
Huntingtin Ubiquitination Mechanisms and Novel Possible Therapies to Decrease the Toxic Effects of Mutated Huntingtin


Abstract
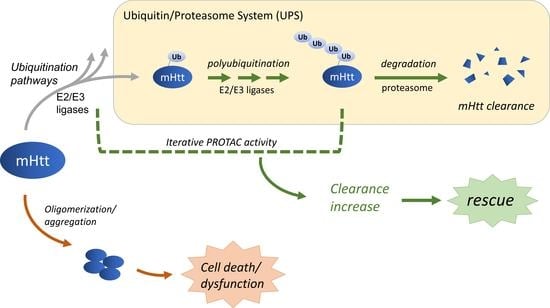
:
1. Introduction
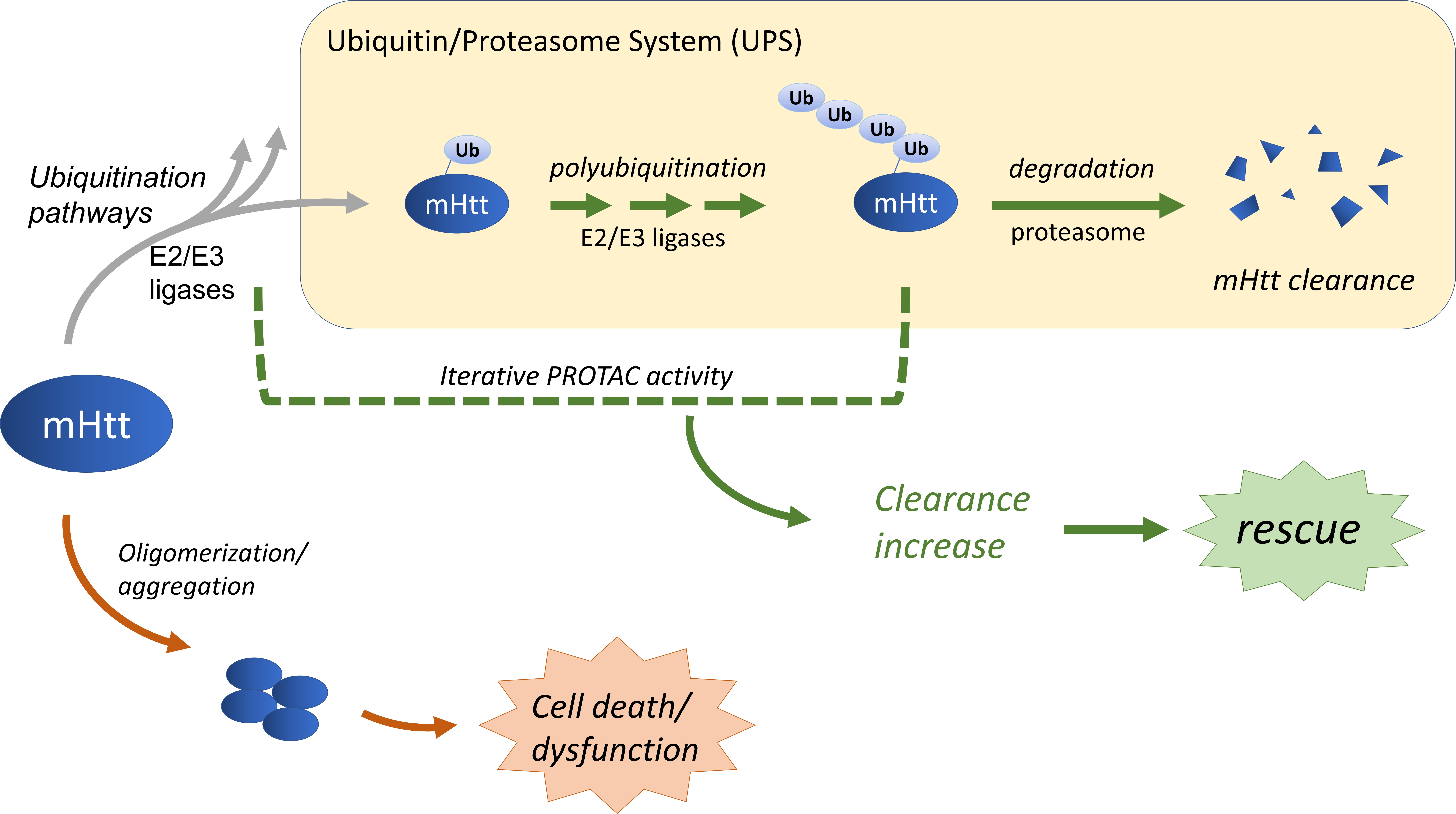
2. Ubiquitin Proteasome System (UPS)
2.1. E1-Activating and E2-Conjugating Enzymes
2.2. E3 Ligases
2.3. Deubiquitinating Enzymes
2.4. Degradation though the Proteasome 26 S
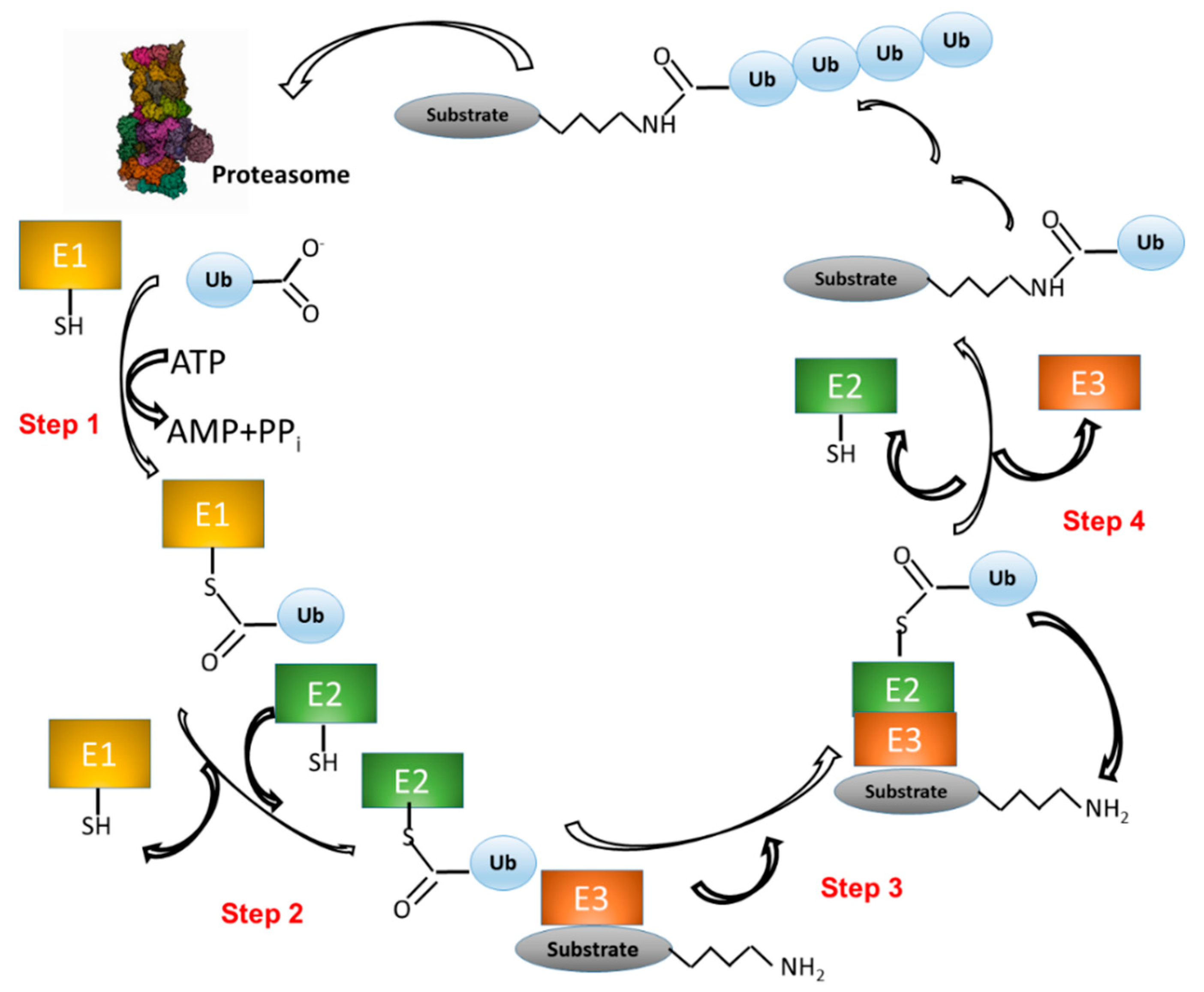
3. The Level of Huntingtin Is Controlled by Ubiquitination
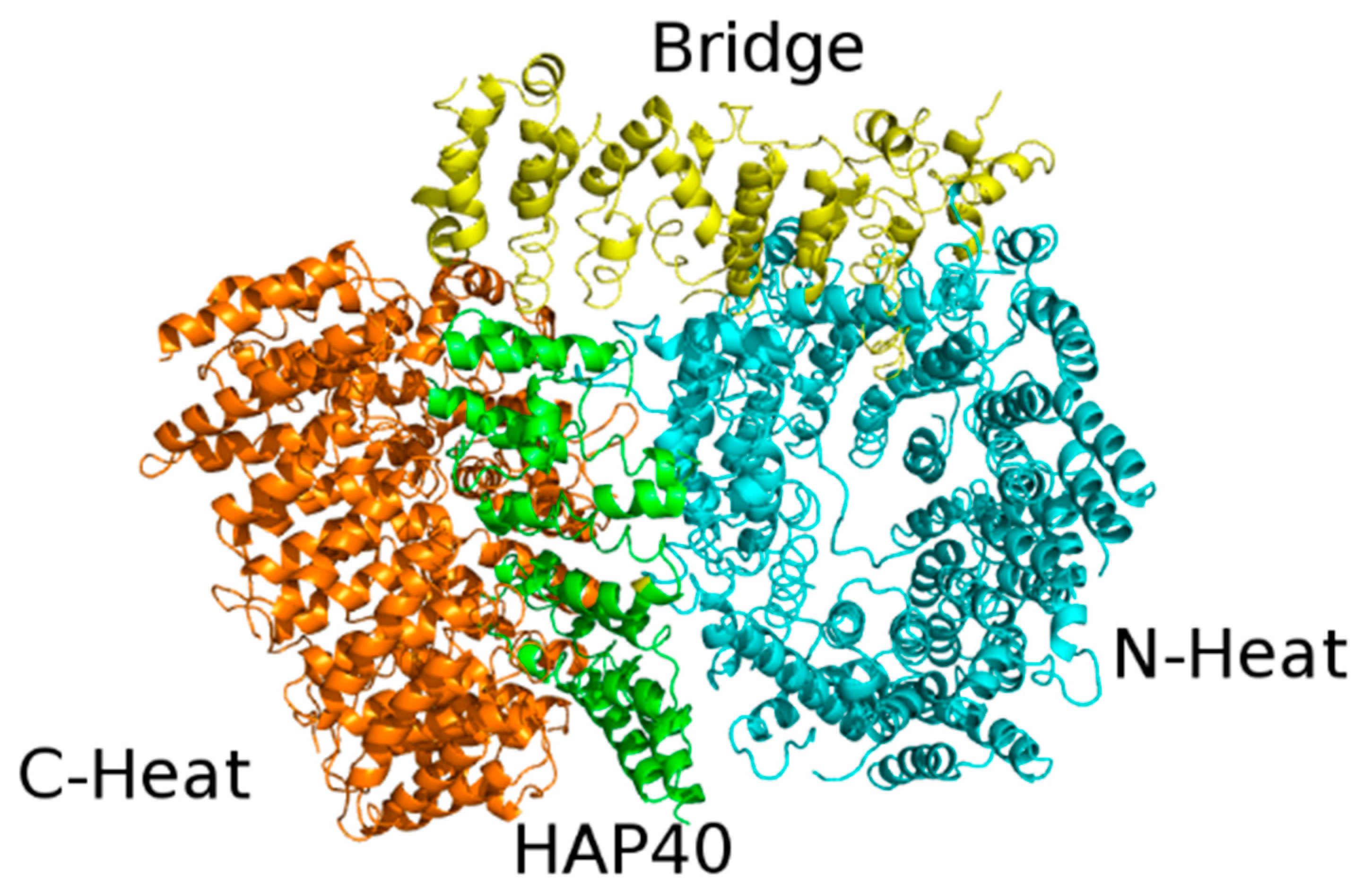
3.1. Htt Structure and Function
3.2. Htt Ubiquitination and SUMOylation
4. E2-Conjugating Enzymes Interacting with mHtt and Htt
4.1. UBE2K/E2-25k Triggers polyQ-Induced Cell Death
4.2. UBE2W Ubiquitinates mHtt at the N-Terminus
5. E3 Ligases Decreasing mHtt Levels in HD
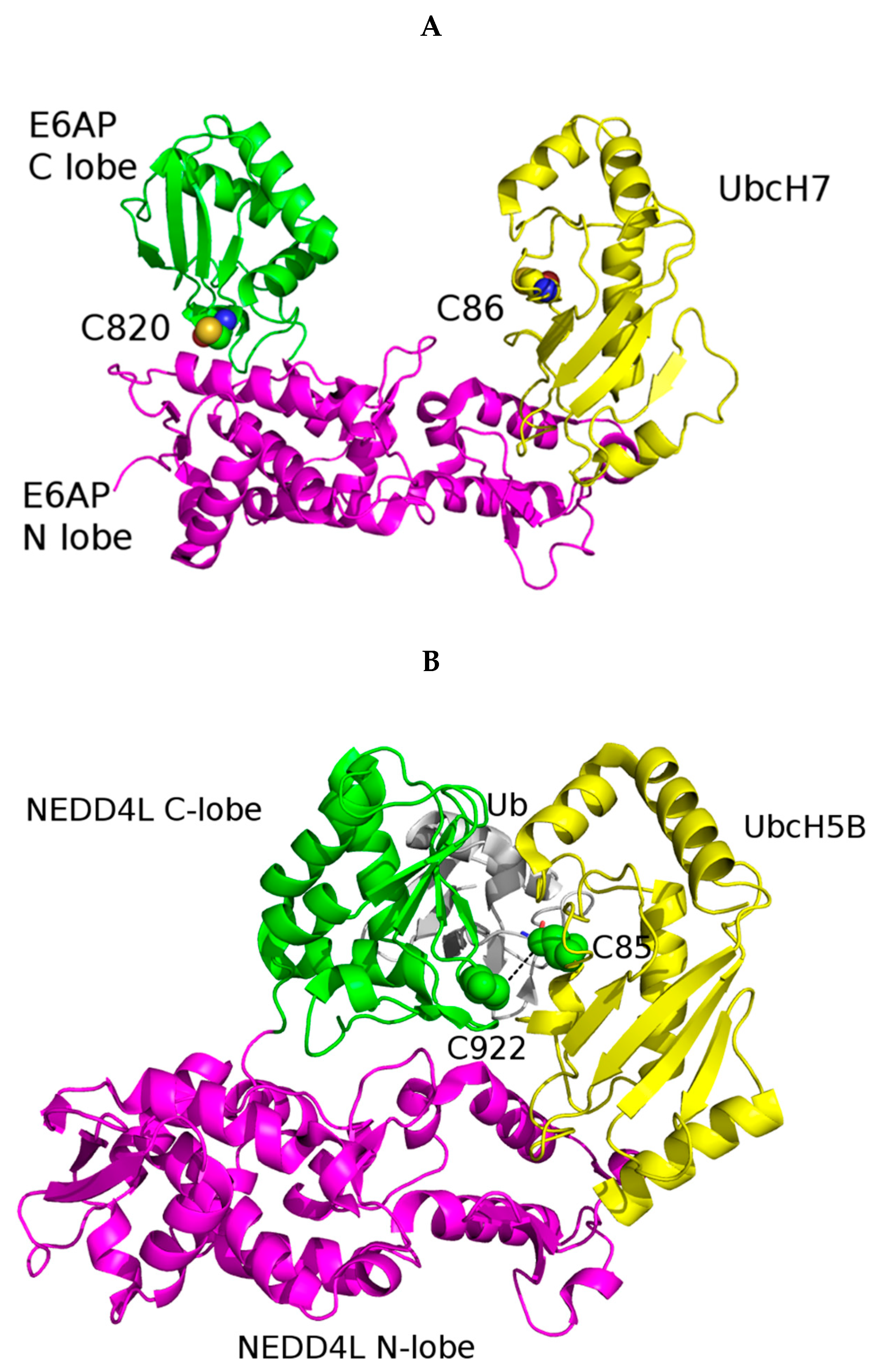
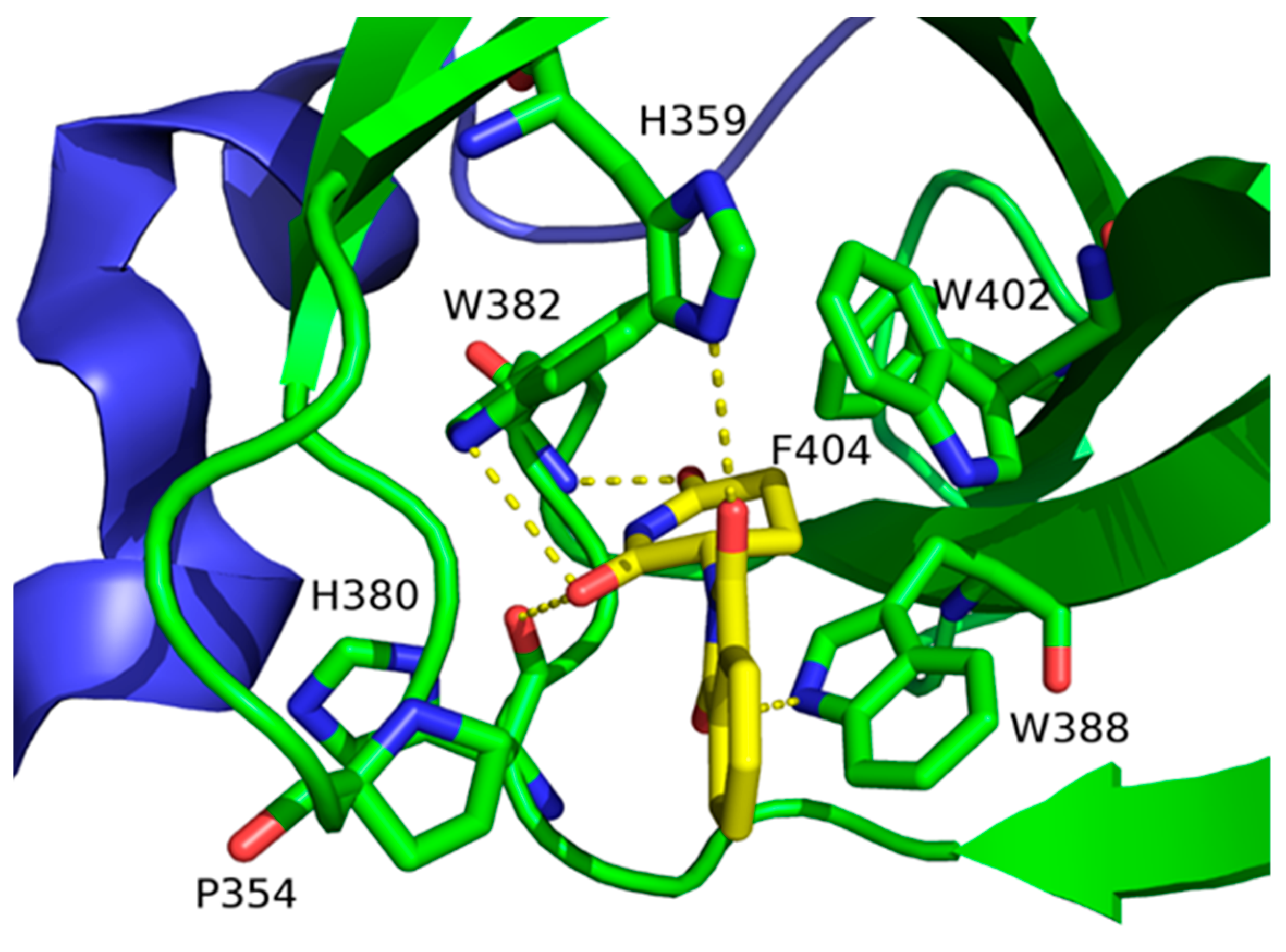
5.1. UBE3A Structure and Its Role in mHtt Ubiquitination
5.2. CHIP Inhibits PolyQ Protein Aggregation
5.3. Hrd1 Protects Cells against Cell Death Induced by mHTT N-Terminal Fragment
5.4. Parkin Suppression Aggravate Motor and Behavioural Deficits in HD Mice
5.5. SCF Complex
6. E3 Ligases Counteracting the Effect of mHtt on Oxidative Metabolism
HACE1 Reduces Oxidative Stress and mHtt Toxicity
7. E3 Ub Ligases Increasing mHtt Levels
7.1. WWP1 Inhibits mHtt Degradation
7.2. TRAF6 Ubiquitinates mHtt Fragments Inducing Aggregate Formation
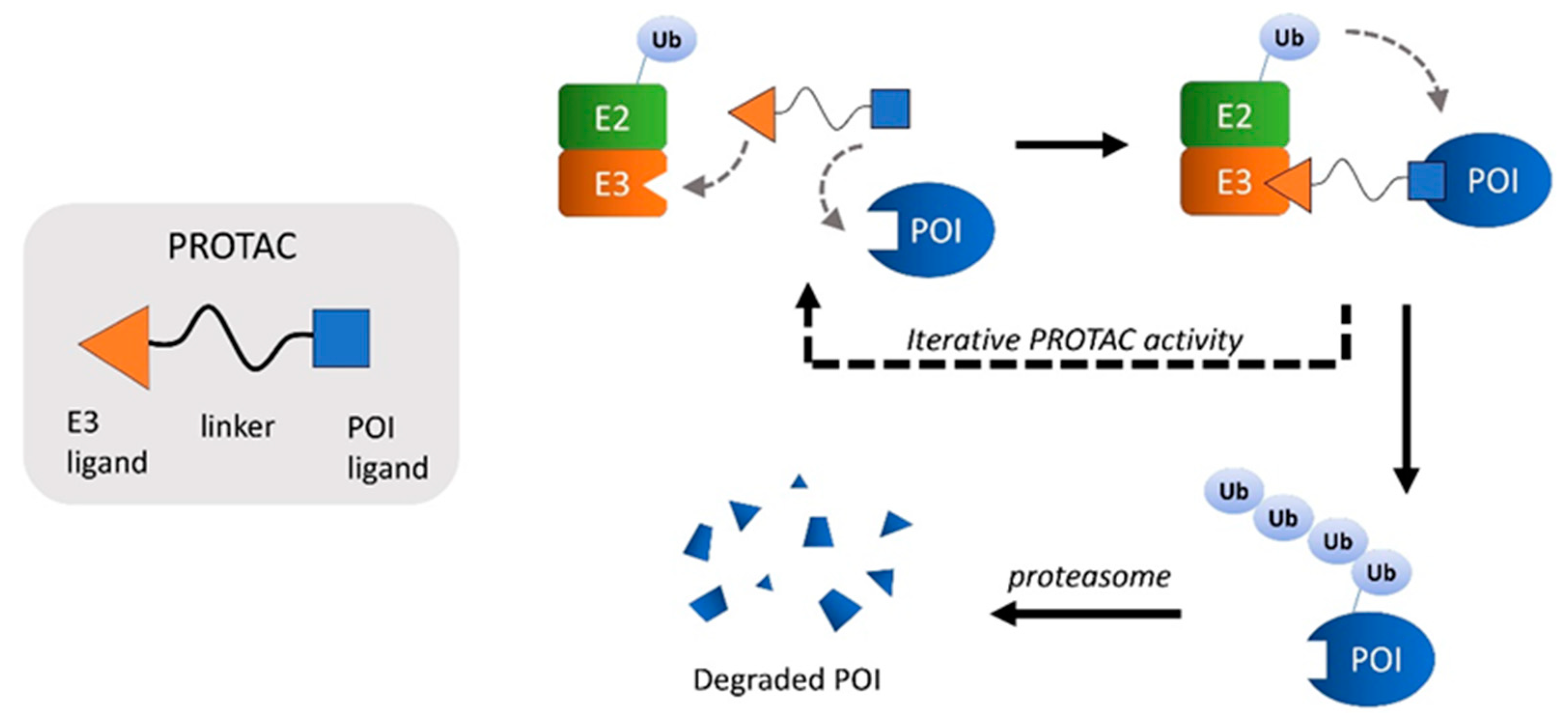
8. PROTACs
8.1. Targeting the Ubiquitination Pathways for the mHtt Clearance
8.2. Targeting the Autophagosomal Pathway to Reduce mHtt
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Fisher, E.R.; Hayden, M.R. Multisource ascertainment of Huntington disease in Canada: Prevalence and population at risk. Mov. Disord. 2013, 29, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Kay, C.; Hayden, M.R.; Leavitt, B.R. Epidemiology of Huntington disease. Handb. Clin. Neurol. 2017, 144, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Crowell, V.; Houghton, R.; Tomar, A.; Fernandes, T.; Squitieri, F. Modeling Manifest Huntington’s Disease Prevalence Using Di-agnosed Incidence and Survival Time. Neuroepidemiology 2021, 55, 361–368. [Google Scholar] [CrossRef]
- Toczek, M.; Zielonka, D.; Zukowska, P.; Marcinkowski, J.; Slominska, E.; Isalan, M.; Smolenski, R.T.; Mielcarek, M. An impaired metabolism of nucleotides underpins a novel mechanism of cardiac remodeling leading to Huntington’s disease related cardiomyopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Mielcarek, M.; Smolenski, R.; Isalan, M. Transcriptional Signature of an Altered Purine Metabolism in the Skeletal Muscle of a Huntington’s Disease Mouse Model. Front. Physiol. 2017, 8, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mielcarek, M. Huntington’s disease is a multi-system disorder. Rare Dis. 2015, 3, e1058464. [Google Scholar] [CrossRef] [PubMed]
- Kremer, B.; Goldberg, P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Lin, B.; Bassett, A.; Almqvist, E.; et al. A Worldwide Study of the Huntington’s Disease Mutation: The Sensitivity and Specificity of Measuring CAG Repeats. N. Engl. J. Med. 1994, 330, 1401–1406. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, C.-A.; Li, S.-H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.-J. Nuclear and Neuropil Aggregates in Huntington’s Disease: Relationship to Neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar] [CrossRef] [PubMed]
- Quarrell, O.; O’Donovan, K.L.; Bandmann, O.; Strong, M. The Prevalence of Juvenile Huntington’s Disease: A Review of the Lit-erature and Meta-Analysis. PLoS Curr. 2012, 4, e4f8606b742ef3. [Google Scholar] [CrossRef] [PubMed]
- Fusilli, C.; Migliore, S.; Mazza, T.; Consoli, F.; De Luca, A.; Barbagallo, G.; Ciammola, A.; Gatto, E.M.; Cesarini, M.; Etcheverry, J.L.; et al. Biological and clinical manifestations of juvenile Huntington’s disease: A retrospective analysis. Lancet Neurol. 2018, 17, 986–993. [Google Scholar] [CrossRef]
- Tydlacka, S.; Wang, C.-E.; Wang, X.; Li, S.; Li, X.-J. Differential Activities of the Ubiquitin-Proteasome System in Neurons versus Glia May Account for the Preferential Accumulation of Misfolded Proteins in Neurons. J. Neurosci. 2008, 28, 13285–13295. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-J.; Li, H.; Li, S. Clearance of mutant huntingtin. Autophagy 2010, 6, 663–664. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, C.-E.; Huang, S.; Xu, X.; Li, X.-J.; Li, H.; Li, S. Inhibiting the ubiquitin–proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum. Mol. Genet. 2010, 19, 2445–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Targeting Proteostasis Through the Protein Quality Control Function of the Hsp90/Hsp70-based Chaperone Machinery for Treatment of Adult Onset Neurodegenerative Diseases. Annu. Rev. Pharm. Toxicol. 2015, 55, 353–371. [Google Scholar] [CrossRef] [Green Version]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.-Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Huang, B.; Cheng, J.; Seefelder, M.; Engler, T. Europe PMC Funders Group The cryo-electron microscopy structure of huntingtin. Nature 2018, 555, 117–120. [Google Scholar] [CrossRef]
- Qi, L.; Zhang, X.D.; Wu, J.C.; Lin, F.; Wang, J.; DiFiglia, M.; Qin, Z.H. The Role of Chaperone-Mediated Autophagy in Huntingtin Deg-radation. PLoS ONE 2012, 7, e46834. [Google Scholar]
- Baldo, B.; Weiss, A.; Parker, C.N.; Bibel, M.; Paganetti, P.; Kaupmann, K. A Screen for Enhancers of Clearance Identifies Huntingtin as a Heat Shock Protein 90 (Hsp90) Client Protein. J. Biol. Chem. 2012, 287, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Ciechanover, A.; Elias, S.; Heller, H.; Hershko, A. "Covalent affinity" purification of ubiquitin-activating enzyme. J. Biol. Chem. 1982, 257, 2537–2542. [Google Scholar] [CrossRef]
- Hershko, A.; Heller, H. Occurrence of a polyubiquitin structure in ubiquitin-protein conjugates. Biochem. Biophys. Res. Commun. 1985, 128, 1079–1086. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Blaser, G.; Horrocks, M.H.; Ruedas-Rama, M.J.; Ibrahim, S.; Zhukov, A.A.; Orte, A.; Klenerman, D.; Jackson, S.E.; Komander, D. Ubiquitin chain conformation regulates recog-nition and activity of interacting proteins. Nature 2012, 492, 266–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grice, G.L.; Lobb, I.; Weekes, M.; Gygi, S.P.; Antrobus, R.; Nathan, J.A. The Proteasome Distinguishes between Heterotypic and Homotypic Lysine-11-Linked Polyubiquitin Chains. Cell Rep. 2015, 12, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, S.E.; Maduka, A.O.; Inada, T.; Silva, G.M. Expanding Role of Ubiquitin in Translational Control. Int. J. Mol. Sci. 2020, 21, 1151. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Bremm, A.; Freund, S.M.V.; Komander, D. Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat. Struct. Mol. Biol. 2010, 17, 939–947. [Google Scholar] [CrossRef] [Green Version]
- Dynek, J.N.; Goncharov, T.; Dueber, E.C.; Fedorova, A.V.; Izrael-Tomasevic, A.; Phu, L.; Helgason, E.; Fairbrother, W.J.; Deshayes, K.; Kirkpatrick, D.; et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 2010, 29, 4198–4209. [Google Scholar] [CrossRef]
- Min, M.; Mevissen, T.E.T.; De Luca, M.; Komander, D.; Lindon, C. Effcient APC/C substrate degradation in cells undergoing mitotic exit depends on K11 ubiquitin linkages. Mol. Biol. Cell 2015, 26, 4325–4332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Horikawa, W.; Ito, K. Inhibiting K63 Polyubiquitination Abolishes No-Go Type Stalled Translation Surveillance in Saccharomyces cerevisiae. PLoS Genet. 2015, 11, e1005197. [Google Scholar] [CrossRef] [Green Version]
- Silva, G.M.; Finley, D.; Vogel, C. K63 polyubiquitination is a new modulator of the oxidative stress response. Nat. Struct. Mol. Biol. 2015, 22, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Z.; Williams, K.M.; Yuan, L.; Atkison, J.H.; Olsen, S.K. Crystal structure of a human ubiquitin E1-ubiquitin complex reveals con-served functional elements essential for activity. J. Biol. Chem. 2018, 293, 18337–18352. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [Green Version]
- George, A.J.; Hoffiz, Y.C.; Charles, A.J.; Zhu, Y.; Mabb, A.M. A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in Neurological Disorders. Front. Genet. 2018, 9, 29. [Google Scholar] [CrossRef]
- Medvar, B.; Raghuram, V.; Pisitkun, T.; Sarkar, A.; Knepper, M.A. Comprehensive database of human E3 ubiquitin ligases: Appli-cation to aquaporin-2 regulation. Physiol. Genom. 2016, 48, 502–512. [Google Scholar] [CrossRef] [Green Version]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Kinnucan, E.; Wang, G.; Beaudenon, S.; Howley, P.M.; Huibregtse, J.M.; Pavletich, N.P. Structure of an E6AP-UbcH7 complex: Insights into ubiquitination by the E2-E3 enzyme cascade. Science 1999, 286, 1321–1326. [Google Scholar] [CrossRef]
- Kamadurai, H.; Souphron, J.; Scott, D.C.; Duda, D.M.; Miller, D.J.; Stringer, D.; Piper, R.; Schulman, B.A. Insights into Ubiquitin Transfer Cascades from a Structure of a UbcH5B∼Ubiquitin-HECTNEDD4L Complex. Mol. Cell 2009, 36, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Wang, P.; Jeffrey, P.D.; Pavletich, N.P. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell 2000, 102, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, S.; I Nakayama, K.-I. U-box proteins as a new family of ubiquitin ligases. Biochem. Biophys. Res. Commun. 2003, 302, 635–645. [Google Scholar] [CrossRef]
- Bard, J.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2018, 565, 49–55. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P.J. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar] [PubMed]
- Arndt, J.R.; Chaibva, M.; Legleiter, J. The emerging role of the first 17 amino acids of huntingtin in Huntington’s disease. Biomol. Concepts 2015, 6, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Yanai, A.; Huang, K.; Kang, R.; Singaraja, R.; Arstikaitis, P.; Gan, L.; Orban, P.C.; Mullard, A.; Cowan, C.M.; Raymond, L.; et al. Palmitoylation of huntingtin by HIP14is essential for its trafficking and function. Nat. Neurosci. 2006, 9, 824–831. [Google Scholar] [CrossRef] [Green Version]
- Seefelder, M.; Alva, V.; Huang, B.; Engler, T.; Baumeister, W.; Guo, Q.; Fernandez-Busnadiego, R.; Lupas, A.N.; Kochanek, S. The evolution of the huntingtin-associated protein 40 (HAP40) in conjunction with huntingtin. BMC Evol. Biol. 2020, 20, 162. [Google Scholar] [CrossRef]
- Huang, B.; Guo, Q.; Niedermeier, M.L.; Cheng, J.; Engler, T.; Maurer, M.; Pautsch, A.; Baumeister, W.; Stengel, F.; Kochanek, S.; et al. Pathological polyQ expansion does not alter the con-formation of the Huntingtin-HAP40 complex. Structure 2021, 29, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nature 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caviston, J.P.; Ross, J.; Antony, S.M.; Tokito, M.; Holzbaur, E.L.F. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Godin, J.D.; Colombo, K.; Molina-Calavita, M.; Keryer, G.; Zala, D.; Charrin, B.C.; Dietrich, P.; Volvert, M.-L.; Guillemot, F.; Dragatsis, I.; et al. Huntingtin Is Required for Mitotic Spindle Orientation and Mammalian Neurogenesis. Neuron 2010, 67, 392–406. [Google Scholar] [CrossRef] [Green Version]
- Zeitlin, S.; Liu, J.-P.; Chapman, D.; Papaioannou, V.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163. [Google Scholar] [CrossRef]
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; et al. Huntingtin Is Required for Normal Excitatory Synapse Development in Cortical and Striatal Circuits. J. Neurosci. 2014, 34, 9455–9472. [Google Scholar] [CrossRef]
- Dragatsis, I.; Levine, M.S.; Zeitlin, S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 2000, 26, 300–306. [Google Scholar] [CrossRef]
- Mehler, M.F.; Petronglo, J.R.; Arteaga-Bracho, E.E.; Gulinello, M.E.; Winchester, M.L.; Pichamoorthy, N.; Young, S.K.; DeJesus, C.D.; Ishtiaq, H.; Gokhan, S.; et al. Loss-of-Huntingtin in Medial and Lateral Ganglionic Lineages Differentially Disrupts Regional Interneuron and Projection Neuron Subtypes and Promotes Huntington’s Disease-Associated Behavioral, Cellular, and Pathological Hallmarks. J. Neurosci. 2019, 39, 1892–1909. [Google Scholar] [CrossRef] [PubMed]
- Morea, V.; Bidollari, E.; Colotti, G.; Fiorillo, A.; Rosati, J.; De Filippis, L.; Squitieri, F.; Ilari, A. Glucose transportation in the brain and its impairment in Huntington disease: One more shade of the energetic metabolism failure? Amino Acids 2017, 49, 1147–1157. [Google Scholar] [CrossRef]
- Tomczyk, M.; Glaser, T.; Ulrich, H.; Slominska, E.M.; Smolenski, R.T. Huntingtin protein maintains balanced energetics in mouse cardiomyocytes. Nucleosides Nucleotides Nucleic Acids 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ismailoglu, I.; Chen, Q.; Popowski, M.; Yang, L.; Gross, S.S.; Brivanlou, A.H. Huntingtin protein is essential for mitochondrial metab-olism, bioenergetics and structure in murine embryonic stem cells. Dev. Biol. 2014, 391, 230–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavitt, B.R.; van Raamsdonk, J.M.; Shehadeh, J.; Fernandes, H.; Murphy, Z.; Graham, R.K.; Wellington, C.L.; Hayden, M.R. Wild-type huntingtin protects neurons from excitotoxicity. J. Neurochem. 2006, 96, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Burrus, C.J.; McKinstry, S.U.; Kim, N.; Ozlu, M.I.; Santoki, A.; Fang, F.Y.; Ma, A.; Karadeniz, Y.B.; Worthington, A.K.; Dragatsis, I.; et al. Striatal Projection Neurons Require Huntingtin for Synaptic Connectivity and Survival. Cell Rep. 2020, 30, 642–657.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Regan, G.C.; Farag, S.H.; Ostroff, G.R.; Tabrizi, S.J.; Andre, R. Wild-type huntingtin regulates human macrophage function. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.-Z.; Cattaneo, E.; et al. SUMO Modification of Huntingtin and Hun-tington’s Disease Pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Udeshi, N.D.; Svinkina, T.; Mertins, P.; Kuhn, E.; Mani, D.R.; Qiao, J.W.; Carr, S.A. Refined preparation and use of anti-diglycine remnant (K-ε-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell Proteom. 2013, 12, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.-K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Callegari, E.; Gloeckner, C.J.; Ueffing, M.; Wang, H. Mass spectrometric identification of novel posttranslational modifi-cation sites in Huntingtin. Proteomics 2012, 12, 2060–2064. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational mod-ifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A proteome-wide, quantitative survey of in vivo ubiq-uitylation sites reveals widespread regulatory roles. Mol. Cell Proteom. 2011, 10, M111.013284. [Google Scholar] [CrossRef] [Green Version]
- Boeing, S.; Williamson, L.; Encheva, V.; Gori, I.; Saunders, R.E.; Instrell, R.; Aygün, O.; Rodriguez-Martinez, M.; Weems, J.C.; Kelly, G.; et al. Multiomic Analysis of the UV-Induced DNA Damage Response. Cell Rep. 2016, 15, 1597–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-wide analysis of ubiquitylation dy-namics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [Google Scholar] [CrossRef]
- Kalchman, M.A.; Graham, R.K.; Xia, G.; Koide, H.B.; Hodgson, J.G.; Graham, K.C.; Goldberg, Y.P.; Gietz, R.D.; Pickart, C.M.; Hayden, M.R. Huntingtin Is Ubiquitinated and Interacts with a Specific Ubiquitin-conjugating Enzyme. J. Biol. Chem. 1996, 271, 19385–19394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a Striatal Specific Protein, Mediates Mutant-Huntingtin Cytotoxicity. Science 2009, 324, 1327–1330. [Google Scholar] [CrossRef] [Green Version]
- Falk, J.D.; Vargiu, P.; Foye, P.E.; Usui, H.; Perez, J.; Danielson, P.E.; Lerner, D.L.; Bernal, J.; Sutcliffe, J.G. Rhes: A striatal-specific Ras homolog related to Dexras. J. Neurosci. Res. 1999, 57, 782–788. [Google Scholar] [CrossRef]
- Carbo, M.; Brandi, V.; Pascarella, G.; Staid, D.S.; Colotti, G.; Polticelli, F.; Ilari, A.; Morea, V. Bioinformatics analysis of Ras homologue enriched in the striatum, a potential target for Huntington’s disease therapy. Int. J. Mol. Med. 2019, 44, 2223–2233. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zeng, L.; Merillat, S.A.; Fischer, S.; Ochaba, J.; Thompson, L.M.; Barmada, S.J.; Scaglione, K.M.; Paulson, H.L. The ubiquitin conjugating enzyme Ube2W regulates solubility of the Huntington’s disease protein, huntingtin. Neurobiol. Dis. 2017, 109, 127–136. [Google Scholar] [CrossRef]
- De Pril, R.; Fischer, D.F.; Roos, R.A.C.; Van Leeuwen, F.W. Ubiquitin-conjugating enzyme E2-25K increases aggregate formation and cell death in polyglutamine diseases. Mol. Cell Neurosci. 2007, 34, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, M.; Shekhar, S.; Singh, B.K.; Jamal, I.; Vatsa, N.; Kumar, V.; Sharma, A.; Jana, N.R.; Kumar, S.S. Deficiency of Ube3a in Huntington’s disease mice brain increases aggregate load and accelerates disease pathology. Hum. Mol. Genet. 2014, 23, 6235–6245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Dikshit, P.; Purkayastha, S.; Sharma, J.; Nukina, N.; Jana, N. E6-AP Promotes Misfolded Polyglutamine Proteins for Proteasomal Degradation and Suppresses Polyglutamine Protein Aggregation and Toxicity. J. Biol. Chem. 2008, 283, 7648–7656. [Google Scholar] [CrossRef] [Green Version]
- Joshi, V.; Amanullah, A.; Upadhyay, A.; Mishra, R.; Kumar, A.; Mishra, A. A decade of boon or burden: What has the chip ever done for cellular protein quality control mechanism implicated in neurodegeneration and aging? Front. Mol. Neurosci. 2016, 9, 93. [Google Scholar] [CrossRef]
- Yang, H.; Zhong, X.; Ballar, P.; Luo, S.; Shen, Y.; Rubinsztein, D.C.; Monteiro, M.J.; Fang, S. Ubiquitin ligase Hrd1 enhances the degradation and sup-presses the toxicity of polyglutamine-expanded huntingtin. Exp. Cell Res. 2007, 313, 538–550. [Google Scholar] [CrossRef]
- Rubio, I.; Rodríguez-Navarro, J.A.; Tomás-Zapico, C.; Ruíz, C.; Casarejos, M.J.; Perucho, J.; Gomez, A.; Rodal, I.; Lucas, J.J.; Mena, M.A.; et al. Effects of partial suppression of parkin on huntingtin mutant R6/1 mice. Brain Res. 2009, 1281, 91–100. [Google Scholar] [CrossRef]
- Bhutani, S.; Das, A.; Maheshwari, M.; Lakhotia, S.; Jana, N. Dysregulation of core components of SCF complex in poly-glutamine disorders. Cell Death Dis. 2012, 3, e428. [Google Scholar] [CrossRef] [PubMed]
- Rotblat, B.; Southwell, A.L.; Ehrnhoefer, D.E.; Skotte, N.H.; Metzler, M.; Franciosi, S.; Leprivier, G.; Somasekharan, S.P.; Barokas, A.; Deng, Y.; et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc. Natl. Acad. Sci. USA 2014, 111, 3032–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Jin, Z.; Tan, H.; Xu, Q.; Peng, T.; Li, H. Atypical ubiquitination by E3 ligase WWP1 inhibits the proteasome-mediated deg-radation of mutant huntingtin. Brain Res. 2016, 1643, 103–112. [Google Scholar] [CrossRef]
- Zucchelli, S.; Marcuzzi, F.; Codrich, M.; Agostoni, E.; Vilotti, S.; Biagioli, M.; Pinto, M.; Carnemolla, A.; Santoro, C.; Gustincich, S.; et al. Tumor Necrosis Factor Receptor-associated Factor 6 (TRAF6) Associates with Huntingtin Protein and Promotes Its Atypical Ubiquitination to Enhance Aggregate Formation. J. Biol. Chem. 2011, 286, 25108–25117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, R.; Tong, Y. Proteostasis in Huntington’s disease: Disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 2018, 39, 754–769. [Google Scholar] [CrossRef] [Green Version]
- Tsou, W.-L.; Ouyang, M.; Hosking, R.R.; Sutton, J.R.; Blount, J.R.; Burr, A.A.; Todi, S.V. The deubiquitinase ataxin-3 requires Rad23 and DnaJ-1 for its neuroprotective role in Drosophila melanogaster. Neurobiol. Dis. 2015, 82, 12–21. [Google Scholar] [CrossRef] [Green Version]
- He, W.-T.; Xue, W.; Gao, Y.; Hong-Wei, Y.; Yue, H.-W.; Jiang, L.-L.; Hu, H.-Y. HSP90 recognizes the N-terminus of huntingtin involved in regulation of huntingtin aggregation by USP. Sci. Rep. 2017, 7, 14797. [Google Scholar] [CrossRef]
- Song, S.; Kim, S.-Y.; Hong, Y.-M.; Jo, D.-G.; Lee, J.-Y.; Shim, S.M.; Chung, C.-W.; Seo, S.J.; Yoo, Y.J.; Koh, J.-Y.; et al. Essential role of E2-25K/Hip-2 in mediating amyloid-beta neu-rotoxicity. Mol. Cell 2003, 12, 553–563. [Google Scholar] [CrossRef]
- Takahashi, T.; Kikuchi, S.; Katada, S.; Nagai, Y.; Nishizawa, M.; Onodera, O. Soluble polyglutamine oligomers formed prior to in-clusion body formation are cytotoxic. Hum. Mol. Genet. 2008, 17, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Ries, L.K.; Liess, A.K.L.; Feiler, C.G.; Spratt, D.; Lowe, E.D.; Lorenz, S. Crystal structure of the catalytic C-lobe of the HECT-type ubiquitin ligase E6AP. Protein Sci. 2020, 29, 1550–1554. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.; Yan, S.; Wang, C.-E.; Li, S.; Li, X.-J. Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc. Natl. Acad. Sci. USA 2014, 111, 5706–5711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jana, N.R.; Dikshit, P.; Goswami, A.; Kotliarova, S.; Murata, S.; Tanaka, K.; Nukina, N. Co-chaperone CHIP Associates with Expanded Polyglutamine Protein and Promotes Their Degradation by Proteasomes. J. Biol. Chem. 2005, 280, 11635–11640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Riley, B.; Lougheed, J.C.; Callaway, K.; Velasquez, M.; Brecht, E.; Nguyen, L.; A Shaler, T.; Walker, D.H.; Yang, Y.; Regnstrom, K.; et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 2013, 4, 1982. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.; Rattner, J. The Centrosome in Higher Organisms: Structure, Composition, and Duplication. Int. Rev. Cytol. 2004, 238, 119–182. [Google Scholar] [CrossRef]
- Zheng, N.; Schulman, B.A.; Song, L.; Miller, J.J.; Jeffrey, P.D.; Wang, P.; Chu, C.; Koepp, D.M.; Elledge, S.J.; Pagano, M.; et al. Structure of the Cul1–Rbx1–Skp1–F box Skp2 SCF ubiquitin ligase complex. Nature 2002, 416, 703–709. [Google Scholar] [CrossRef]
- Kang, Y.; Guo, J.; Yang, T.; Li, W.; Zhang, S. Regulation of the human ether-a-go-go-related gene (hERG) potassium channel by Nedd4 family interacting proteins (Ndfips). Biochem. J. 2015, 472, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Anta, B.; Martín-Rodríguez, C.; Gomis-Perez, C.; Calvo, L.; López-Benito, S.; Calderón-García, A.A.; Vicente-Garcia, C.; Villarroel, A.; Arevalo, J.C. Ubiquitin-specific protease 36 (USP36) controls neuronal precursor cell-expressed developmentally down-regulated 4-2 (Nedd4-2) actions over the neu-rotrophin receptor TrkA and potassium voltage-gated channels 7.2/3 (kv7.2/3). J. Biol. Chem. 2016, 291, 19132–19145. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Lamothe, B.; Besse, A.; Campos, A.D.; Webster, W.K.; Wu, H.; Darnay, B.G. Site-specific Lys-63-linked tumor necrosis factor recep-tor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J. Biol. Chem. 2007, 282, 4102–4112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, T.-M.; Shen, C.; Li, Q.; Zhang, P.; Wu, H. Mechanism of ubiquitin transfer promoted by TRAF. Proc. Natl. Acad. Sci. USA 2018, 115, 1783–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: New molecular signals. “Protein Modifications: Beyond the Usual Suspects” Review Series. EMBO Rep. 2008, 9, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [Green Version]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Degradation of huntingtin mediated by a hybrid molecule composed of IAP antagonist linked to phenyldiazenyl benzothiazole derivative. Bioorg. Med. Chem. Lett. 2018, 28, 707–710. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein Knockdown Using Methyl Bestatin−Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Hon, W.C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.W.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydrox-yproline in HIF-1α by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef]
- Min, J.H.; Yang, H.; Ivan, M.; Gertler, F.; Kaelin, W.G.; Pavietich, N.P. Structure of an HIF-1α-pVHL complex: Hydroxyproline recognition in signaling. Science 2002, 296, 1886–1889. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, J.S.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.; Crews, C.M. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef] [Green Version]
- Galdeano, C.; Gadd, M.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57, 8657–8663. [Google Scholar] [CrossRef]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1–CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, P.P.; Lopez-Girona, A.; Miller, K.; Carmel, G.; Pagarigan, B.; Chie-Leon, B.; Rychak, E.; Corral, L.G.; Ren, Y.J.; Wang, M.; et al. Structure of the human Cere-blon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 2014, 21, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrylak, D.P.; Gao, X.; Vogelzang, N.J.; Garfield, M.H.; Taylor, I.; Moore, M.D.; Peck, R.A.; Burris, H.A. First-in-human phase I study of ARV-110, an androgen receptor (AR) PROTAC degrader in patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC) following enzalutamide (ENZ) and/or abiraterone (ABI). J. Clin. Oncol. 2020, 38, 3500. [Google Scholar] [CrossRef]
- Flanagan, J.; Qian, Y.; Gough, S.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019, 79, P5-04-18. [Google Scholar] [CrossRef]
- Okuhira, K.; Ohoka, N.; Sai, K.; Nishimaki-Mogami, T.; Itoh, Y.; Ishikawa, M.; Hashimoto, Y.; Naito, M. Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein. FEBS Lett. 2011, 585, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Hu, L. Nrf2 activation through the inhibition of Keap1–Nrf2 protein–protein interaction. Med. Chem. Res. 2020, 29, 846–867. [Google Scholar] [CrossRef]
- Lu, M.; Liu, T.; Jiao, Q.; Ji, J.; Tao, M.; Liu, Y.; You, Q.; Jiang, Z. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiq-uitination-proteasome degradation pathway. Eur. J. Med. Chem. 2018, 146, 251–259. [Google Scholar] [CrossRef]
- Tong, B.; Luo, M.; Xie, Y.; Spradlin, J.N.; Tallarico, J.A.; McKenna, J.M.; Schirle, M.; Maimone, T.J.; Nomura, D.K. Bardoxolone conjugation enables targeted protein deg-radation of BRD. Sci. Rep. 2020, 10, 1–8. [Google Scholar]
- Ohoka, N.; Tsuji, G.; Shoda, T.; Fujisato, T.; Kurihara, M.; Demizu, Y.; Naito, M. Development of Small Molecule Chimeras That Recruit AhR E3 Ligase to Target Proteins. ACS Chem. Biol. 2019, 14, 2822–2832. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.C.; Kleinman, J.I.; Brittain, S.M.; Lee, P.S.; Chung, C.Y.S.; Kim, K.; Petri, Y.; Thomas, J.R.; Tallarico, J.A.; McKenna, J.M.; et al. Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol. 2019, 14, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Spradlin, J.N.; Hu, X.; Ward, C.C.; Brittain, S.M.; Jones, M.D.; Ou, L.; To, M.; Proudfoot, A.; Ornelas, E.; Woldegiorgis, M.; et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 2019, 15, 747–755. [Google Scholar] [CrossRef]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Discovery of Small Molecules that Induce the Degradation of Huntingtin. Angew. Chem. Int. Ed. 2017, 56, 11530–11533. [Google Scholar] [CrossRef]
- Yamashita, H.; Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Application of protein knockdown strategy targeting β-sheet structure to multiple disease-associated polyglutamine proteins. Bioorg. Med. Chem. 2019, 28, 115175. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, C.; Wang, Z.; Zhu, C.; Li, J.; Sha, T.; Ma, L.; Gao, C.; Yang, Y.; Sun, Y.; et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature 2019, 575, 203–209. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
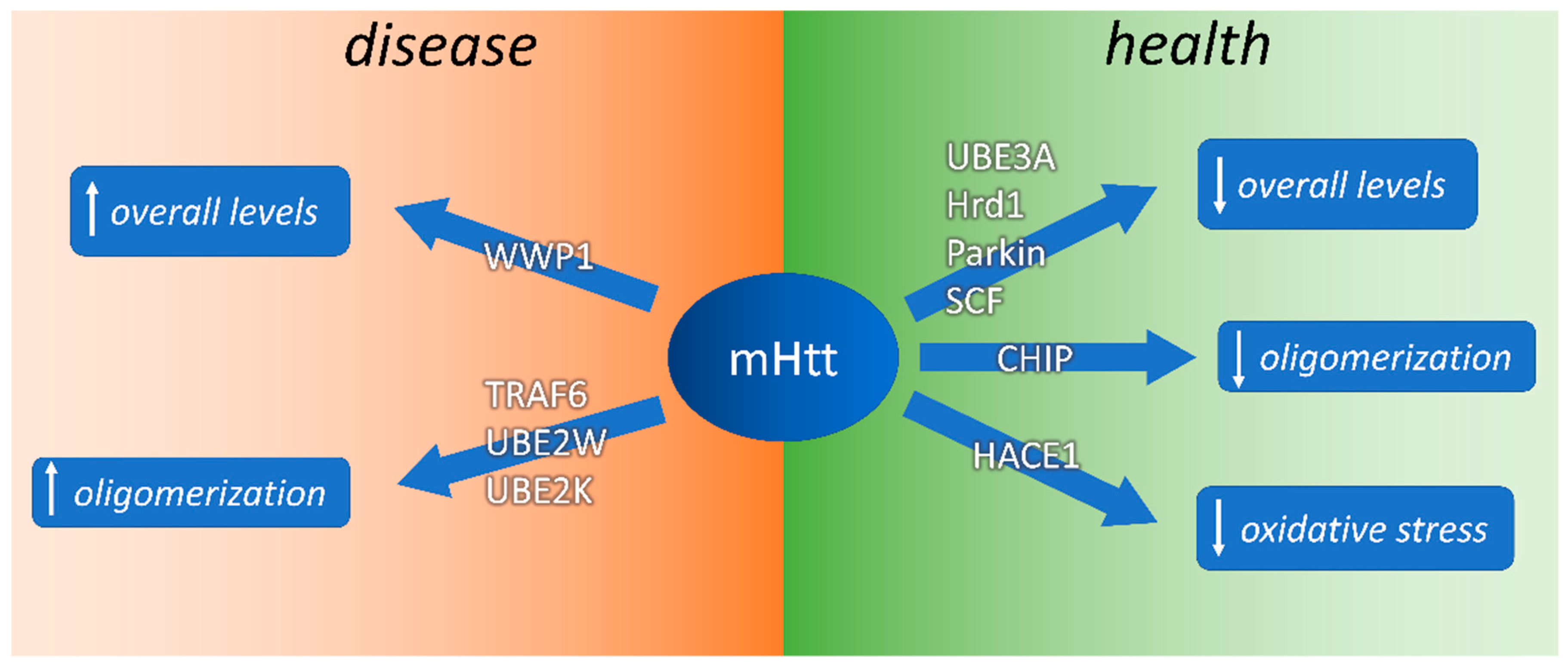{kind=link}
| Residue (Human Htt) | Demonstrated Modification | Reference |
|---|---|---|
| Lys 6 | SUMOylation | [66] |
| Lys 9 | SUMOylation | [66] |
| Lys 253 | Ubiquitination | [67] |
| Lys 335 | Ubiquitination | [68] |
| Lys 442 | Ubiquitination | [69] |
| Lys 662 | Ubiquitination | [68] |
| Lys 667 | Ubiquitination | [67] |
| Lys 698 | Ubiquitination | [67] |
| Lys 813 | Ubiquitination | [67] |
| Lys 902 | Ubiquitination | [67] |
| Lys 937 | Ubiquitination | [67] |
| Lys 941 | Ubiquitination | [70] |
| Lys 1121 | Ubiquitination | [68] |
| Lys 1223 | Ubiquitination | [68] |
| Lys 1244 | Ubiquitination | [68] |
| Lys 1262 | Ubiquitination | [71] |
| Lys 1337 | Ubiquitination | [67] |
| Lys 1402 | Ubiquitination | [67,68,70,71,72] |
| Lys 1410 | Ubiquitination | [67,68,70] |
| Lys 1415 | Ubiquitination | [68] |
| Lys 1431 | Ubiquitination | [68,70,73] |
| Lys 1885 | Ubiquitination | [67] |
| Lys 2417 | Ubiquitination | [71] |
| Lys 2423 | Ubiquitination | [68] |
| Lys 2443 | Ubiquitination | [68] |
| Lys 2537 | Ubiquitination | [67] |
| Lys 2564 | Ubiquitination | [67] |
| Lys 2757 | Ubiquitination | [67] |
| Lys 2901 | Ubiquitination | [67] |
| Lys 2967 | Ubiquitination | [67] |
| Protein | Function/Type | Effect on mHtt | Protein Length | PDB Codes |
|---|---|---|---|---|
| UBE2K/E2-25k | E2 | mHtt oligomerization | 200 residues | 1YLA (1–200), 2O25 (1–200), 3E46 (1–200), 3F92 (1–200), 3K9O (1–200), 3K9P (1–200), 5DFL (1–200), 6IF1 (1–199), 6JB6 (1–200), 6JB7 (1–200) |
| UBE2W | E2 | mHtt oligomerization | 151 residues | 2A7L (1–117), 2MT6 (1–151) |
| UBE3A/E6AP | HECT-type E3 | mHtt clearance | 875 residues | 1C4Z (518–875), 1D5F (518–875), 1EQX (401–418), 2KR1 (24–87), 4GIZ (403–414), 4XR8 (406–417), 6SJV (403–417), 6SLM (403–417), 6TGK (765–869), 6U19 (24–87) |
| CHIP/STUB1 | U-box E3 | mHtt oligomerization inhibition | 303 residues | 4KBQ (21–154), 6EFK (23–154), 6NSV (23–152) |
| HRD1/SYVN1 | RING-type E3 | httN clearance | 617 residues | 6A3Z (279–334), 6JB7 (1–200) |
| Parkin | RING/HECT hybrid E3 | mHtt clearance | 465 residues | 1IYF (1–76), 2JMO (308–384), 4BM9 (137–465), 4I1F (141–465), 5C1Z (1–465), 5C23 (1–465), 5C9V (137–465), 5N2W (1–465), 5N38 (1–465), 5TR5 (1–76), 6GLC (1–382), 6HUE (1–465), N13 (144–465) |
| SCF complex (formed by Rbx1, Cul1, Skp1) | E3 complex | mHtt clearance | 1LDJ | |
| HACE1 | HECT-type E3 | NRF2-mediated antioxidative stress | 909 residues | No structure |
| WWP1 | NEDD4-like E3 | Increase in mHtt in the cell | 922 residues | 1ND7 (546–917), 2OP7 (494–532), HPS (537–917), 5HPT (537–917), 6J1X (379–922), 6J1Y (410–922) |
| TRAF6 | RING-type E3 | Increase in mHtt fragment aggregates | 522 residues | 1LB4 (348–504), 1LB5 (347–504), 1LB6 (347–504), 2ECI (50–128), 2JMD (67–124), 3HCS (50–211), 3HCT (50–159), 3HCU (50–159), 4Z8M (346–504), 5ZUJ (350–501), 6A33 (350–501), 7L3L (52–158) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorillo, A.; Morea, V.; Colotti, G.; Ilari, A. Huntingtin Ubiquitination Mechanisms and Novel Possible Therapies to Decrease the Toxic Effects of Mutated Huntingtin. J. Pers. Med. 2021, 11, 1309. https://doi.org/10.3390/jpm11121309
Fiorillo A, Morea V, Colotti G, Ilari A. Huntingtin Ubiquitination Mechanisms and Novel Possible Therapies to Decrease the Toxic Effects of Mutated Huntingtin. Journal of Personalized Medicine. 2021; 11(12):1309. https://doi.org/10.3390/jpm11121309
Chicago/Turabian StyleFiorillo, Annarita, Veronica Morea, Gianni Colotti, and Andrea Ilari. 2021. "Huntingtin Ubiquitination Mechanisms and Novel Possible Therapies to Decrease the Toxic Effects of Mutated Huntingtin" Journal of Personalized Medicine 11, no. 12: 1309. https://doi.org/10.3390/jpm11121309