4.1. Bioinformatics Pipeline
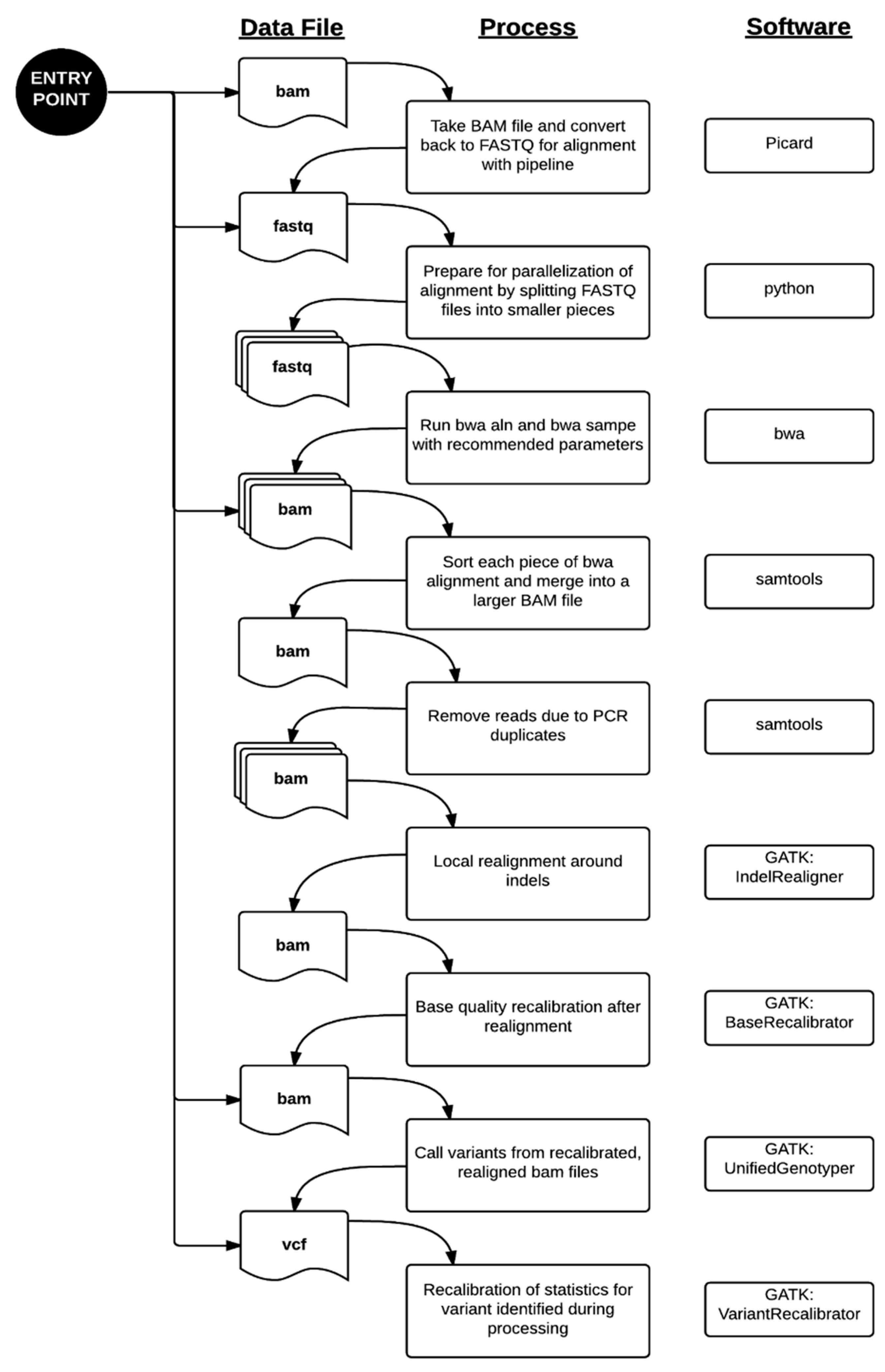
The bioinformatics pipeline processes data from the hard drives delivered by Illumina Clinical Services Laboratory (San Diego, CA, USA) to the variant report files generated for clinical interpretation in a fairly automated fashion. As these hard drives are received, they are accessioned in the laboratory and uploaded to our high performance computing (HPC) environment. The BAM files delivered on the hard drives contain reads aligned by the pipeline developed at Illumina using CASAVA (or ISAAC in recent data deliveries). These files contained both aligned and unaligned reads, so the conversion from BAM back to FASTQ format to decouple mapping information from the read sequence does not incur loss of sequencing information.
Data processing from the FASTQ file to the VCF file was performed primarily using the parameters recommended by each respective software package [
10,
11,
12,
13]. Paired-end alignment of the sequencing reads to the hg19 reference genome was performed using bwa 0.6.1-r104. The aligned reads were sorted and PCR duplicates removed using samtools 0.1.18. Local indel realignment, base quality recalibration, variant calling by UnifiedGenotyper, and variant recalibration were performed using Genome Analysis ToolKit (GATK) 2.2.5 and the recommendations in the Best Practices Workflow by the GATK development team at the Broad Institute (Cambridge, MA, USA). The entire workflow is summarized in
Figure 1. For more efficient computing, our validated process includes parallelization by dividing files into smaller pieces for even distribution across the cluster. This method was described in our application of this pipeline to the MedSeq Project, a randomized clinical trial assessing the impact of genome sequencing in clinical practice [
14] and to the discovery of a putative locus causing a novel recessive syndrome presenting with skeletal malformation and malignant lymphoproliferative disease [
15]. Similar pipelines have been used successfully in other clinical labs using NGS previously [
4,
6,
16,
17,
18].
Successive variant annotation is accomplished through a set of scripts that independently annotate the dataset with a wide collection of information, including: (1) transcript and gene annotations from Alamut (Interactive Biosoftware, Rouen, France); (2) gene annotations from variant effect predictor (VEP); (3) variant annotations from 1000 Genomes Project, ClinVar, and Exome Sequencing Project (ESP); and (4) clinical interpretation from our laboratory maintained in GeneInsight (Cambridge, MA, USA) [
19,
20,
21,
22]. As a clinical laboratory, we are focused on genomic regions that are known or likely to be associated with disease. Thus, we have limited the annotation of variants called on the genome to only those that lie in our target regions. The target region contains the RefSeq coding sequence for all genes mapped to hg19, pharmacogenomic variants, and association or risk variants taken from the NHGRI GWAS catalog [
23]. These regions are buffered by 50 base pairs (bp). The VCF file is limited to only this target region prior to variant annotation. Additionally, during filtration and coverage analysis, we focus in on the regions most likely to contain clinically reportable disease causing variants. Thus, we created clinical region of interest (ROI) files containing all coding regions of all exons for each gene ±15 bp or ±2 bp for filtration and coverage, respectively.
The sample data and variant annotations are uploaded to an Oracle SQL database. The variant filtration process was designed to query this database and return the variants of interest in an excel spreadsheet for careful clinical review. Variant filtration has been an evolving process, and filtration specifications vary depending on the prevalence, penetrance, and predicted mode of inheritance of the genetic disorder.
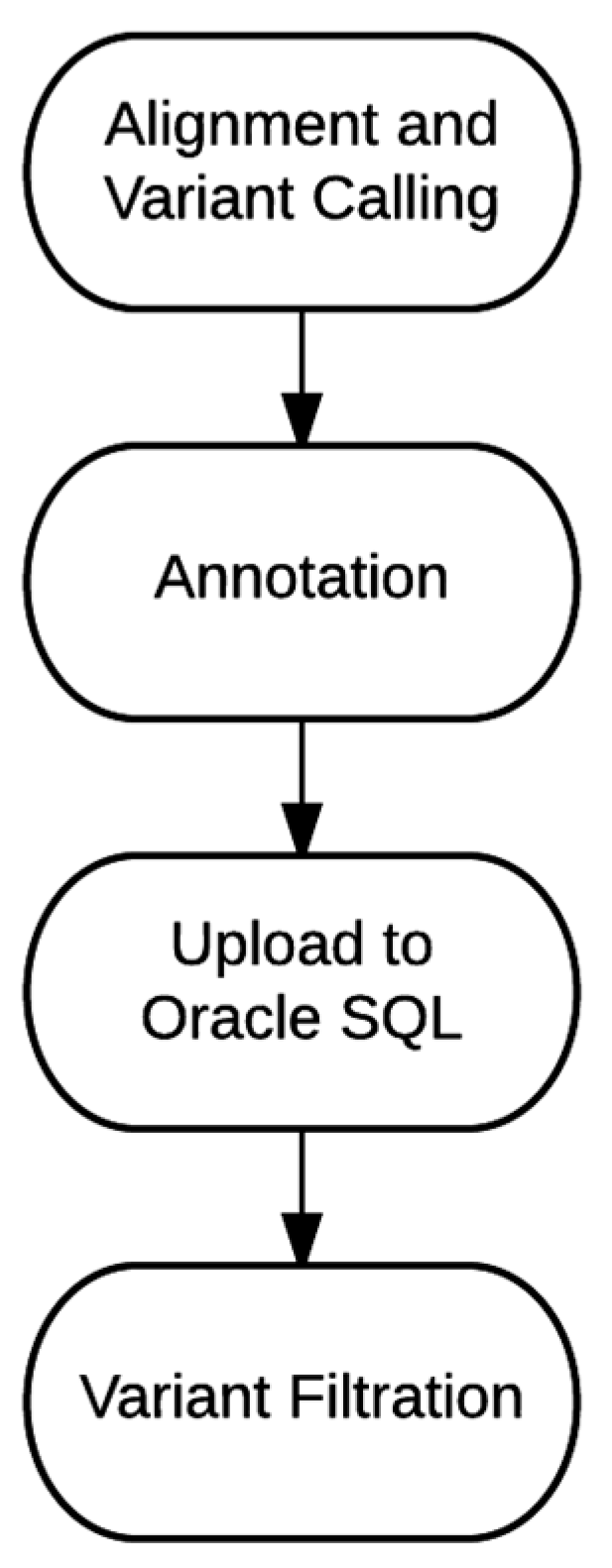
Table 4 lists some of the common filtrations used to generate the final report. Often, the final variant report is generated using Boolean logic to combine more than one of these filters. The overview of the entire bioinformatics process, from alignment and variant calling to final report generation is outlined in
Figure 2.
4.2. Bioinformatics Validation
The WGS dataset of NA12878 retrieved from Coriell Cell Repositories (Camden, NJ, USA) and sequenced at Illumina was used to validate the efficacy of this pipeline. The pipeline was run twice on the exact same dataset to test the robustness of the pipeline, proving it can consistently deliver the same results for a given dataset. For this test, the pipeline was triggered from two different upstream entry points, Illumina-aligned BAM files and FASTQ files, and the final variant output files of this test were compared. Another source of validation comes from understanding the sensitivity and specificity of the variant calling pipeline. These metrics were estimated using different standard datasets that have assessed the genome level variation on NA12878. The variant calls were first compared to internally generated sequencing data. NA12878 has been the validation standard of many laboratories, and we have sequenced about 700 kb of sequence across 195 genes from our cardiomyopathy, hearing loss, respiratory, Noonan syndrome, and Marfan syndrome tests using a combination of orthogonal technologies including Sanger sequencing, array-based sequencing, and targeted next-generation sequencing [
2,
24,
25]. Consequently, we estimated sensitivity and specificity of our WGS workflow using only this specific region of interest. As recommended for earlier versions of GATK, we varied the QD and FS filters for variants called to find the optimal threshold. We additionally checked the concordance rate of our genome-level data against the variants in NA12878 reported by the 1000 Genomes Project dataset of high coverage WGS (>30× average coverage) [
19]. The variants were assessed by their presence in each VCF file (variant concordance) as well as the concordance of the genotype called for each variant (genotype concordance).
4.3. Characterization of Poorly-Covered Regions
We selected 15 genomes sequenced at Illumina Clinical Services Laboratory and processed through our bioinformatics pipeline in FY2014 to generate a coverage list for the coding sequence of RefSeq genes plus 2 bp of buffer to encapsulate splice sites. We customized GATK 2.2.5 to run CallableLoci to determine the callability of every coding base pair, where “callable” is defined as a base having at least 8X of coverage from quality reads and less than 10% of the total reads in that region have poor mapping quality. The average of the callable base pairs for each gene across 15 genomes was calculated for this coverage list. Our pipeline did not perform variant calling on chrMT, so mitochondrial genes were excluded from this list.
Next, we wanted to assess the clinical relevance of the poorly covered regions identified in the coverage list. To do so, we selected the ClinVar dataset of reported variants, a public archive of variants and their clinical significance as contributed by the medical genetics community [
26]. We selected “Pathogenic” and “Likely pathogenic” variants interpreted by the clinical significance element in the XML release of the ClinVar dataset (August 2015 release). ClinVar has several categorizations of variants, and we define a unique variant as one having a unique MeasureSet ID. To identify clinically relevant genes that are currently assayed in clinical sequencing laboratories, we focused on variants submitted by laboratories and working groups. To do this, we removed variant classifications from large, research-grade databases, including OMIM. MeasureSet IDs with submissions of conflicting clinical significance, such as being reported as pathogenic by one laboratory but benign in a different laboratory, were also excluded. However, an assertion of pathogenic in one laboratory but uncertain significance in another laboratory would be consistent and therefore not counted as a conflicted variant.
4.4. Cost Analysis and Scalability
WGS is more computationally intensive and demanding of storage than other current sequencing solutions. It incurs additional costs as clinical sequencing requires the data to be preserved for a set amount of time. The computational costs were estimated by the price of purchasing the compute nodes adjusted for the amount of CPU-hours necessary for a genome run and the lifespan of the hardware, which is guaranteed for five years at Partners HealthCare. The hands-on time was estimated by the price we charge per hour for bioinformatics analysis multiplied by the time necessary to successfully trigger the pipeline, deliver the variants to the geneticists, and archive the data. We also calculated the cost of storage based on current internal prices offered. The cluster resources used to run the genome sequencing pipeline is shared across many bioinformatics processes. At present, we have 10 compute nodes, each with 128 GB RAM and 16-core Intel Xeon 2.60 GHz processors running CentOS 6.5 and IBM LSF job scheduler.


 ,
, 


{kind=link}
{kind=link}