
Immunosenescence and Aging: Neuroinflammation Is a Prominent Feature of Alzheimer’s Disease and Is a Likely Contributor to Neurodegenerative Disease Pathogenesis


{kind=link}
{kind=link}
Abstract
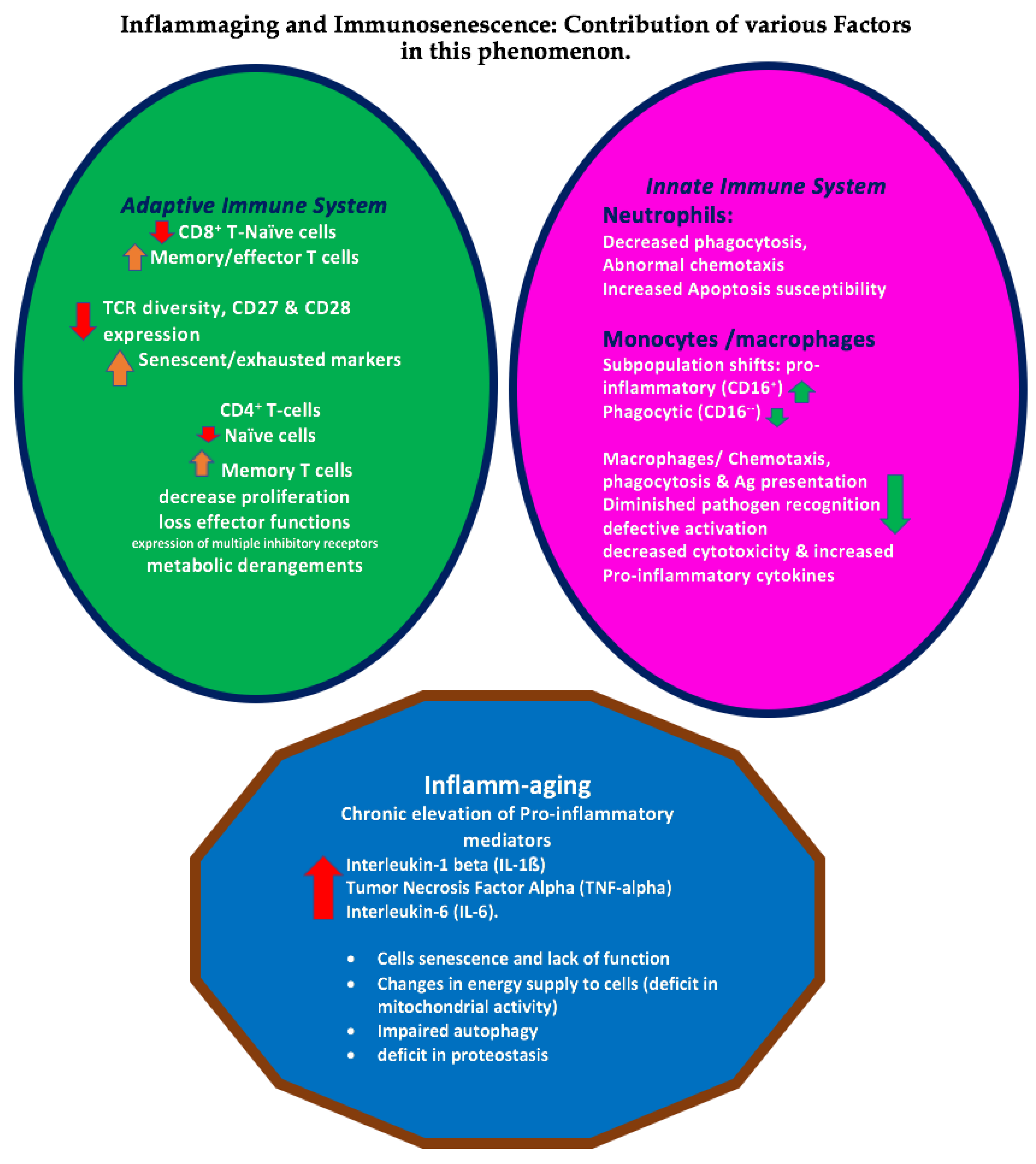
:1. Introduction
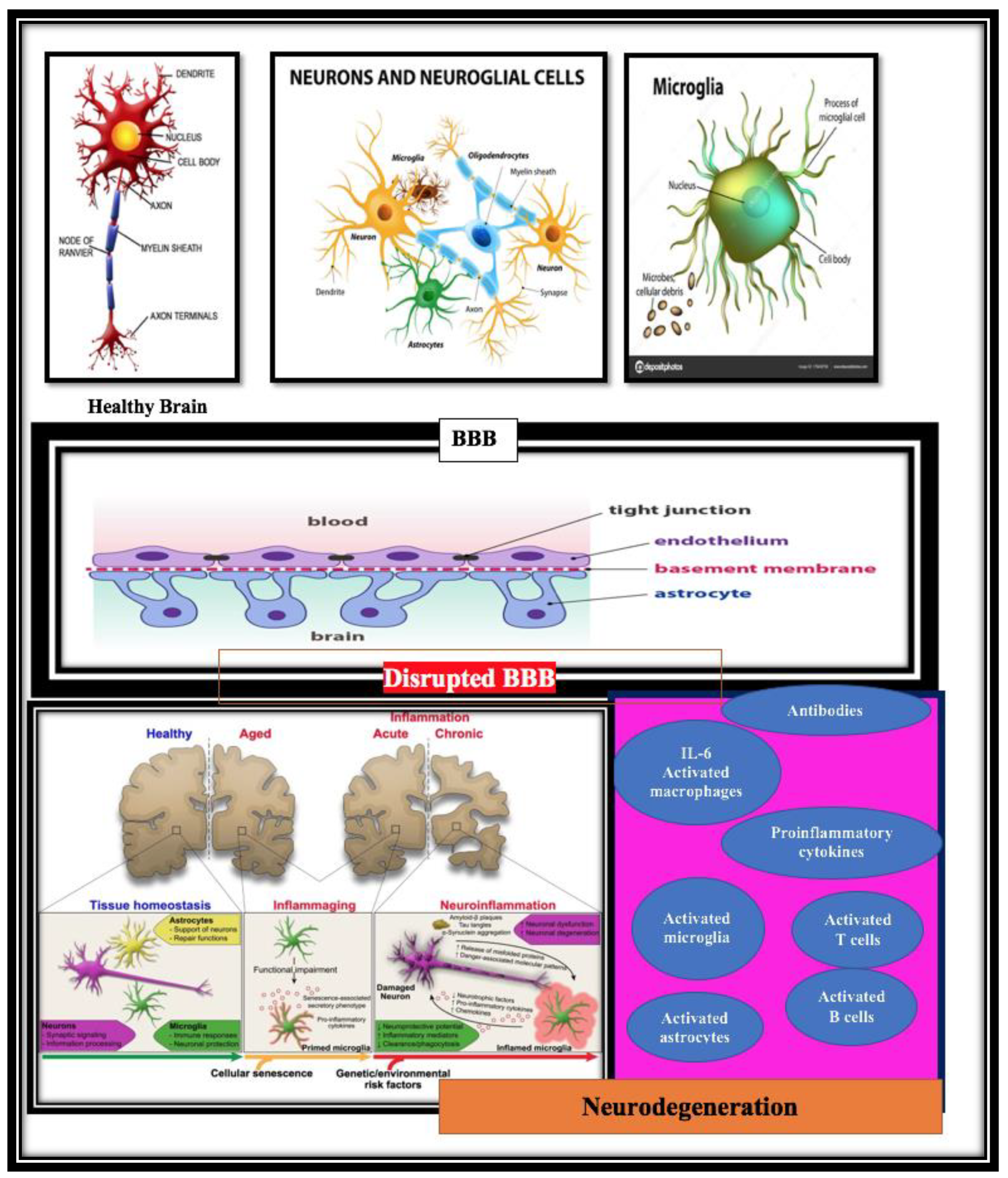
2. Neuroimmune Involvement in the of Pathogenesis of AD
3. Immune System and Alzheimer’s Diseases (AD)—The Microglia
4. Astrocytes
5. Lymphocytes
6. Cytokines
7. Monocytes and Macrophages
8. Discussion
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Alzheimer’s disease | (AD) |
| Amyloid-beta | (Aβ) |
| Neurofibrillary tangles | (NFT) |
| Central nervous system | (CNS) |
| Cluster of differentiation | (CD) |
| Blood–brain barrier | (BBB) |
| Interferon gamma | (IFN-γ) |
| Tumor necrosis factor | (TNF) |
| Hydrogen peroxide | (H2O2) |
| Helper T cells | (Th) |
| Polymorphonuclear leukocytes | (PMN) |
| Hematopoietic stem cell | (HSC) |
| Human cytomegalovirus | (HCMV) |
| Immunoglobulin G | IgG |
| Immunoglobulin A | IgA |
| Interleukin | (IL) |
| Transforming growth factor-β | (TGFβ) |
References
- Alzheimer, A. Über einen eigenartigen schweren Erkrankungsprozeb der Hirnrincle. Neurol. Central. 1906, 25, 146–148. [Google Scholar]
- Möller, H.J.; Graeber, M.B. The case described by Alois Alzheimer in 1911. Eur. Arch. Psychiatry Clin. Neurosci. 1998, 248, 111–122. [Google Scholar]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Samanta, S.; Ramesh, M.; Govindaraju, T. Alzheimer’s is a Multifactorial Disease. In Alzheimer’s Disease: Recent Findings in Pathophysiology, Diagnostic and Therapeutic Modalities; Royal Society of Chemistry: London, UK, 2022; pp. 1–34. [Google Scholar]
- Avila, J. Intracellular and extracellular tau. Front. Neurosci. 2010, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Griffin, W.S.T.; Braak, H. Parenchymal and vascular Aβ-deposition and its effects on the degeneration of neurons and cognition in Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 1848–1862. [Google Scholar] [CrossRef] [Green Version]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar]
- Zotova, E.; Nicoll, J.A.; Kalaria, R.; Holmes, C.; Boche, D. Inflammation in Alzheimer’s disease: Relevance to pathogenesis and therapy. Alzheimer’s Res. Ther. 2010, 2, 1. [Google Scholar] [CrossRef]
- Dineen, R.A.; Vilisaar, J.; Hlinka, J.; Bradshaw, C.M.; Morgan, P.; Constantinescu, C.; Auer, D. Disconnection as a mechanism for cognitive dysfunction in multiple sclerosis. Brain 2009, 132, 239–249. [Google Scholar] [CrossRef]
- Delbeuck, X.; Van Der Linden, M.; Collette, F. Alzheimer’ Disease as a Disconnection Syndrome? Neuropsychol. Rev. 2003, 13, 79–92. [Google Scholar] [CrossRef]
- Tajima, H.; Kita, Y. Neuronal Cell Death in Alzheimers Disease and a Neuroprotective Factor, Humanin. Curr. Neuropharmacol. 2006, 4, 139–147. [Google Scholar] [CrossRef]
- Alifragis, P.; Marsh, J. Synaptic dysfunction in Alzheimer’s disease: The effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen. Res. 2018, 13, 616–623. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Terry, A.V.; Buccafusco, J.J. The Cholinergic Hypothesis of Age and Alzheimer’s Disease-Related Cognitive Deficits: Recent Challenges and Their Implications for Novel Drug Development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of Amyloid Precursor Protein in the Mitochondrial Import Channels of Human Alzheimer’s Disease Brain Is Associated with Mitochondrial Dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [Green Version]
- Frozza, R.L.; Lourenco, M.; De Felice, F.G. Challenges for Alzheimer’s Disease Therapy: Insights from Novel Mechanisms Beyond Memory Defects. Front. Neurosci. 2018, 12, 37. [Google Scholar] [CrossRef]
- Tatulian, S.A. Challenges and hopes for Alzheimer’s disease. Drug Discov. Today 2022, 27, 1027–1043. [Google Scholar]
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. J. Cereb. Blood Flow Metab. 2019, 176, 3447–3463. [Google Scholar] [CrossRef]
- Atri, A. The Alzheimer’s disease clinical spectrum: Diagnosis and management. Med. Clin. 2019, 103, 263–293. [Google Scholar]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frolich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in alzheimer’s disease: The path to 2025. Alzheimers Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Central Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, F.; Guo, J.; Xu, C.; Cao, Y.; Fang, Z.; Wang, Q. Pharmacological Mechanisms Underlying the Neuroprotective Effects of Alpinia oxyphylla Miq. on Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2071. [Google Scholar] [CrossRef] [Green Version]
- Frost, G.R.; Jonas, L.A.; Li, Y.M. Friend, foe or both? Immune activity in Alzheimer’s disease. Front. Aging Neurosci. 2019, 11, 337. [Google Scholar]
- Golde, T.E. Alzheimer’s disease—The journey of a healthy brain into organ failure. Mol. Neurodegener. 2022, 17, 1–19. [Google Scholar]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Sengoku, R. Aging and Alzheimer’s disease pathology. Neuropathology 2019, 40, 22–29. [Google Scholar] [CrossRef]
- Price, M.; Bellwood, P.; Kitson, N.; Davies, I.; Weber, J.; Lau, F. Conditions potentially sensitive to a Personal Health Record (PHR) intervention, a systematic review. BMC Med. Inform. Decis. Mak. 2015, 15, 32. [Google Scholar] [CrossRef]
- Kormas, P.; Moutzouri, A. Current Psychological Approaches in Neurodegenerative Diseases. In Handbook of Computational Neurodegeneration; Springer International Publishing: Cham, Switzerland, 2022; pp. 1–29. [Google Scholar] [CrossRef]
- Velardi, E.; Tsai, J.J.; van den Brink, M.R. T cell regeneration after immunological injury. Nat. Rev. Immunol. 2020, 21, 277–291. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef]
- Van Eldik, L.J.; Carrillo, M.C.; Cole, P.E.; Feuerbach, D.; Greenberg, B.D.; Hendrix, J.A.; Kennedy, M.; Kozauer, N.; Margolin, R.A.; Molinuevo, J.L.; et al. The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 99–109. [Google Scholar]
- Lutshumba, J.; Nikolajczyk, B.S.; Bachstetter, A.D. Dysregulation of Systemic Immunity in Aging and Dementia. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Kopf, M. On the Role of the Innate Immunity in Autoimmune Disease. J. Exp. Med. 2001, 193, F47–F50. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C.A., Jr. Innate immunity: Impact on the adaptive immune response. Curr. Opin. Immunol. 1997, 9, 4–9. [Google Scholar]
- Huang, Y.; Chen, Z. Inflammatory bowel disease related innate immunity and adaptive immunity. Am. J. Transl. Res. 2016, 8, 2490–2497. [Google Scholar]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate Immunity and Neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef]
- Kumar, V. Macrophages: The Potent Immunoregulatory Innate Immune Cells. In Macrophage Activation—Biology and Disease; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef] [Green Version]
- Linehan, E.; Fitzgerald, D. Ageing and the immune system: Focus on macrophages. Eur. J. Microbiol. Immunol. 2015, 5, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlang, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Shastri, A.; Bonifati, D.M.; Kishore, U. Innate Immunity and Neuroinflammation. Mediat. Inflamm. 2013, 2013, 342931. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Matejuk, A.; Vandenbark, A.A.; Offner, H. Cross-Talk of the CNS With Immune Cells and Functions in Health and Disease. Front. Neurol. 2021, 12, 672455. [Google Scholar] [CrossRef]
- Bachiller, S.; Ferrer, I.J.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [Green Version]
- Harry, G.J.; Kraft, A.D. Microglia in the developing brain: A potential target with lifetime effects. NeuroToxicology 2012, 33, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The Role of Microglia and Macrophages in CNS Homeostasis, Autoimmunity, and Cancer. J. Immunol. Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef] [Green Version]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Gómez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of Microglial Proliferation during Chronic Neurodegeneration. J. Neurosci. 2013, 33, 2481–2493. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Nicola, D.; Perry, V.H. Microglial dynamics and role in the healthy and diseased brain: A paradigm of functional plasticity. Neuroscientist 2015, 21, 169–184. [Google Scholar]
- Cuadros, M.A.; Navascués, J. The origin and differentiation of microglial cells during development. Prog. Neurobiol. 1998, 56, 173–189. [Google Scholar]
- Jimenez-Ferrer, I.; Jewett, M.; Tontanahal, A.; Romero-Ramos, M.; Swanberg, M. Allelic difference in Mhc2ta confers altered microglial activation and susceptibility to α-synuclein-induced dopaminergic neurodegeneration. Neurobiol. Dis. 2017, 106, 279–290. [Google Scholar] [CrossRef]
- Ventura, M.T.; Casciaro, M.; Gangemi, S.; Buquicchio, R. Immunosenescence in aging: Between immune cells depletion and cytokines up-regulation. Clin. Mol. Allergy 2017, 15, 21. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Hirokawa, K.; Cohen, A.A.; Witkowski, J.M. Immunosenescence is both functional/adaptive and dysfunctional/maladaptive. Semin. Immunopathol. 2020, 42, 521–536. [Google Scholar] [CrossRef]
- Encarnacion, M.R.; Beata, B.M.; Kenneth, D. Cause, consequences, and reversal of immune system aging. J. Clin. Investig. 2013, 123, 958–965. [Google Scholar]
- Crooke, S.N.; Ovsyannikova, I.G.; Poland, G.A.; Kennedy, R.B. Immunosenescence and human vaccine immune responses. Immun. Ageing 2019, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Rolandi, E.; Zaccaria, D.; Vaccaro, R.; Abbondanza, S.; Pettinato, L.; Davin, A.; Guaita, A. Estimating the potential for dementia prevention through modifiable risk factors elimination in the real-world setting: A population-based study. Alzheimer’s Res. Ther. 2020, 12, 94. [Google Scholar] [CrossRef]
- Teresa, N.; Linda, P. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar]
- Lee, K.-A.; Flores, R.R.; Jang, I.H.; Saathoff, A.; Robbins, P.D. Immune Senescence, Immunosenescence and Aging. Front. Aging 2022, 3, 900028. [Google Scholar] [CrossRef]
- Bossù, P.; Toppi, E.; Sterbini, V.; Spalletta, G. Implication of Aging Related Chronic Neuroinflammation on COVID-19 Pandemic. J. Pers. Med. 2020, 10, 102. [Google Scholar] [CrossRef]
- Benedetto, S.D.; Müller, L. Aging, Immunity, and Neuroinflammation: The Modulatory Potential of Nutrition. In Nutrition and Immunity; Springer: Cham, Switzerland, 2019; pp. 301–322. [Google Scholar]
- Luo, X.-G.; Ding, J.-Q.; Chen, S.-D. Microglia in the aging brain: Relevance to neurodegeneration. Mol. Neurodegener. 2010, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.G. Defining activation states of microglia in human brain tissue: An unresolved issue for Alzheimer’s disease. Neuroimmunol. Neuroinflamm. 2020, 7, 194–214. [Google Scholar] [CrossRef]
- Chen, Y.; Hong, T.; Chen, F.; Sun, Y.; Wang, Y.; Cui, L. Interplay Between Microglia and Alzheimer’s Disease—Focus on the Most Relevant Risks: APOE Genotype, Sex and Age. Front. Aging Neurosci. 2021, 13, 631827. [Google Scholar]
- Heavener, K.S.; Bradshaw, E.M. The aging immune system in Alzheimer’s and Parkinson’s diseases. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2022; pp. 1–9. [Google Scholar]
- Mayne, K.; White, J.A.; Mcmurran, C.E.; Rivera, F.J.; De La Fuente, A.G. Aging and Neurodegenerative Disease: Is the Adaptive Immune System a Friend or Foe? Front. Aging Neurosci. 2020, 12, 572090. [Google Scholar] [CrossRef]
- Epelman, S.; LaVine, K.J.; Randolph, G.J. Origin and Functions of Tissue Macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Hammond, T.R.; Robinton, D.; Stevens, B. Microglia and the Brain: Complementary Partners in Development and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 523–544. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef]
- Streit, W.J.; Braak, H.; Xue, Q.-S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Shahidehpour, R.K.; Higdon, R.E.; Crawford, N.G.; Neltner, J.H.; Ighodaro, E.T.; Patel, E.; Price, D.; Nelson, P.T.; Bachstetter, A.D. Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol. Aging 2021, 99, 19–27. [Google Scholar] [CrossRef]
- St-Pierre, M.-K.; Carrier, M.; Ibáñez, F.G.; Šimončičová, E.; Wallman, M.-J.; Vallières, L.; Parent, M.; Tremblay, M. Ultrastructural characterization of dark microglia during aging in a mouse model of Alzheimer’s disease pathology and in human post-mortem brain samples. J. Neuroinflamm. 2022, 19, 1–22. [Google Scholar] [CrossRef]
- Caldeira, C.; Cunha, C.; Vaz, A.R.; Falcão, A.S.; Barateiro, A.; Seixas, E.; Fernandes, A.; Brites, D. Key Aging-Associated Alterations in Primary Microglia Response to Beta-Amyloid Stimulation. Front. Aging Neurosci. 2017, 9, 277. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Fryatt, G.L.; Ghorbani, M.; Obst, J.; Menassa, D.A.; Martin-Estebane, M.; Muntslag, T.A.; Olmos-Alonso, A.; Guerrero-Carrasco, M.; Thomas, D.; et al. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Aβ pathology. Cell Rep. 2021, 35, 109228. [Google Scholar] [CrossRef]
- Yu, H.-M.; Zhao, Y.-M.; Luo, X.-G.; Feng, Y.; Ren, Y.; Shang, H.; He, Z.-Y.; Chen, S.-D.; Wang, X.-Y. Repeated Lipopolysaccharide Stimulation Induces Cellular Senescence in BV2 Cells. Neuroimmunomodulation 2012, 19, 131–136. [Google Scholar] [CrossRef]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar]
- Bradshaw, E.M.; Initiative, T.A.D.N.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s Disease Risk Gene CD33 Inhibits Microglial Uptake of Amyloid Beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Salih, D.A.; Bayram, S.; Guelfi, S.; Reynolds, R.H.; Shoai, M.; Ryten, M.; Brenton, J.W.; Zhang, D.; Matarin, M.; Botia, J.A.; et al. Genetic variability in response to amyloid beta deposition influences Alzheimer’s disease risk. Brain Commun. 2019, 1, fcz022. [Google Scholar] [CrossRef] [Green Version]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Galatro, T.; Holtman, I.R.; Lerario, A.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Olah, M.; Patrick, E.; Villani, A.-C.; Xu, J.; White, C.C.; Ryan, K.J.; Piehowski, P.; Kapasi, A.; Nejad, P.; Cimpean, M.; et al. A transcriptomic atlas of aged human microglia. Nat. Commun. 2018, 9, 539. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.E9. [Google Scholar]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [Green Version]
- Nordengen, K.; Kirsebom, B.E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S.B.; Grøntvedt, G.R.; Aarsland, D.; Nilsson, L.N.; et al. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflamm. 2019, 16, 46. [Google Scholar]
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 2002, 124, 83–92. [Google Scholar] [CrossRef]
- Merlini, M.; Kirabali, T.; Kulic, L.; Nitsch, R.M.; Ferretti, M.T. Extravascular CD3+ T Cells in Brains of Alzheimer Disease Patients Correlate with Tau but Not with Amyloid Pathology: An Immunohistochemical Study. Neurodegener. Dis. 2018, 18, 49–56. [Google Scholar] [CrossRef]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Cohen, S.; Doyle, W.J.; Skoner, D.P.; Rabin, B.S.; Gwaltney, J.M. Social ties and susceptibility to the common cold. JAMA 1997, 277, 1940–1944. [Google Scholar]
- Michell-Robinson, M.; Touil, H.; Healy, L.; Owen, D.; Durafourt, B.; Bar-Or, A.; Antel, J.; Moore, C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef] [Green Version]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar]
- Boullerne, A.I.; Feinstein, D.L. History of Neuroscience I. Pío del Río-Hortega (1882–1945): The discoverer of microglia and oligodendroglia. ASN Neuro 2020, 12, 1759091420953259. [Google Scholar]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar]
- Florent, G.; Melanie, G.; Marylene, L.; Sayan, N.; Peter, S.; Solen, G.; Mehler, M.F.; Conway, S.J.; Guan, N.L.; Richard, S.E. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar]
- Sousa, C.; Biber, K.; Michelucci, A. Cellular and Molecular Characterization of Microglia: A Unique Immune Cell Population. Front. Immunol. 2017, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Majumdar, A.; Cruz, D.; Asamoah, N.; Buxbaum, A.; Sohar, I.; Lobel, P.; Maxfield, F.R. Activation of Microglia Acidifies Lysosomes and Leads to Degradation of Alzheimer Amyloid Fibrils. Mol. Biol. Cell 2007, 18, 1490–1496. [Google Scholar] [CrossRef] [Green Version]
- Button, E.B.; Mitchell, A.S.; Domingos, M.M.; Chung, J.H.-J.; Bradley, R.M.; Hashemi, A.; Marvyn, P.M.; Patterson, A.C.; Stark, K.D.; Quadrilatero, J.; et al. Microglial Cell Activation Increases Saturated and Decreases Monounsaturated Fatty Acid Content, but Both Lipid Species are Proinflammatory. Lipids 2014, 49, 305–316. [Google Scholar] [CrossRef]
- De Biase, L.M.; Schuebel, K.E.; Fusfeld, Z.H.; Jair, K.; Hawes, I.A.; Cimbro, R.; Zhang, H.Y.; Liu, Q.R.; Shen, H.; Xi, Z.X.; et al. Local cues establish and maintain region-specific phenotypes of basal ganglia microglia. Neuron 2017, 95, 341–356. [Google Scholar]
- Chiu, I.M.; Morimoto, E.T.; Goodarzi, H.; Liao, J.T.; O’Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; et al. A Neurodegeneration-Specific Gene-Expression Signature of Acutely Isolated Microglia from an Amyotrophic Lateral Sclerosis Mouse Model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.-C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Harry, G.J.; Kraft, A.D. Neuroinflammation and microglia: Considerations and approaches for neurotoxicity assessment. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1265–1277. [Google Scholar] [CrossRef]
- Eyo, U.; Dailey, M.E. Microglia: Key Elements in Neural Development, Plasticity, and Pathology. J. Neuroimmune Pharmacol. 2013, 8, 494–509. [Google Scholar] [CrossRef] [Green Version]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.; Tang, Y. Phagocytosis of Microglia in the Central Nervous System Diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [Green Version]
- Ousman, S.S.; Kubes, P. Immune surveillance in the central nervous system. Nat. Neurosci. 2012, 15, 1096–1101. [Google Scholar] [CrossRef]
- Amor, S.; Woodroofe, M.N. Innate and adaptive immune responses in neurodegeneration and repair. Immunology 2014, 141, 287–291. [Google Scholar] [CrossRef]
- McGeer, E.G.; McGeer, P.L. The importance of inflammatory mechanisms in alzheimer disease. Exp. Gerontol. 1998, 33, 371–378. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; Van Der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Radenovic, L.; Nenadic, M.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J.; Andjus, P.R.; Pluta, R. Heterogeneity in brain distribution of activated microglia and astrocytes in a rat ischemic model of Alzheimer’s disease after 2 years of survival. Aging 2020, 12, 12251–12267. [Google Scholar] [CrossRef]
- Westergard, T.; Rothstein, J.D. Astrocyte Diversity: Current Insights and Future Directions. Neurochem. Res. 2020, 45, 1298–1305. [Google Scholar] [CrossRef]
- Patabendige, A.; Singh, A.; Jenkins, S.; Sen, J.; Chen, R. Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke. Int. J. Mol. Sci. 2021, 22, 4280. [Google Scholar] [CrossRef]
- Weber, B.; Barros, L.F. The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb. Perspect. Biol. 2015, 7, a020396. [Google Scholar] [CrossRef] [Green Version]
- Davis, N.; Mota, B.C.; Stead, L.; Palmer, E.O.C.; Lombardero, L.; Rodríguez-Puertas, R.; de Paola, V.; Barnes, S.J.; Sastre, M. Pharmacological ablation of astrocytes reduces Aβ degradation and synaptic connectivity in an ex vivo model of Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 73. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation. Cells 2022, 11, 2728. [Google Scholar]
- Xiao, M.; Hu, G. Involvement of Aquaporin 4 in Astrocyte Function and Neuropsychiatric Disorders. CNS Neurosci. Ther. 2014, 20, 385–390. [Google Scholar] [CrossRef]
- Li, C.; Zhao, R.; Gao, K.; Wei, Z.; Yin, M.Y.; Lau, L.T.; Chui, D.; Yu, A.C.H. Astrocytes: Implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Pellerin, L. Astrocytes Couple Synaptic Activity to Glucose Utilization in the Brain. News Physiol. Sci. 1999, 14, 177–182. [Google Scholar] [CrossRef]
- Magistretti, P.J. Neuron-glia metabolic coupling and plasticity. Exp. Physiol. 2011, 96, 407–410. [Google Scholar] [CrossRef] [Green Version]
- Falkowska, A.; Gutowska, I.; Goschorska, M.; Nowacki, P.; Chlubek, D.; Baranowska-Bosiacka, I. Energy Metabolism of the Brain, Including the Cooperation between Astrocytes and Neurons, Especially in the Context of Glycogen Metabolism. Int. J. Mol. Sci. 2015, 16, 25959–25981. [Google Scholar] [CrossRef] [Green Version]
- Desgranges, B.; Baron, J.C.; de la Sayette, V.; Petit-Taboué, M.C.; Benali, K.; Landeau, B.; Lechevalier, B.; Eustache, F. The neural substrates of memory systems impairment in Alzheimer’s disease. A PET study of resting brain glucose utilization. Brain J. Neurol. 1998, 121, 611–631. [Google Scholar]
- Mosconi, L.; Tsui, W.-H.; De Santi, S.; Li, J.; Rusinek, H.; Convit, A.; Li, Y.; Boppana, M.; de Leon, M.J. Reduced hippocampal metabolism in MCI and AD: Automated FDG-PET image analysis. Neurology 2005, 64, 1860–1867. [Google Scholar] [CrossRef]
- Mosconi, L.; Pupi, A.; de Leon, M.J. Brain Glucose Hypometabolism and Oxidative Stress in Preclinical Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef]
- Soucek, T.; Cumming, R.; Dargusch, R.; Maher, P.; Schubert, D. The Regulation of Glucose Metabolism by HIF-1 Mediates a Neuroprotective Response to Amyloid Beta Peptide. Neuron 2003, 39, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Schubert, D.; Soucek, T.; Blouw, B. The induction of HIF-1 reduces astrocyte activation by amyloid beta peptide. Eur. J. Neurosci. 2009, 29, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Merlini, M.; Meyer, E.P.; Ulmann-Schuler, A.; Nitsch, R.M. Vascular β-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAβ mice. Acta Neuropathol. 2011, 122, 293–311. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, M.; Lehtonen, S.; Jaronen, M.; Goldsteins, G.; Hämäläinen, R.H.; Koistinaho, J. Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell. Mol. Life Sci. 2019, 76, 2739–2760. [Google Scholar]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar]
- Acioglu, C.; Li, L.; Elkabes, S. Contribution of astrocytes to neuropathology of neurodegenerative diseases. Brain Res. 2021, 1758, 147291. [Google Scholar] [CrossRef]
- Kim, S.; Son, Y. Astrocytes Stimulate Microglial Proliferation and M2 Polarization In Vitro through Crosstalk between Astrocytes and Microglia. Int. J. Mol. Sci. 2021, 22, 8800. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Buffo, A.; Rolando, C.; Ceruti, S. Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 2010, 79, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-L.; Xu, C.-J. Astrocytes autophagy in aging and neurodegenerative disorders. Biomed. Pharmacother. 2020, 122, 109691. [Google Scholar] [CrossRef]
- Solana, R.; Tarazona, R.; Gayoso, I.; Lesur, O.; Dupuis, G.; Fulop, T. Innate immunosenescence: Effect of aging on cells and receptors of the innate immune system in humans. Semin. Immunol. 2012, 24, 331–341. [Google Scholar] [CrossRef]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef] [Green Version]
- Berent-Maoz, B.; Montecino-Rodriguez, E.; Dorshkind, K. Genetic regulation of thymocyte progenitor aging. Semin. Immunol. 2012, 24, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Zheng, H. Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–17. [Google Scholar] [CrossRef]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Fernández, P.; De La Fuente, M. Role of the immune system in aging and longevity. Curr. Aging Sci. 2011, 4, 78–100. [Google Scholar] [CrossRef]
- Shalit, F.; Sredni, B.; Brodie, C.; Kott, E.; Huberman, M. T Lymphocyte Subpopulations and Activation Markers Correlate with Severity of Alzheimer’s Disease. Clin. Immunol. Immunopathol. 1995, 75, 246–250. [Google Scholar] [CrossRef]
- Sommer, A.; Winner, B.; Prots, I. The Trojan horse—Neuroinflammatory impact of T cells in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Bigazzi, P.E.; Yoshida, T. Similarities of T cell function in cell-mediated immunity and antibody production. Cell. Immunol. 1974, 12, 150–159. [Google Scholar] [CrossRef]
- Janeway, C.; Murphy, K.P.; Travers, P.; Walport, M. Janeway’s Immuno Biology; Garland Science: New York, NY, USA, 2008. [Google Scholar]
- Julier, Z.; Park, A.J.; Briquez, P.S.; Martino, M.M. Promoting tissue regeneration by modulating the immune system. Acta Biomater. 2017, 53, 13–28. [Google Scholar] [CrossRef]
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve—An integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638. [Google Scholar]
- Lewis, D.E.; Blutt, S.E. Organization of the immune system. In Clinical Immunology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 19–38. [Google Scholar]
- Zhang, J.M.; An, J. Cytokines, inflammation and pain. Int. Anesthesiol. Clin. 2007, 45, 27. [Google Scholar]
- Okada, H.; Murakami, S. Cytokine Expression in Periodontal Health and Disease. Crit. Rev. Oral Biol. Med. 1998, 9, 248–266. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Proinflammatory cytokines. Chest 2000, 118, 503–508. [Google Scholar]
- Tedgui, A.; Mallat, Z. Cytokines in Atherosclerosis: Pathogenic and Regulatory Pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef] [Green Version]
- Rai, V.; Dilisio, M.F.; Samadi, F.; Agrawal, D.K. Counteractive Effects of IL-33 and IL-37 on Inflammation in Osteoarthritis. Int. J. Environ. Res. Public Health 2022, 19, 5690. [Google Scholar] [CrossRef]
- Ng, T.H.S.; Britton, G.; Hill, E.V.; Everhagen, J.; Burton, B.R.; Wraith, D.C. Regulation of Adaptive Immunity; The Role of Interleukin-10. Front. Immunol. 2013, 4, 129. [Google Scholar] [CrossRef] [Green Version]
- Lubowicka, E.; Przylipiak, A.; Zajkowska, M.; Piskór, B.M.; Malinowski, P.; Fiedorowicz, W.; Ławicki, S. Plasma Chemokine CCL2 and Its Receptor CCR2 Concentrations as Diagnostic Biomarkers for Breast Cancer Patients. BioMed Res. Int. 2018, 2018, 2124390. [Google Scholar] [CrossRef]
- Liu, C.; Cui, G.; Zhu, M.; Kang, X.; Guo, H. Neuroinflammation in Alzheimer’s disease: Chemokines produced by astrocytes and chemokine receptors. Int. J. Clin. Exp. Pathol. 2014, 7, 8342–8355. [Google Scholar]
- Garden, G.A.; Campbell, B.M. Glial biomarkers in human central nervous system disease. Glia 2016, 64, 1755–1771. [Google Scholar] [CrossRef] [Green Version]
- Nikolich-Žugich, J. The twilight of immunity: Emerging concepts in aging of the immune system review-article. Nat. Immunol. 2018. ahead of print. [Google Scholar]
- High, K.P.; Akbar, A.N.; Nikolich-Zugich, J. Translational research in immune senescence: Assessing the relevance of current models. Semin. Immunol. 2012, 24, 373–382. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef]
- Parihar, A.; Eubank, T.D.; Doseff, A.I. Monocytes and Macrophages Regulate Immunity through Dynamic Networks of Survival and Cell Death. J. Innate Immun. 2010, 2, 204–215. [Google Scholar] [CrossRef] [Green Version]
- Auffray, C.; Sieweke, M.H.; Geissmann, F. Blood Monocytes: Development, Heterogeneity, and Relationship with Dendritic Cells. Annu. Rev. Immunol. 2009, 27, 669–692. [Google Scholar] [CrossRef] [Green Version]
- Fahy, R.J.; Doseff, A.I.; Wewers, M.D. Spontaneous human monocyte apoptosis utilizes a caspase-3-dependent pathway that is blocked by endotoxin and is independent of caspase-1. J. Immunol. 1999, 163, 1755–1762. [Google Scholar]
- Wiktor-Jedrzejczak, W.; Gordon, S. Cytokine regulation of the macrophage (M phi) system studied using the colony stimulating factor-1-deficient op/op mouse. Physiol. Rev. 1996, 76, 927–947. [Google Scholar] [CrossRef]
- Goyal, A.; Wang, Y.; Graham, M.M.; Doseff, A.I.; Bhatt, N.Y.; Marsh, C.B. Monocyte Survival Factors Induce Akt Activation and Suppress Caspase-3. Am. J. Respir. Cell Mol. Biol. 2002, 26, 224–230. [Google Scholar] [CrossRef]
- De Maeyer, R.P.; Chambers, E.S. The impact of ageing on monocytes and macrophages. Immunol. Letters. 2021, 230, 1–10. [Google Scholar]
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Cardona, A.E. The myeloid cells of the central nervous system parenchyma. Nature 2010, 468, 253–262. [Google Scholar] [CrossRef]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Graeber, M.B. Changing Face of Microglia. Science 2010, 330, 783–788. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial Physiology: Unique Stimuli, Specialized Responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Thériault, P.; ElAli, A.; Rivest, S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar]
- Zhang, R.; Miller, R.G.; Madison, C.; Jin, X.; Honrada, R.; Harris, W.; Katz, J.; Forshew, D.A.; McGrath, M.S. Systemic immune system alterations in early stages of Alzheimer’s disease. J. Neuroimmunol. 2013, 256, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2012, 61, 71–90. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer Disease--A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harb. Perspect. Med. 2011, 2, a006346. [Google Scholar] [CrossRef]
- Shechter, R.; Schwartz, M. Harnessing monocyte-derived macrophages to control central nervous system pathologies: No longer ‘if’but ‘how’. J. Pathol. 2013, 229, 332–346. [Google Scholar]
- Garré, J.M.; Yang, G. Contributions of monocytes to nervous system disorders. Klin. Wochenschr. 2018, 96, 873–883. [Google Scholar] [CrossRef]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef]
- Sutherland, K.; Li, T.; Cao, C. Alzheimer’s Disease and the Immune System. SOJ Neurol 2 (1), 1-11. Alzheimer’s Dis. Immune Syst. 2015, 30–60. [Google Scholar]
- Kölliker-Frers, R.; Udovin, L.; Otero-Losada, M.; Kobiec, T.; Herrera, M.I.; Palacios, J.; Razzitte, G.; Capani, F. Neuroinflammation: An Integrating Overview of Reactive-Neuroimmune Cell Interactions in Health and Disease. Mediat. Inflamm. 2021, 2021, 9999146. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Moyse, E.; Krantic, S.; Djellouli, N.; Roger, S.; Angoulvant, D.; Debacq, C.; Leroy, V.; Fougere, B.; Aidoud, A. Neuroinflammation: A Possible Link Between Chronic Vascular Disorders and Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 827263. [Google Scholar] [CrossRef]
- Cervellati, C.; Trentini, A.; Pecorelli, A.; Valacchi, G. Inflammation in Neurological Disorders: The Thin Boundary Between Brain and Periphery. Antioxidants Redox Signal. 2020, 33, 191–210. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2017, 18, 225–242. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Tauber, A.I. Immunity: The Evolution of an Idea; Oxford University Press: New York, NY, USA, 2017. [Google Scholar]
- Vaz, N.; Varela, F. Self and non-sense: An organism-centered approach to immunology. Med. Hypotheses 1978, 4, 231–267. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar]
- Hoebe, K.; Janssen, E.; Beutler, B. The interface between innate and adaptive immunity. Nat. Immunol. 2004, 5, 971–974. [Google Scholar] [CrossRef]
- Delpedro, A.D.; Barjavel, M.J.; Mamdouh, Z.; Faure, S.; Bakouche, O. Signal Transduction in LPS-Activated Aged and Young Monocytes. J. Interf. Cytokine Res. 1998, 18, 429–437. [Google Scholar] [CrossRef]
- Panda, A.; Arjona, A.; Sapey, E.; Bai, F.; Fikrig, E.; Montgomery, R.; Lord, J.M.; Shaw, A.C. Human innate immunosenescence: Causes and consequences for immunity in old age. Trends Immunol. 2009, 30, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef]
- van den Biggelaar, A.H.; Huizinga, T.W.; de Craen, A.J.; Gussekloo, J.; Heijmans, B.T.; Frölich, M.; Westendorp, R.G. Impaired innate immunity predicts frailty in old age. The Leiden 85-plus study. Exp. Gerontol. 2004, 39, 1407–1414. [Google Scholar]
- Larbi, A.; Franceschi, C.; Mazzatti, D.; Solana, R.; Wikby, A.; Pawelec, G. Aging of the Immune System as a Prognostic Factor for Human Longevity. Physiology 2008, 23, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Weng, N.-P. Aging of the Immune System: How Much Can the Adaptive Immune System Adapt? Immunity 2006, 24, 495–499. [Google Scholar] [CrossRef] [Green Version]
- Saurwein-Teissl, M.; Lung, T.L.; Marx, F.; Gschösser, C.; Asch, E.; Blasko, I.; Parson, W.; Böck, G.; Schönitzer, D.; Trannoy, E.; et al. Lack of Antibody Production Following Immunization in Old Age: Association with CD8+CD28− T Cell Clonal Expansions and an Imbalance in the Production of Th1 and Th2 Cytokines. J. Immunol. 2002, 168, 5893–5899. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- D’adda Di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Robert, L.; Fulop, T. (Eds.) Aging: Facts and Theories; Karger Medical and Scientific Publishers: Basel, Switzerland, 2014; Volume 39, pp. 163–176. [Google Scholar]
- Walford, R.L. The immunologic theory of aging. Immunol. Rev. 1969, 2, 171. [Google Scholar] [CrossRef]
- Franceschi, C.; Monti, D.; Barbier, D.; Salvioli, S.; Grassilli, E.; Capri, M.; Troiano, L.; Guido, M.; Bonafè, M.; Tropea, F.; et al. Successful immunosenescence and the remodelling of immune responses with ageing. Nephrol. Dial. Transplant. 1996, 11, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Candore, G.; Caruso, C.; Jirillo, E.; Magrone, T.; Vasto, S. Low Grade Inflammation as a Common Pathogenetic Denominator in Age-Related Diseases: Novel Drug Targets for Anti-Ageing Strategies and Successful Ageing Achievement. Curr. Pharm. Des. 2010, 16, 584–596. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef] [Green Version]
- Capri, M.; Monti, D.; Salvioli, S.; Lescai, F.; Pierini, M.; Altilia, S.; Sevini, F.; Valensin, S.; Ostan, R.; Bucci, L.; et al. Complexity of Anti-immunosenescence Strategies in Humans. Artif. Organs 2006, 30, 730–742. [Google Scholar] [CrossRef]
- Effros, R.B. Roy Walford and the immunologic theory of aging. Immun. Ageing 2005, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Caruso, C.; Candore, G.; Colonna-Romano, G.; Lio, D.; Franceschi, C. Inflammation and Life-Span. Science 2005, 307, 208–209. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Understanding immunosenescence to improve responses to vaccines. Nat. Immunol. 2013, 14, 428–436. [Google Scholar] [CrossRef] [Green Version]
- Pawelec, G. Hallmarks of human “immunosenescence”: Adaptation or dysregulation? Immun. Ageing 2012, 9, 15. [Google Scholar]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: Emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef]
- Ponnappan, S.; Ponnappan, U. Aging and Immune Function: Molecular Mechanisms to Interventions. Antioxidants Redox Signal. 2011, 14, 1551–1585. [Google Scholar] [CrossRef]
- Borgoni, S.; Kudryashova, K.S.; Burka, K.; de Magalhães, J.P. Targeting immune dysfunction in aging. Ageing Res. Rev. 2021, 70, 101410. [Google Scholar] [CrossRef]
- Avery, P.; Barzilai, N.; Benetos, A.; Bilianou, H.; Capri, M.; Caruso, C.; Franceschi, C.; Katsiki, N.; Mikhailidis, D.; Panotopoulos, G.; et al. Editorial: Ageing, Longevity, Exceptional Longevity and Related Genetic and Non Genetics Markers: Panel Statement. Curr. Vasc. Pharmacol. 2014, 12, 659–661. [Google Scholar] [CrossRef]
- Bruunsgaard, H.; Andersen-Ranberg, K.; Hjelmborg, J.V.; Pedersen, B.K.; Jeune, B. Elevated levels of tumor necrosis factor alpha and mortality in centenarians. Am. J. Med. 2003, 115, 278–283. [Google Scholar] [CrossRef]
- Ershler, W.B.; Keller, E.T. Age-Associated Increased Interleukin-6 Gene Expression, Late-Life Diseases, and Frailty. Annu. Rev. Med. 2000, 51, 245–270. [Google Scholar] [CrossRef]
- O’Mahony, L.; Holland, J.; Jackson, J.; Feighery, C.; Hennessy, T.P.J.; Mealy, K. Quantitative intracellular cytokine measurement: Age-related changes in proinflammatory cytokine production. Clin. Exp. Immunol. 1998, 113, 213–219. [Google Scholar] [CrossRef]
- Cunha, L.L.; Perazzio, S.F.; Azzi, J.; Cravedi, P.; Riella, L.V. Remodeling of the Immune Response With Aging: Immunosenescence and Its Potential Impact on COVID-19 Immune Response. Front. Immunol. 2020, 11, 1748. [Google Scholar] [CrossRef]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Stephenson, J.; Nutma, E.; Van Der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [Green Version]
- Mapunda, J.A.; Tibar, H.; Regragui, W.; Engelhardt, B. How Does the Immune System Enter the Brain? Front. Immunol. 2022, 13, 805657. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Gate, D.; Town, T. CNS Infiltration of Peripheral Immune Cells: D-Day for Neurodegenerative Disease? J. Neuroimmune Pharmacol. 2009, 4, 462–475. [Google Scholar] [CrossRef] [Green Version]
- Kamma, E.; Lasisi, W.; Libner, C.; Ng, H.S.; Plemel, J.R. Central nervous system macrophages in progressive multiple sclerosis: Relationship to neurodegeneration and therapeutics. J. Neuroinflamm. 2022, 19, 45. [Google Scholar] [CrossRef]
- Jevtic, S.; Sengar, A.; Salter, M.W.; McLaurin, J. The role of the immune system in Alzheimer disease: Etiology and treatment. Ageing Res. Rev. 2017, 40, 84–94. [Google Scholar] [CrossRef]
- Burgaletto, C.; Munafò, A.; Di Benedetto, G.; De Francisci, C.; Caraci, F.; Di Mauro, R.; Bucolo, C.; Bernardini, R.; Cantarella, G. The immune system on the TRAIL of Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 298. [Google Scholar] [CrossRef]
- Greenhalgh, A.D.; David, S.; Bennett, F.C. Immune cell regulation of glia during CNS injury and disease. Nat. Rev. Neurosci. 2020, 21, 139–152. [Google Scholar] [CrossRef]
- Ferro, A.; Auguste, Y.S.S.; Cheadle, L. Microglia, Cytokines, and Neural Activity: Unexpected Interactions in Brain Development and Function. Front. Immunol. 2021, 12, 703527. [Google Scholar] [CrossRef]
- Kanmiki, E.W.; Bawah, A.A.; Phillips, J.F.; Awoonor-Williams, J.K.; Kachur, S.P.; Asuming, P.O.; Agula, C.; Akazili, J. Out-of-pocket payment for primary healthcare in the era of national health insurance: Evidence from northern Ghana. PLoS ONE 2019, 14, e0221146. [Google Scholar] [CrossRef]
- Walker, K.A.; Gottesman, R.F.; Wu, A.; Knopman, D.S.; Gross, A.L.; Mosley, T.H.; Selvin, E.; Windham, B.G. Systemic inflammation during midlife and cognitive change over 20 years: The ARIC Study. Neurology 2019, 92, e1256–e1267. [Google Scholar]
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and central immune system crosstalk in Alzheimer disease—A research prospectus. Nat. Rev. Neurol. 2021, 17, 689–701. [Google Scholar] [CrossRef]
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [Green Version]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef]
- Singh, A.; Raju, R.; Münch, G. Potential anti-neuroinflammatory compounds from Australian plants—A review. Neurochem. Int. 2020, 142, 104897. [Google Scholar] [CrossRef]
- Ramos-Martínez, I.E.; Rodríguez, M.C.; Cerbón, M.; Ramos-Martínez, J.C.; Ramos-Martínez, E.G. Role of the Cholinergic Anti-Inflammatory Reflex in Central Nervous System Diseases. Int. J. Mol. Sci. 2021, 22, 13427. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Lin, K.-J.; Lin, P.-C.; Huang, C.-C.; Chang, C.-C.; Lee, Y.-C.; Hsiao, I.-T.; Yen, T.-C.; Huang, W.-S.; Yang, B.-H.; et al. Plasma amyloid assay as a pre-screening tool for amyloid positron emission tomography imaging in early stage Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 111. [Google Scholar] [CrossRef]
- Cisbani, G.; Rivest, S. Targeting innate immunity to protect and cure Alzheimer’s disease: Opportunities and pitfalls. Mol. Psychiatry 2021, 26, 5504–5515. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowirrat, A. Immunosenescence and Aging: Neuroinflammation Is a Prominent Feature of Alzheimer’s Disease and Is a Likely Contributor to Neurodegenerative Disease Pathogenesis. J. Pers. Med. 2022, 12, 1817. https://doi.org/10.3390/jpm12111817
Bowirrat A. Immunosenescence and Aging: Neuroinflammation Is a Prominent Feature of Alzheimer’s Disease and Is a Likely Contributor to Neurodegenerative Disease Pathogenesis. Journal of Personalized Medicine. 2022; 12(11):1817. https://doi.org/10.3390/jpm12111817
Chicago/Turabian StyleBowirrat, Abdalla. 2022. "Immunosenescence and Aging: Neuroinflammation Is a Prominent Feature of Alzheimer’s Disease and Is a Likely Contributor to Neurodegenerative Disease Pathogenesis" Journal of Personalized Medicine 12, no. 11: 1817. https://doi.org/10.3390/jpm12111817