetramethoxyphenyl)octane (14)
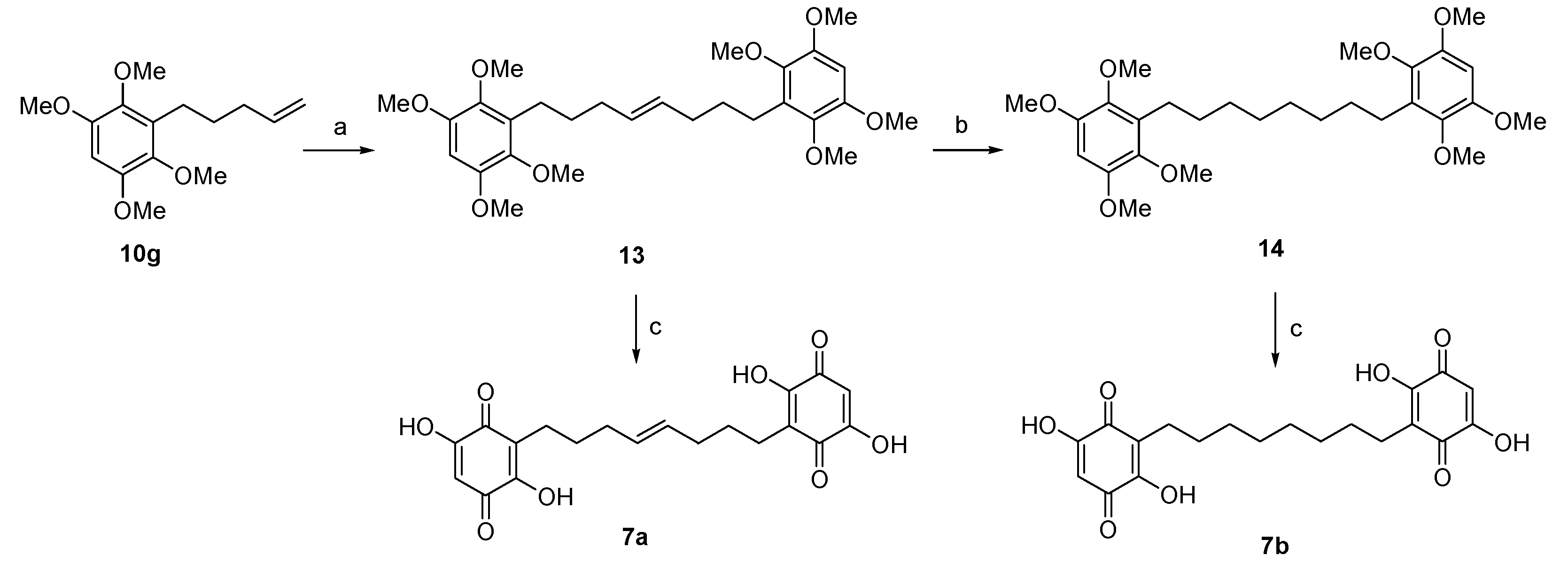
2.8.18. (E)-3,3’-(oct-4-ene-1,8-diyl)bis(2,5-dihydroxycyclohexa-2,5-diene-1,4-dione) (7a)
2.8.19. 3,3’-(octane-1,8-diyl)bis(2,5-dihydroxycyclohexa-2,5-diene-1,4-dione) (7b)
3. Results
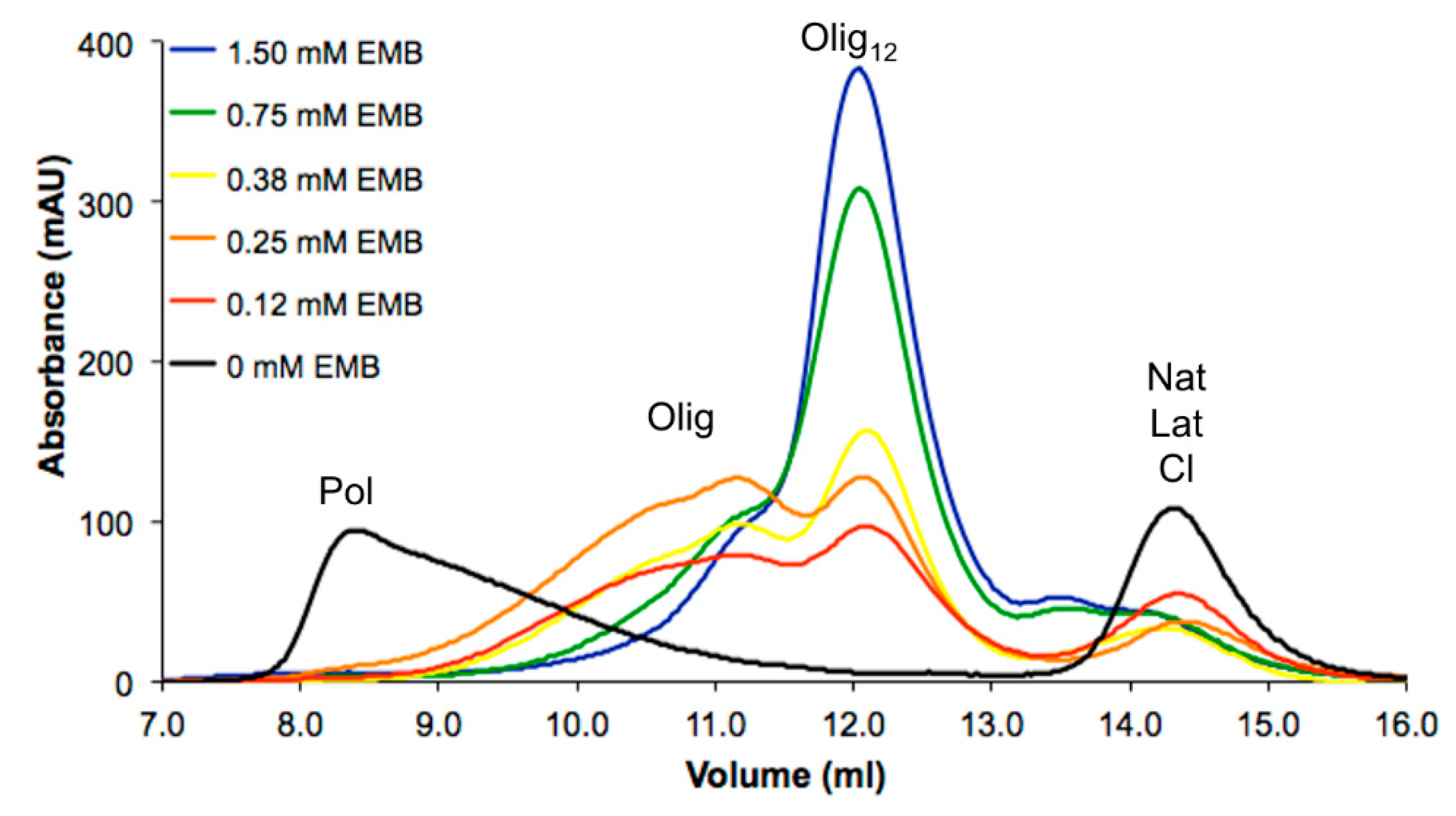
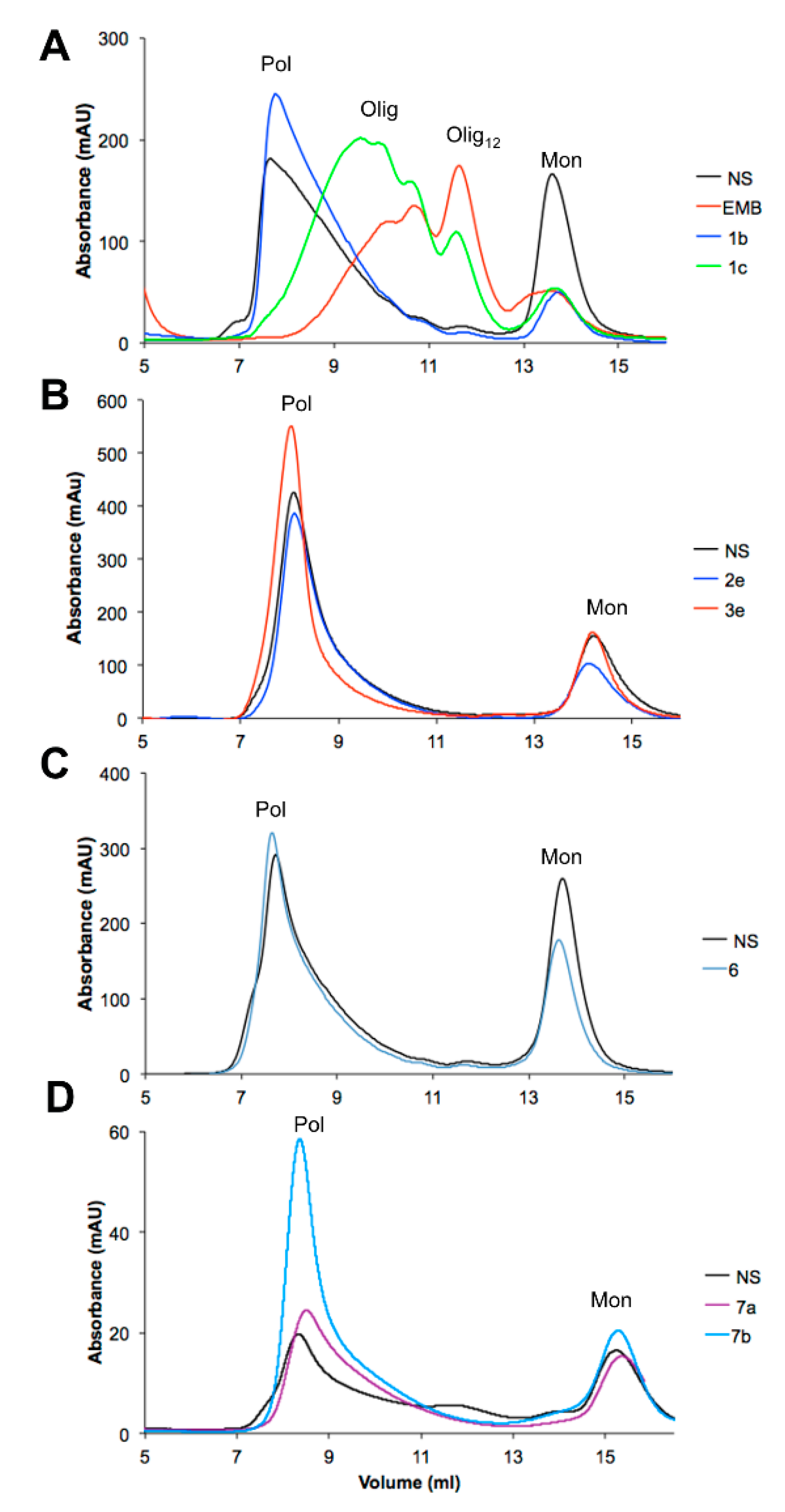
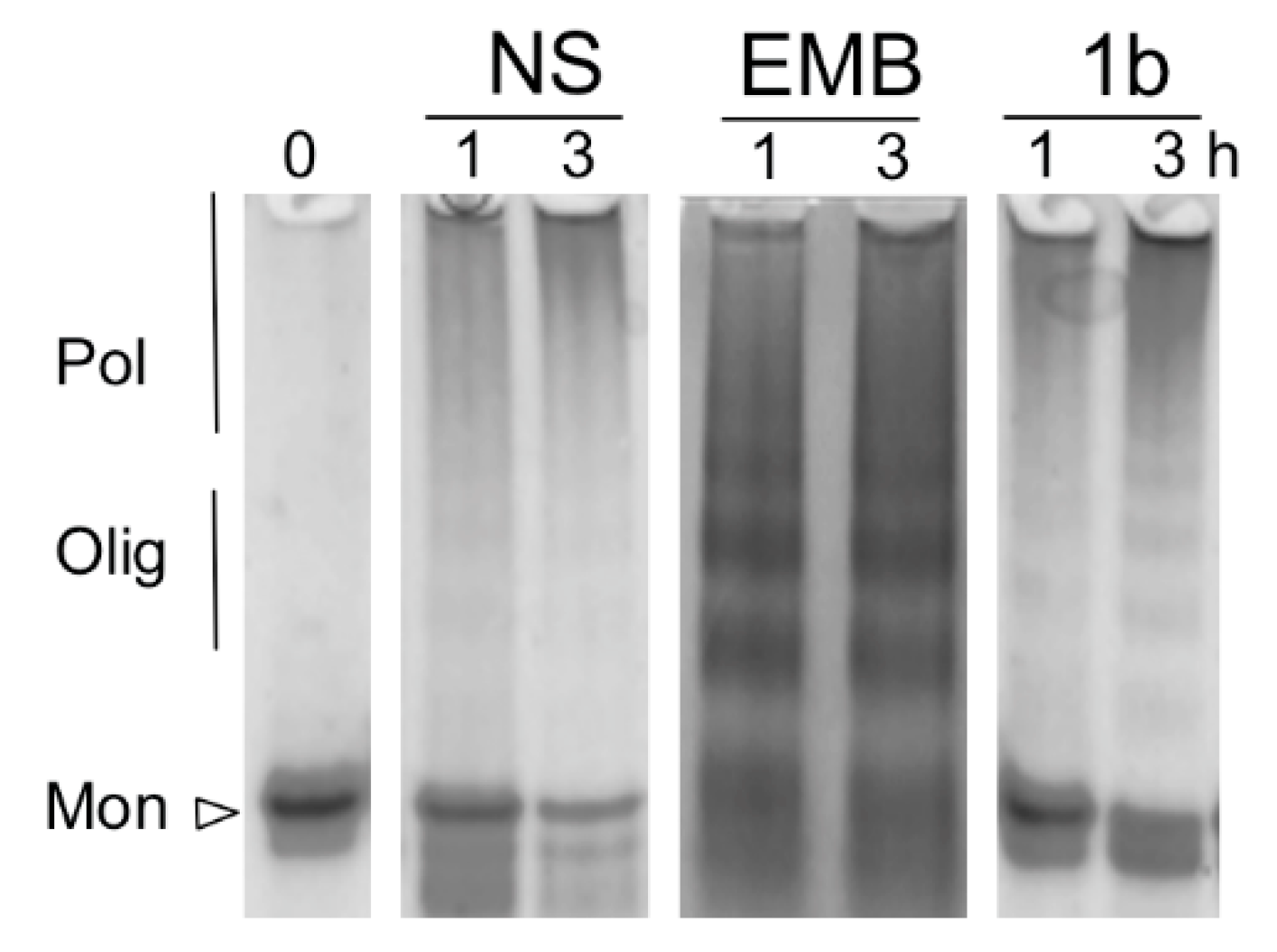
3.1. NS Polymerization at Different Concentrations of EMB
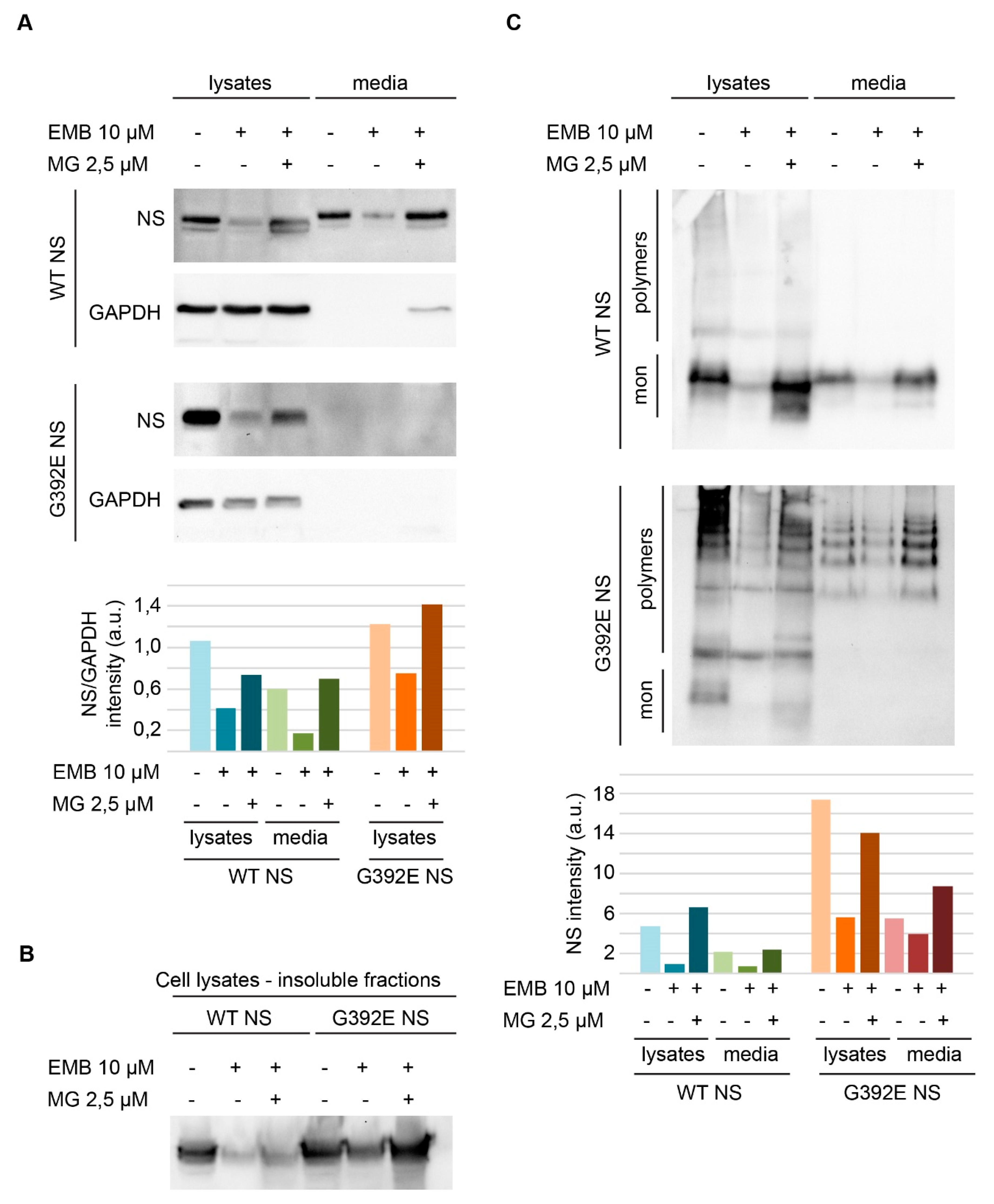
3.2. EMB Promotes Proteasomal Degradation in Cell Lines Expressing NS
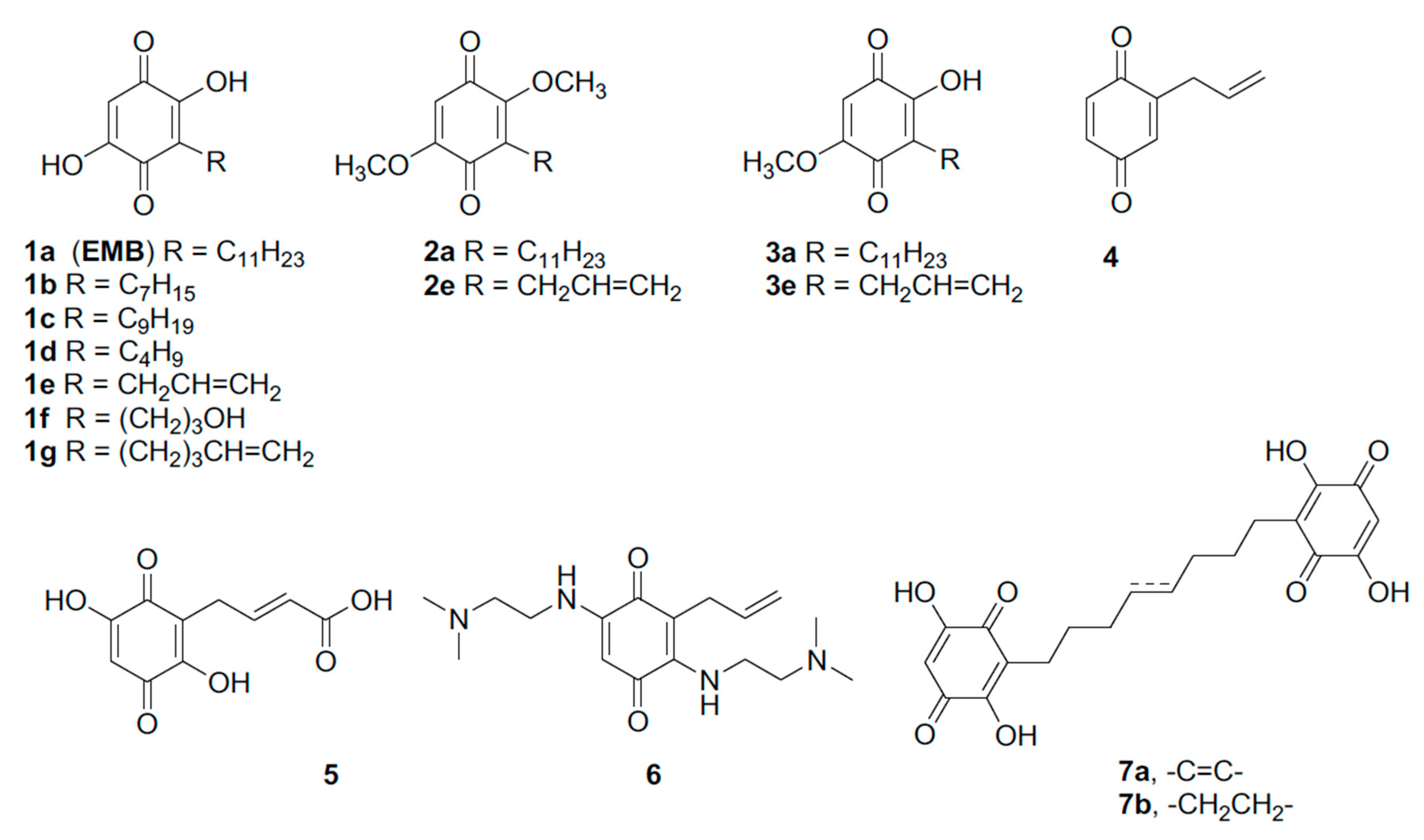
3.3. Design and Synthesis of EMB Analogs
3.4. Effects of the EMB Analogs on NS Polymerization
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fra, A.; D’Acunto, E.; Laffranchi, M.; Miranda, E. Cellular Models for the Serpinopathies. Methods Mol. Biol. Clifton NJ 2018, 1826, 109–121. [Google Scholar] [CrossRef]
- Davis, R.L.; Shrimpton, A.E.; Holohan, P.D.; Bradshaw, C.; Feiglin, D.; Collins, G.H.; Sonderegger, P.; Kinter, J.; Becker, L.M.; Lacbawan, F.; et al. Familial dementia caused by polymerization of mutant neuroserpin. Nature 1999, 401, 376–379. [Google Scholar] [CrossRef]
- Davis, R.L.; Holohan, P.D.; Shrimpton, A.E.; Tatum, A.H.; Daucher, J.; Collins, G.H.; Todd, R.; Bradshaw, C.; Kent, P.; Feiglin, D.; et al. Familial encephalopathy with neuroserpin inclusion bodies. Am. J. Pathol. 1999, 155, 1901–1913. [Google Scholar] [CrossRef]
- Davis, R.L.; Shrimpton, A.E.; Carrell, R.W.; Lomas, D.A.; Gerhard, L.; Baumann, B.; Lawrence, D.A.; Yepes, M.; Kim, T.S.; Ghetti, B.; et al. Association between conformational mutations in neuroserpin and onset and severity of dementia. Lancet Lond. Engl. 2002, 359, 2242–2247. [Google Scholar] [CrossRef]
- Coutelier, M.; Andries, S.; Ghariani, S.; Dan, B.; Duyckaerts, C.; van Rijckevorsel, K.; Raftopoulos, C.; Deconinck, N.; Sonderegger, P.; Scaravilli, F.; et al. Neuroserpin Mutation Causes Electrical Status Epilepticus of Slow-Wave Sleep. Neurology 2008, 71, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Hagen, M.C.; Murrell, J.R.; Delisle, M.B.; Andermann, E.; Andermann, F.; Guiot, M.C.; Ghetti, B. Encephalopathy with Neuroserpin Inclusion Bodies Presenting as Progressive Myoclonus Epilepsy and Associated with a Novel Mutation in the Proteinase Inhibitor 12 Gene. Brain Pathol. Zurich Switz. 2011, 21, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, E.; MacLeod, I.; Davies, M.J.; Pérez, J.; Römisch, K.; Crowther, D.C.; Lomas, D.A. The intracellular accumulation of polymeric neuroserpin explains the severity of the dementia FENIB. Hum. Mol. Genet. 2008, 17, 1527–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, E.; Lomas, D.A. Neuroserpin: A serpin to think about. Cell Mol. Life Sci. CMLS 2006, 63, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Hastings, G.A.; Coleman, T.A.; Haudenschild, C.C.; Stefansson, S.; Smith, E.P.; Barthlow, R.; Cherry, S.; Sandkvist, M.; Lawrence, D.A. Neuroserpin, a brain-associated inhibitor of tissue plasminogen activator is localized primarily in neurons. Implications for the regulation of motor learning and neuronal survival. J. Biol. Chem. 1997, 272, 33062–33067. [Google Scholar] [CrossRef] [Green Version]
- Yepes, M.; Lawrence, D.A. Tissue-type plasminogen activator and neuroserpin: A well-balanced act in the nervous system? Trends Cardiovasc. Med. 2004, 14, 173–180. [Google Scholar] [CrossRef]
- Galliciotti, G.; Sonderegger, P. Neuroserpin. Front. Biosci J. Virtual Libr 2006, 11, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Caccia, S.; Ricagno, S.; Bolognesi, M. Molecular bases of neuroserpin function and pathology. Biomol. Concepts 2010, 1, 117–130. [Google Scholar] [CrossRef]
- Huntington, J.A.; Read, R.J.; Carrell, R.W. Structure of a serpin-protease complex shows inhibition by deformation. Nature 2000, 407, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Barker-Carlson, K.; Lawrence, D.A.; Schwartz, B.S. Acyl-enzyme complexes between tissue-type plasminogen activator and neuroserpin are short-lived in vitro. J. Biol. Chem. 2002, 277, 46852–46857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricagno, S.; Caccia, S.; Sorrentino, G.; Antonini, G.; Bolognesi, M. Human neuroserpin: Structure and time-dependent inhibition. J. Mol. Biol. 2009, 388, 109–121. [Google Scholar] [CrossRef]
- Gooptu, B.; Lomas, D.A. Conformational pathology of the serpins: Themes, variations, and therapeutic strategies. Annu. Rev. Biochem 2009, 78, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Moriconi, C.; Ordonez, A.; Lupo, G.; Gooptu, B.; Irving, J.A.; Noto, R.; Martorana, V.; Manno, M.; Timpano, V.; Guadagno, N.A.; et al. Interactions between N-linked glycosylation and polymerisation of neuroserpin within the endoplasmic reticulum. FEBS J. 2015, 282, 4565–4579. [Google Scholar] [CrossRef] [PubMed]
- Visentin, C.; Broggini, L.; Sala, B.M.; Russo, R.; Barbiroli, A.; Santambrogio, C.; Nonnis, S.; Dubnovitsky, A.; Bolognesi, M.; Miranda, E.; et al. Glycosylation Tunes Neuroserpin Physiological and Pathological Properties. Int. J. Mol. Sci. 2020, 21, 3235. [Google Scholar] [CrossRef]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef]
- Yamasaki, M.; Li, W.; Johnson, D.J.D.; Huntington, J.A. Crystal structure of a stable dimer reveals the molecular basis of serpin polymerization. Nature 2008, 455, 1255–1258. [Google Scholar] [CrossRef]
- Yamasaki, M.; Sendall, T.J.; Pearce, M.C.; Whisstock, J.C.; Huntington, J.A. Molecular basis of α1-antitrypsin deficiency revealed by the structure of a domain-swapped trimer. EMBO Rep. 2011, 12, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Noto, R.; Santangelo, M.G.; Ricagno, S.; Mangione, M.R.; Levantino, M.; Pezzullo, M.; Martorana, V.; Cupane, A.; Bolognesi, M.; Manno, M. The tempered polymerization of human neuroserpin. PLoS ONE 2012, 7, e32444. [Google Scholar] [CrossRef]
- Santangelo, M.G.; Noto, R.; Levantino, M.; Cupane, A.; Ricagno, S.; Pezzullo, M.; Bolognesi, M.; Mangione, M.R.; Martorana, V.; Manno, M. On the molecular structure of human neuroserpin polymers. Proteins 2012, 80, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Noto, R.; Santangelo, M.G.; Levantino, M.; Cupane, A.; Mangione, M.R.; Parisi, D.; Ricagno, S.; Bolognesi, M.; Manno, M.; Martorana, V. Functional and dysfunctional conformers of human neuroserpin characterized by optical spectroscopies and Molecular Dynamics. Biochim. Biophys. Acta 2015, 1854, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saga, G.; Sessa, F.; Barbiroli, A.; Santambrogio, C.; Russo, R.; Sala, M.; Raccosta, S.; Martorana, V.; Caccia, S.; Noto, R.; et al. Embelin binds to human neuroserpin and impairs its polymerisation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, N.; Gnanamani, A.; Prasad, N.R.; Mandal, A.B. Inhibition of UVB-induced oxidative damage and apoptotic biochemical changes in human lymphocytes by 2,5-dihydroxy-3-undecyl-1,4-benzoquinone (embelin). Int. J. Radiat. Biol. 2012, 88, 575–582. [Google Scholar] [CrossRef]
- Kundap, U.P.; Bhuvanendran, S.; Kumari, Y.; Othman, I.; Shaikh, M.F. Plant Derived Phytocompound, Embelin in CNS Disorders: A Systematic Review. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef]
- Pathan, S.A.; Iqbal, Z.; Zaidi, S.; Talegaonkar, S.; Vohra, D.; Jain, G.K.; Azeem, A.; Jain, N.; Lalani, J.R.; Khar, R.K.; et al. CNS drug delivery systems: Novel approaches. Recent. Pat. Drug Deliv. Formul. 2009, 3, 71–89. [Google Scholar] [CrossRef]
- Filosa, R.; Peduto, A.; Aparoy, P.; Schaible, A.M.; Luderer, S.; Krauth, V.; Petronzi, C.; Massa, A.; de Rosa, M.; Reddanna, P.; et al. Discovery and biological evaluation of novel 1,4-benzoquinone and related resorcinol derivatives that inhibit 5-lipoxygenase. Eur. J. Med. Chem. 2013, 67, 269–279. [Google Scholar] [CrossRef]
- Filosa, R.; Peduto, A.; Schaible, A.M.; Krauth, V.; Weinigel, C.; Barz, D.; Petronzi, C.; Bruno, F.; Roviezzo, F.; Spaziano, G.; et al. Novel series of benzoquinones with high potency against 5-lipoxygenase in human polymorphonuclear leukocytes. Eur. J. Med. Chem. 2015, 94, 132–139. [Google Scholar] [CrossRef]
- Martín-Acosta, P.; Haider, S.; Amesty, Á.; Aichele, D.; Jose, J.; Estévez-Braun, A. A new family of densely functionalized fused-benzoquinones as potent human protein kinase CK2 inhibitors. Eur. J. Med. Chem. 2018, 144, 410–423. [Google Scholar] [CrossRef]
- Lamblin, M.; Sallustrau, A.; Commandeur, C.; Cresteil, T.; Felpin, F.-X.; Dessolin, J. Synthesis and biological evaluation of hydrophilic embelin derivatives. Tetrahedron 2012, 68, 4655–4663. [Google Scholar] [CrossRef]
- Ogata, T.; Doe, M.; Matsubara, A.; Torii, E.; Nishiura, C.; Nishiuchi, A.; Kobayashi, Y.; Kimachi, T. Studies on the oxidative cyclization of 3-hydroxyalkyl-1,2,4-trialkoxynaphthalenes and synthetic application for the biologically active natural compound rhinacanthone. Tetrahedron 2014, 70, 502–509. [Google Scholar] [CrossRef]
- Kwon, Y.-J.; Sohn, M.-J.; Kim, C.-J.; Koshino, H.; Kim, W.-G. Flavimycins A and B, Dimeric 1,3-Dihydroisobenzofurans with Peptide Deformylase Inhibitory Activity from Aspergillus flavipes. J. Nat. Prod. 2012, 75, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Pan, S.C. Total synthesis of the novel, biologically active epoxyquinone dimer (+/-)-torreyanic acid: A biomimetic approach. Org. Lett. 2004, 6, 3985–3988. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Jensen, J.K.; Hong, Z.; Shi, X.; Hu, L.; Andreasen, P.A.; Huang, M. Structural insight into inactivation of plasminogen activator inhibitor-1 by a small-molecule antagonist. Chem. Biol. 2013, 20, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Zhang, G.; Hong, Z.; Lin, Z.; Lei, M.; Huang, M.; Hu, L. Design, synthesis, and SAR of embelin analogues as the inhibitors of PAI-1 (plasminogen activator inhibitor-1). Bioorg. Med. Chem. Lett. 2014, 24, 2379–2382. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gao, M.; Jian, R.; Hong, W.D.; Tang, X.; Li, Y.; Zhao, D.; Zhang, K.; Chen, W.; Zheng, X.; et al. Design, synthesis and α-glucosidase inhibition study of novel embelin derivatives. J. Enzyme Inhib. Med. Chem. 2020, 35, 565–573. [Google Scholar] [CrossRef]
- Miranda, E.; Römisch, K.; Lomas, D.A. Mutants of neuroserpin that cause dementia accumulate as polymers within the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 28283–28291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belorgey, D.; Crowther, D.C.; Mahadeva, R.; Lomas, D.A. Mutant Neuroserpin (S49P) that causes familial encephalopathy with neuroserpin inclusion bodies is a poor proteinase inhibitor and readily forms polymers in vitro. J. Biol. Chem. 2002, 277, 17367–17373. [Google Scholar] [CrossRef] [Green Version]
- Irving, J.A.; Ekeowa, U.I.; Belorgey, D.; Haq, I.; Gooptu, B.; Miranda, E.; Pérez, J.; Roussel, B.D.; Ordóñez, A.; Dalton, L.E.; et al. The serpinopathies studying serpin polymerization in vivo. Methods Enzymol. 2011, 501, 421–466. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.S.; Rimando, A.M.; Duke, S.O. Phytotoxic activity of quinones and resorcinolic lipid derivatives. J. Agric. Food Chem. 2010, 58, 4353–4355. [Google Scholar] [CrossRef] [PubMed]
- Trapella, C.; Fischetti, C.; Lazzari, I.; Guerrini, R.; Rizzi, A.; Camarda, V.; Lambert, D.G.; McDonald, J.; Regoli, D.; Salvadori, S. Structure-activity studies on the nociceptin/orphanin FQ receptor antagonist 1-benzyl-N-{3-[spiroisobenzofuran-1(3H),4’-piperidin-1-yl]propyl} pyrrolidine-2-carboxamide. Bioorg. Med. Chem. 2009, 17, 5080–5095. [Google Scholar] [CrossRef] [PubMed]
- Reinaud, O.; Capdevielle, P.; Maumy, M. Synthesis of New 3-(-2-Alkenyl)-2-hydroxy-5-methoxy-p-benzoquinones via Claisen Rearrangement of Original 5-Methoxy-4-(2-propenyloxy)-o-benzoquinones. Synthesis 1988, 1988, 293–300. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pol | Olig | Olig12 | Mon | |
|---|---|---|---|---|
| Control | 73% | 0% | 5% | 22% |
| EMB, 1a | 2% | 37% | 48% | 14% |
| 1b | 80% | 0% | 3% | 17% |
| 1c | 10% | 50% | 27% | 13% |
| 1d | 54% | 0% | 0% | 46% |
| 1e | 66% | 0% | 0% | 34% |
| 1f | 69% | 0% | 0% | 31% |
| 1g | 52% | 0% | 0% | 48% |
| 2a | 3% | 44% | 23% | 30% |
| 3a | 7% | 35% | 38% | 20% |
| 2e | 79% | 0% | 0% | 21% |
| 3e | 77% | 0% | 0% | 23% |
| 4 | 73% | 0% | 11% | 16% |
| 5 | 76% | 0% | 0% | 24% |
| 6 | 66% | 0% | 0% | 34% |
| 7a | 56% | 8% | 0% | 35% |
| 7b | 70% | 06% | 0% | 24% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visentin, C.; Musso, L.; Broggini, L.; Bonato, F.; Russo, R.; Moriconi, C.; Bolognesi, M.; Miranda, E.; Dallavalle, S.; Passarella, D.; et al. Embelin as Lead Compound for New Neuroserpin Polymerization Inhibitors. Life 2020, 10, 111. https://doi.org/10.3390/life10070111
Visentin C, Musso L, Broggini L, Bonato F, Russo R, Moriconi C, Bolognesi M, Miranda E, Dallavalle S, Passarella D, et al. Embelin as Lead Compound for New Neuroserpin Polymerization Inhibitors. Life. 2020; 10(7):111. https://doi.org/10.3390/life10070111
Chicago/Turabian StyleVisentin, Cristina, Loana Musso, Luca Broggini, Francesca Bonato, Rosaria Russo, Claudia Moriconi, Martino Bolognesi, Elena Miranda, Sabrina Dallavalle, Daniele Passarella, and et al. 2020. "Embelin as Lead Compound for New Neuroserpin Polymerization Inhibitors" Life 10, no. 7: 111. https://doi.org/10.3390/life10070111