
Improved In Vivo Delivery of Small RNA Based on the Calcium Phosphate Method

 , ,
, , 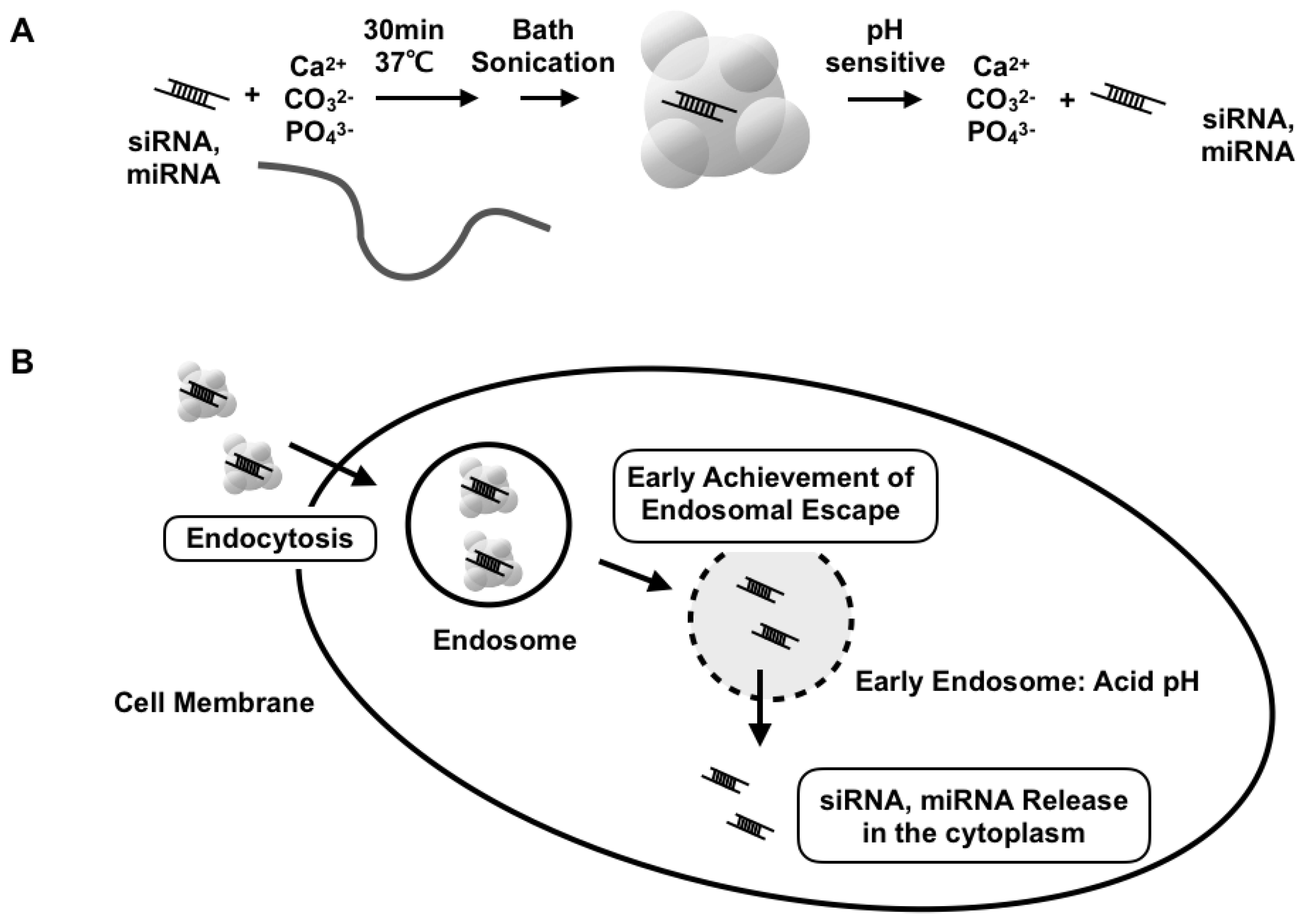{kind=link}
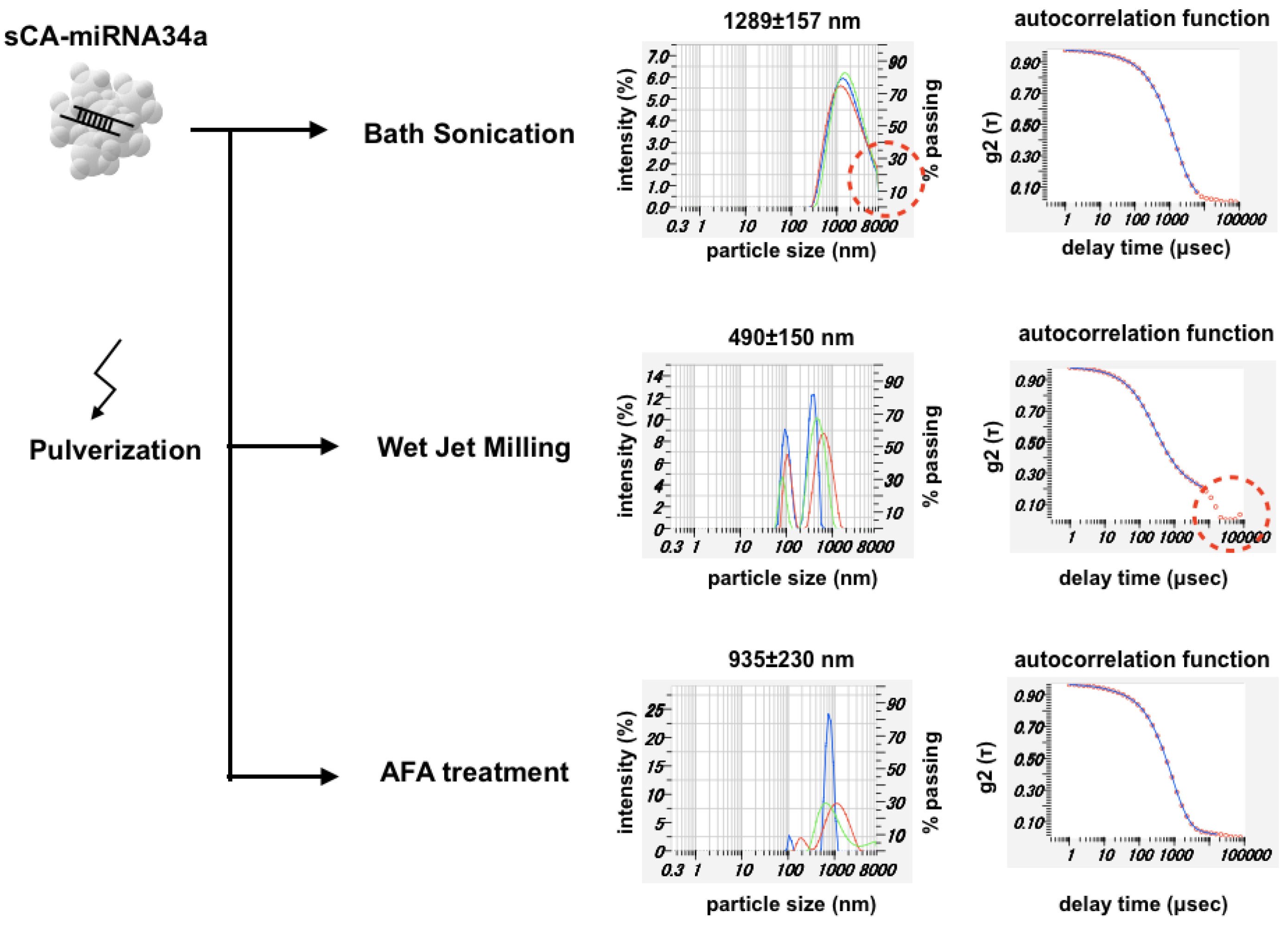{kind=link}
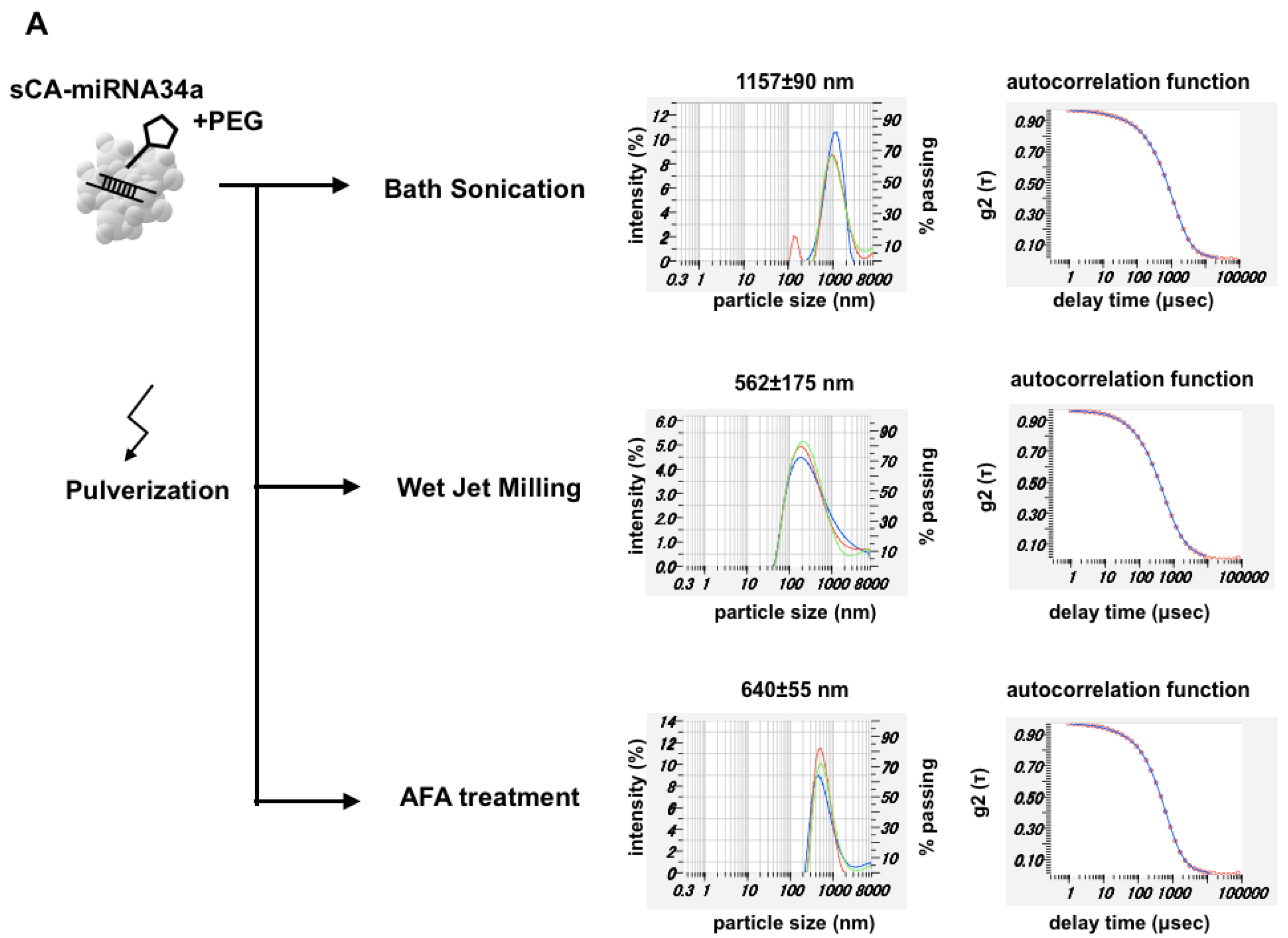{kind=link}
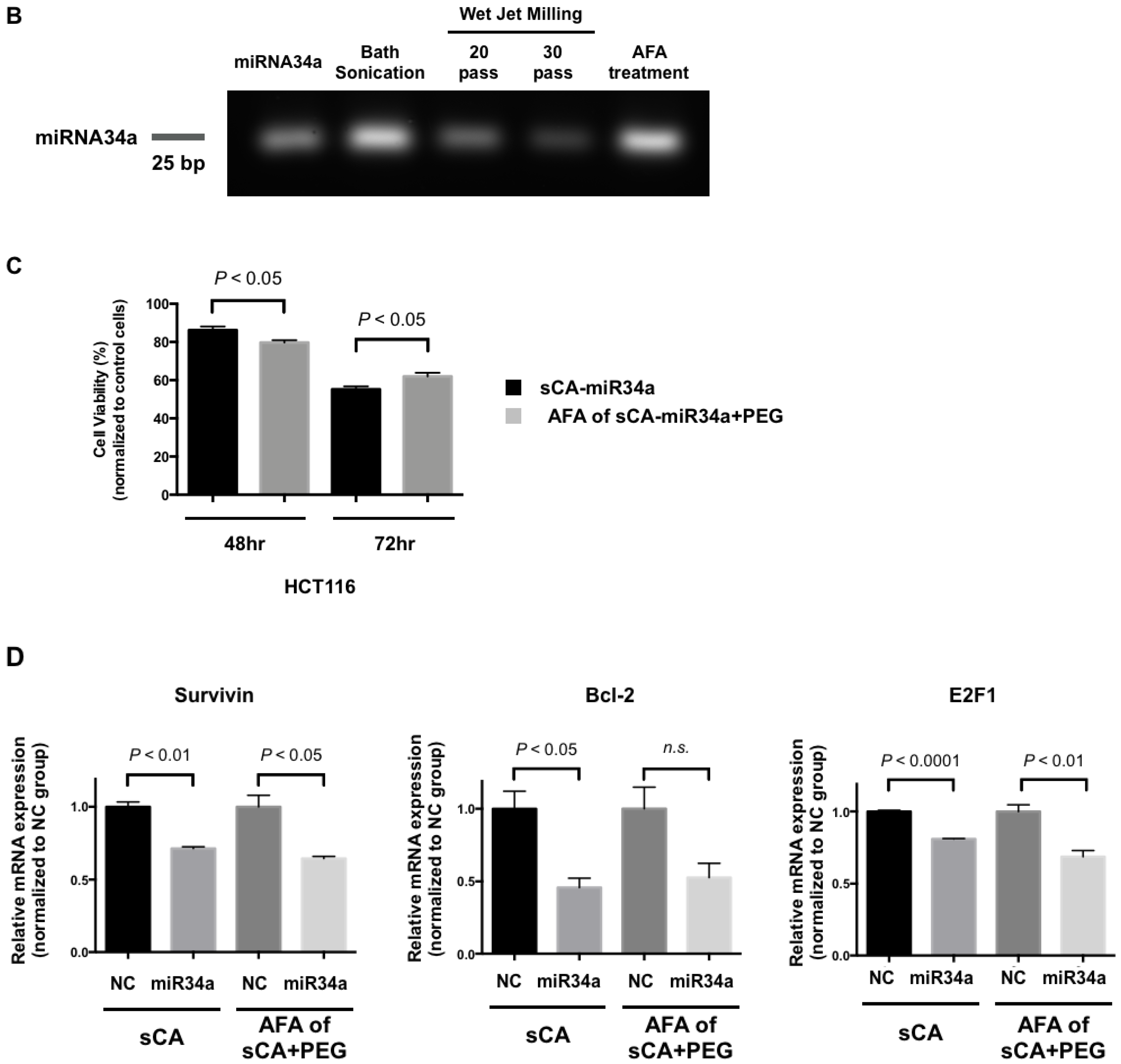{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
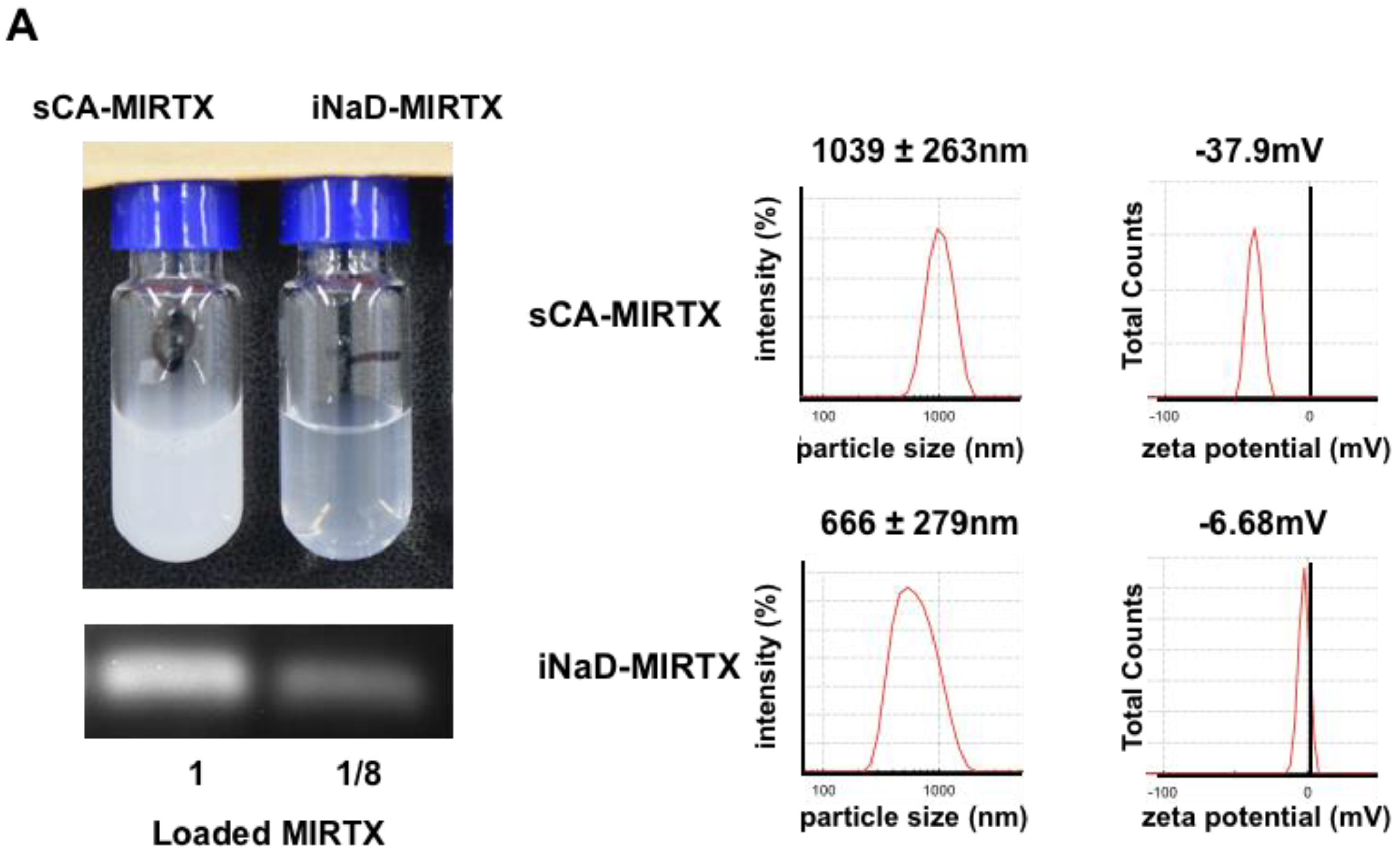
2.2. Production of sCA and iNaD
2.3. Assaying Nanoparticle Features
2.4. miRNA Electrophoresis
2.5. Cell Proliferation Assay
2.6. Quantitative Real-Time RT-PCR Analysis of mRNA Expression
2.7. Western Blot Analysis
2.8. Animals
2.9. Cell Line-Derived Xenograft Models
2.10. Rheumatoid Arthritis Models
2.11. Statistical Analysis
3. Results
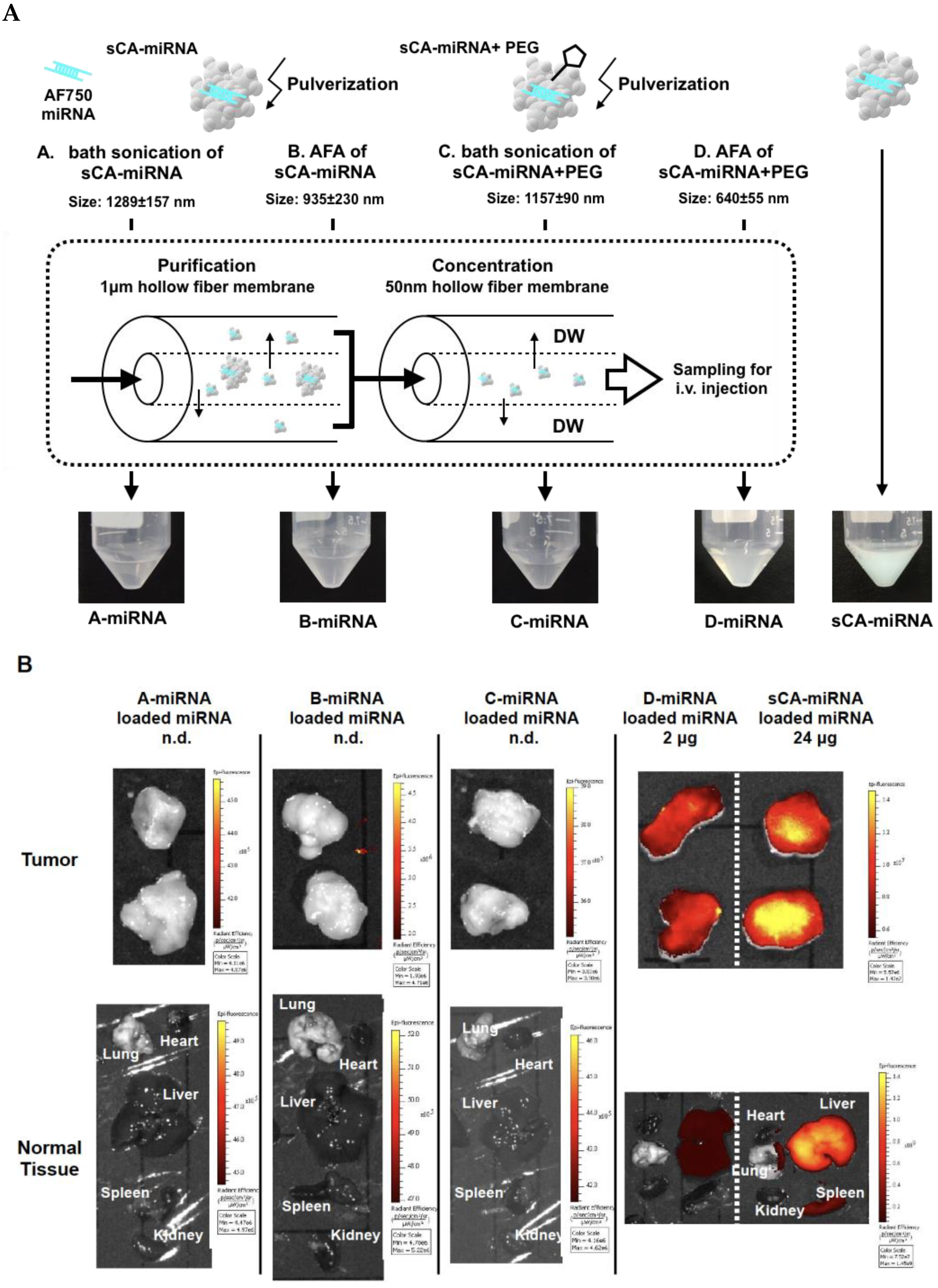
3.1. Mechanical Pulverization of sCA-miRNA34a
3.2. Production of PEG-Blended sCA Followed by Mechanical Pulverization
3.3. Purification and Concentration
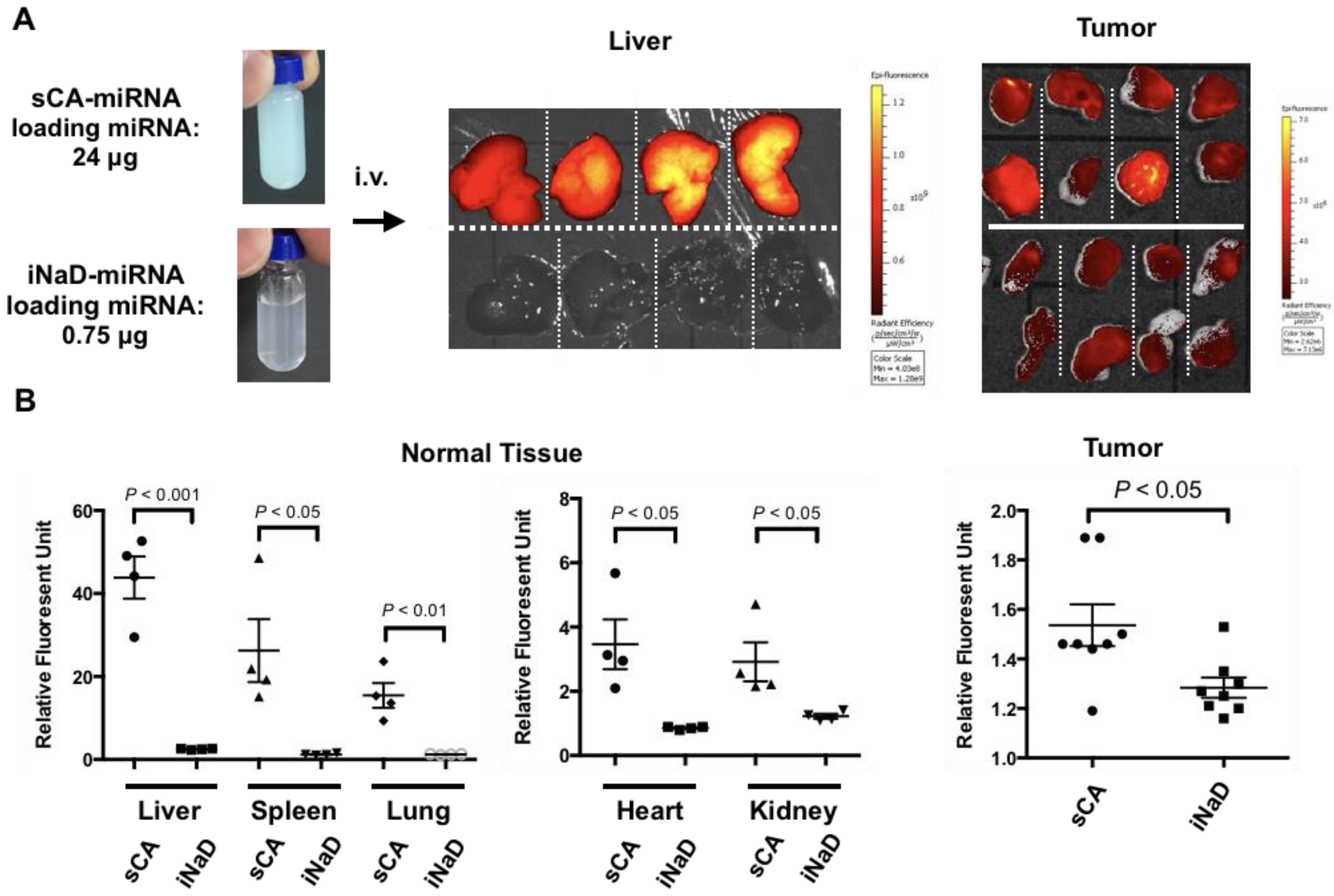
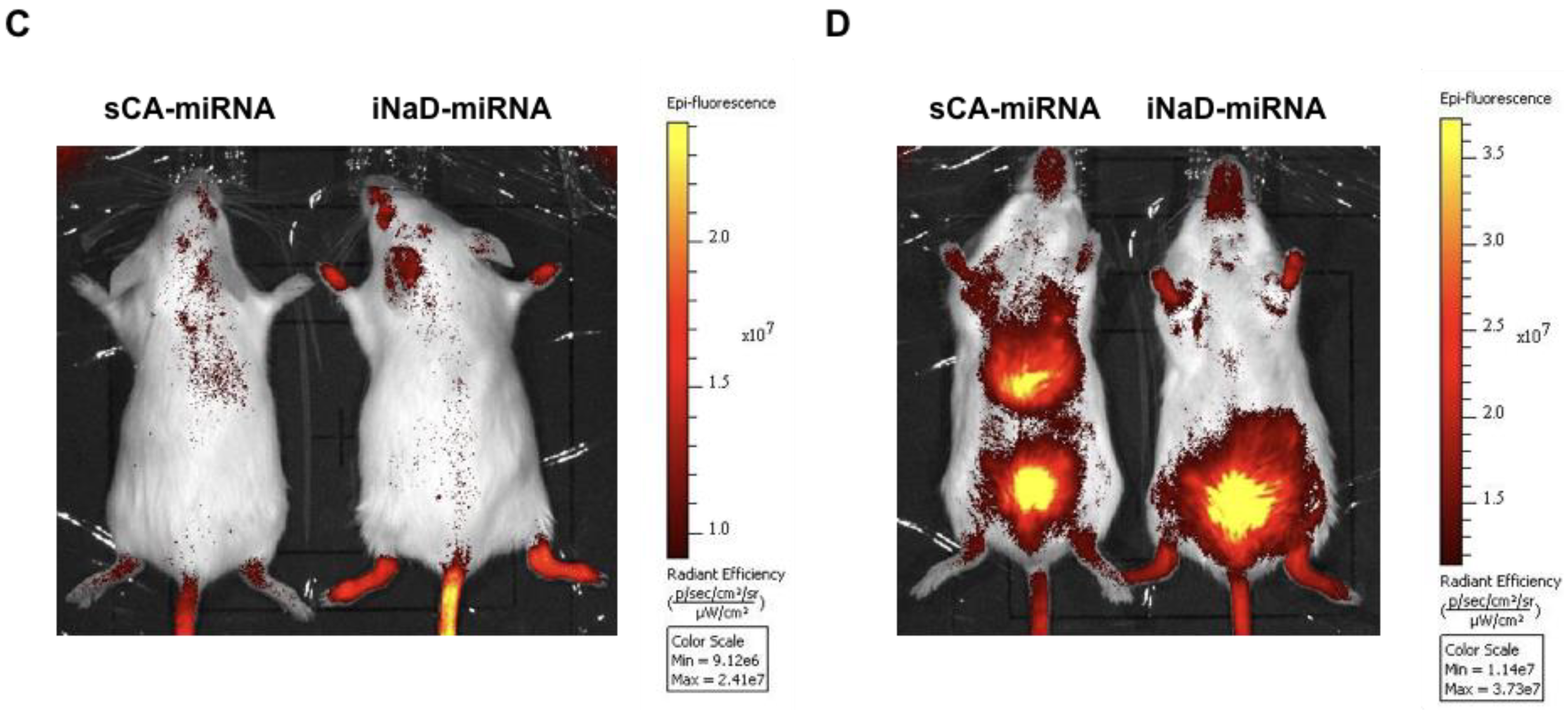
3.4. Bio-Distribution of New Nanoparticles as iNaD
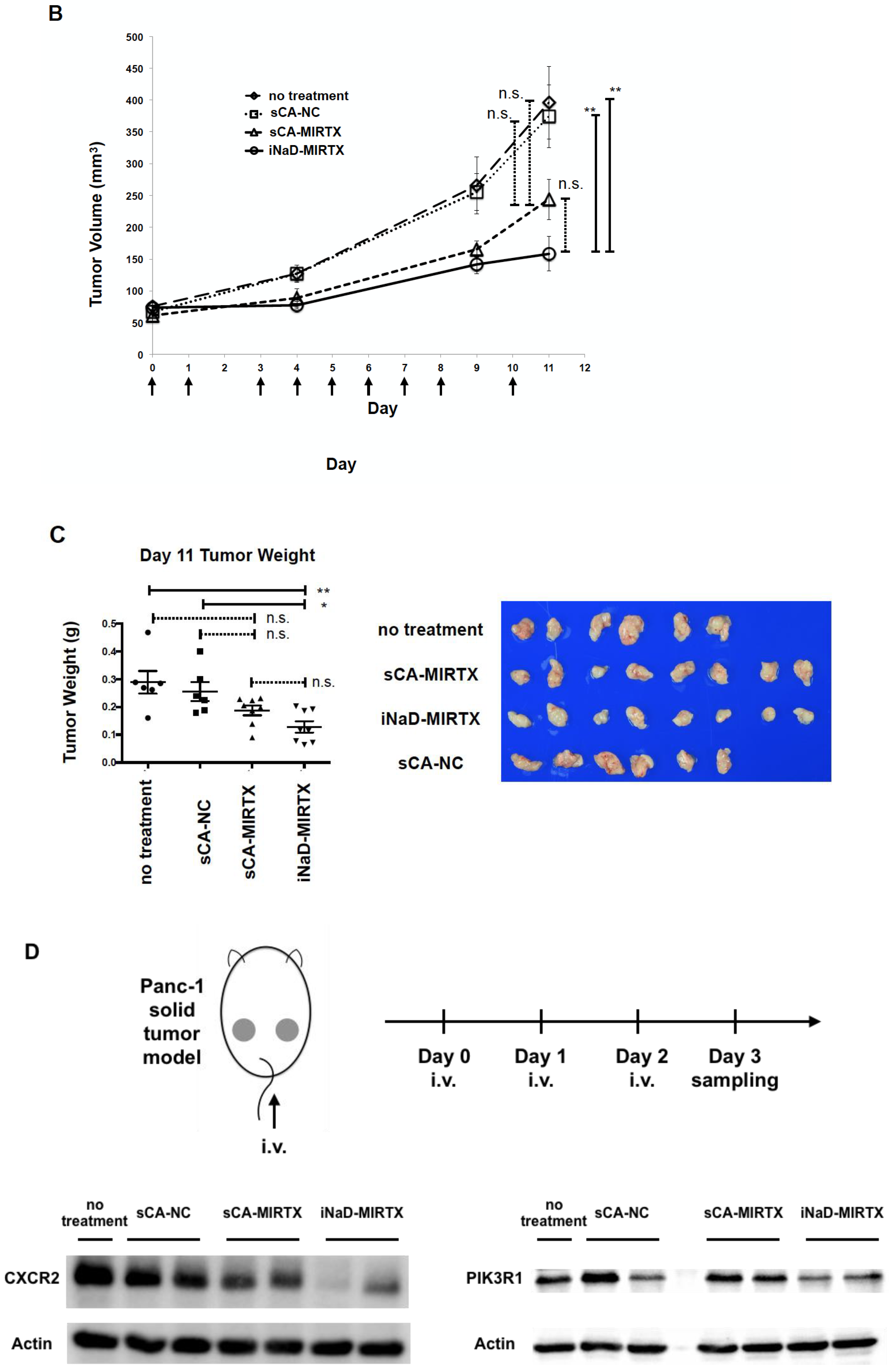
3.5. Anti-Tumor Effect of iNaD-MIRTX
3.6. Bio-Distribution in Rheumatoid Arthritis Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46 Pt 1, 6387–6392. [Google Scholar]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Rivas-García, L.; Baptista, P.V.; Fernandes, A.R. Gene Therapy in Cancer Treatment: Why Go Nano? Pharmaceutics 2020, 12, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mainini, F.; Eccles, M.R. Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy. Molecules 2020, 25, 2692. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.P.; Dwyer, R.M. Nanoparticle-Based Delivery of Tumor Suppressor microRNA for Cancer Therapy. Cells 2020, 9, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Yamamoto, H.; Nakanishi, H.; Yamamoto, Y.; Inoue, A.; Tei, M.; Hirose, H.; Uemura, M.; Nishimura, J.; Hata, T.; et al. Innovative delivery of siRNA to solid tumors by super carbonate apatite. PLoS ONE 2015, 10, e0116022. [Google Scholar] [CrossRef] [Green Version]
- Takeyama, H.; Yamamoto, H.; Yamashita, S.; Wu, X.; Takahashi, H.; Nishimura, J.; Haraguchi, N.; Miyake, Y.; Suzuki, R.; Murata, K.; et al. Decreased miR-340 expression in bone marrow is associated with liver metastasis of colorectal cancer. Mol. Cancer Ther. 2014, 13, 976–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Nishimura, J.; Kagawa, Y.; Kano, Y.; Takahashi, Y.; Wu, X.; Hiraki, M.; Hamabe, A.; Konno, M.; Haraguchi, N.; et al. Significance of Polypyrimidine Tract-Binding Protein 1 Expression in Colorectal Cancer. Mol. Cancer Ther. 2015, 14, 1705–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, H.; Wu, X.; Kawamoto, K.; Nishida, N.; Konno, M.; Koseki, J.; Matsui, H.; Noguchi, K.; Gotoh, N.; Yamamoto, T.; et al. MicroRNAs Induce Epigenetic Reprogramming and Suppress Malignant Phenotypes of Human Colon Cancer Cells. PLoS ONE 2015, 10, e0127119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraki, M.; Nishimura, J.; Takahashi, H.; Wu, X.; Takahashi, Y.; Miyo, M.; Nishida, N.; Uemura, M.; Hata, T.; Takemasa, I.; et al. Concurrent Targeting of KRAS and AKT by MiR-4689 Is a Novel Treatment Against Mutant KRAS Colorectal Cancer. Mol. Ther. Nucleic Acids 2015, 4, e231. [Google Scholar] [CrossRef]
- Inoue, A.; Mizushima, T.; Wu, X.; Okuzaki, D.; Kambara, N.; Ishikawa, S.; Wang, J.; Qian, Y.; Hirose, H.; Yokoyama, Y.; et al. A miR-29b Byproduct Sequence Exhibits Potent Tumor-Suppressive Activities via Inhibition of NF-kappaB Signaling in KRAS-Mutant Colon Cancer Cells. Mol. Cancer Ther. 2018, 17, 977–987. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, Y.; Mizushima, T.; Wu, X.; Okuzaki, D.; Yokoyama, Y.; Inoue, A.; Hata, T.; Hirose, H.; Qian, Y.; Wang, J.; et al. miR-4711-5p regulates cancer stemness and cell cycle progression via KLF5, MDM2 and TFDP1 in colon cancer cells. Br. J. Cancer 2020, 122, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Fukata, T.; Mizushima, T.; Nishimura, J.; Okuzaki, D.; Wu, X.; Hirose, H.; Yokoyama, Y.; Kubota, Y.; Nagata, K.; Tsujimura, N.; et al. The Supercarbonate Apatite-MicroRNA Complex Inhibits Dextran Sodium Sulfate-Induced Colitis. Mol. Ther. Nucleic Acids 2018, 12, 658–671. [Google Scholar] [CrossRef] [Green Version]
- Aoki, M.; Aoki, H.; Mukhopadhyay, P.; Tsuge, T.; Yamamoto, H.; Matsumoto, N.M.; Toyohara, E.; Okubo, Y.; Ogawa, R.; Takabe, K. Sphingosine-1-Phosphate Facilitates Skin Wound Healing by Increasing Angiogenesis and Inflammatory Cell Recruitment with Less Scar Formation. Int. J. Mol. Sci. 2019, 20, 3381. [Google Scholar] [CrossRef] [Green Version]
- Aoki, M.; Matsumoto, N.M.; Dohi, T.; Kuwahawa, H.; Akaishi, S.; Okubo, Y.; Ogawa, R.; Yamamoto, H.; Takabe, K. Direct Delivery of Apatite Nanoparticle-Encapsulated siRNA Targeting TIMP-1 for Intractable Abnormal Scars. Mol. Ther. Nucleic Acids 2020, 22, 50–61. [Google Scholar] [CrossRef]
- Takahashi, H.; Misato, K.; Aoshi, T.; Yamamoto, Y.; Kubota, Y.; Wu, X.; Kuroda, E.; Ishii, K.J.; Yamamoto, H.; Yoshioka, Y. Carbonate Apatite Nanoparticles Act as Potent Vaccine Adjuvant Delivery Vehicles by Enhancing Cytokine Production Induced by Encapsulated Cytosine-Phosphate-Guanine Oligodeoxynucleotides. Front. Immunol. 2018, 9, 783. [Google Scholar] [CrossRef]
- Tamai, K.; Mizushima, T.; Wu, X.; Inoue, A.; Ota, M.; Yokoyama, Y.; Miyoshi, N.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; et al. Photodynamic Therapy Using Indocyanine Green Loaded on Super Carbonate Apatite as Minimally Invasive Cancer Treatment. Mol. Cancer Ther. 2018, 17, 1613–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merhautova, J.; Demlova, R.; Slaby, O. MicroRNA-Based Therapy in Animal Models of Selected Gastrointestinal Cancers. Front. Pharmacol. 2016, 7, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, R.U.; Prieto-Vila, M.; Kohama, I.; Ochiya, T. Development of miRNA-based therapeutic approaches for cancer patients. Cancer Sci. 2019, 110, 1140–1147. [Google Scholar] [CrossRef] [Green Version]
- Abd-Aziz, N.; Kamaruzman, N.I.; Poh, C.L. Development of MicroRNAs as Potential Therapeutics against Cancer. J. Oncol. 2020, 2020, 8029721. [Google Scholar] [CrossRef] [PubMed]
- Forterre, A.; Komuro, H.; Aminova, S.; Harada, M. A Comprehensive Review of Cancer MicroRNA Therapeutic Delivery Strategies. Cancers 2020, 12, 1852. [Google Scholar] [CrossRef] [PubMed]
- Pecot, C.V.; Calin, G.A.; Coleman, R.L.; Lopez-Berestein, G.; Sood, A.K. RNA interference in the clinic: Challenges and future directions. Nat. Rev. Cancer 2011, 11, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Perrault, S.D.; Walkey, C.; Jennings, T.; Fischer, H.C.; Chan, W.C. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009, 9, 1909–1915. [Google Scholar] [CrossRef]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.R.; Miyazono, K.; Uesaka, M.; et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815–823. [Google Scholar] [CrossRef]
- Tejera-Garcia, R.; Ranjan, S.; Zamotin, V.; Sood, R.; Kinnunen, P.K. Making unilamellar liposomes using focused ultrasound. Langmuir 2011, 27, 10088–10097. [Google Scholar] [CrossRef]
- Hermeking, H. p53 enters the microRNA world. Cancer Cell 2007, 12, 414–418. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Knight, R.A. miR-34: From bench to bedside. Oncotarget 2014, 5, 872–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tazawa, H.; Tsuchiya, N.; Izumiya, M.; Nakagama, H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15472–15477. [Google Scholar] [CrossRef] [Green Version]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- MacDiarmid, J.A.; Mugridge, N.B.; Weiss, J.C.; Phillips, L.; Burn, A.L.; Paulin, R.P.; Haasdyk, J.E.; Dickson, K.A.; Brahmbhatt, V.N.; Pattison, S.T.; et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell 2007, 11, 431–445. [Google Scholar] [CrossRef] [Green Version]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Yoshitomi, H.; Sakaguchi, N.; Kobayashi, K.; Brown, G.D.; Tagami, T.; Sakihama, T.; Hirota, K.; Tanaka, S.; Nomura, T.; Miki, I.; et al. A role for fungal {beta}-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. J. Exp. Med. 2005, 201, 949–960. [Google Scholar] [CrossRef]
- Kakizawa, Y.; Furukawa, S.; Kataoka, K. Block copolymer-coated calcium phosphate nanoparticles sensing intracellular environment for oligodeoxynucleotide and siRNA delivery. J. Control. Release 2004, 97, 345–356. [Google Scholar] [CrossRef]
- Giger, E.V.; Puigmartí-Luis, J.; Schlatter, R.; Castagner, B.; Dittrich, P.S.; Leroux, J.C. Gene delivery with bisphosphonate-stabilized calcium phosphate nanoparticles. J. Control. Release 2011, 150, 87–93. [Google Scholar] [CrossRef]
- Pittella, F.; Miyata, K.; Maeda, Y.; Suma, T.; Watanabe, S.; Chen, Q.; Christie, R.J.; Osada, K.; Nishiyama, N.; Kataoka, K. Pancreatic cancer therapy by systemic administration of VEGF siRNA contained in calcium phosphate/charge-conversional polymer hybrid nanoparticles. J. Control. Release 2012, 161, 868–874. [Google Scholar] [CrossRef]
- Wang, R.; Murali, V.S.; Draper, R. Detecting Sonolysis of Polyethylene Glycol Upon Functionalizing Carbon Nanotubes. Methods Mol. Biol. 2017, 1530, 147–164. [Google Scholar]
- Kawasaki, H.; Takeda, Y.; Arakawa, R. Mass spectrometric analysis for high molecular weight synthetic polymers using ultrasonic degradation and the mechanism of degradation. Anal. Chem. 2007, 79, 4182–4187. [Google Scholar] [CrossRef]
- Suslick, K.S. Sonochemistry. Science 1990, 247, 1439–1445. [Google Scholar] [CrossRef]
- Wang, J.; Pelletier, M.; Zhang, H.; Xia, H.; Zhao, Y. High-frequency ultrasound-responsive block copolymer micelle. Langmuir 2009, 25, 13201–13205. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Yang, M.; Li, X.M.; Jiang, P.; Baranov, E.; Li, S.; Xu, M.; Penman, S.; Hoffman, R.M. Tumor-targeting bacterial therapy with amino acid auxotrophs of GFP-expressing Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 2005, 102, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Yang, M.; Ma, H.; Li, X.; Tan, X.; Li, S.; Yang, Z.; Hoffman, R.M. Targeted therapy with a Salmonella typhimurium leucine-arginine auxotroph cures orthotopic human breast tumors in nude mice. Cancer Res. 2006, 66, 7647–7652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Liao, L.; Yin, H.; Nakamura, H.; Shin, T.; Maeda, H. Enhanced bacterial tumor delivery by modulating the EPR effect and therapeutic potential of Lactobacillus casei. J. Pharm. Sci. 2014, 103, 3235–3243. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Islam, W.; Maeda, H. Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers. Adv. Drug Deliv. Rev. 2020, 157, 142–160. [Google Scholar] [CrossRef]
- He, H.; Liu, L.; Morin, E.E.; Liu, M.; Schwendeman, A. Survey of Clinical Translation of Cancer Nanomedicines-Lessons Learned from Successes and Failures. Acc. Chem. Res. 2019, 52, 2445–2461. [Google Scholar] [CrossRef]
- Islam, W.; Fang, J.; Imamura, T.; Etrych, T.; Subr, V.; Ulbrich, K.; Maeda, H. Augmentation of the Enhanced Permeability and Retention Effect with Nitric Oxide-Generating Agents Improves the Therapeutic Effects of Nanomedicines. Mol. Cancer Ther. 2018, 17, 2643–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Jeong, J.H.; Lee, S.H.; Kim, S.W.; Park, T.G. Local and systemic delivery of VEGF siRNA using polyelectrolyte complex micelles for effective treatment of cancer. J. Control. Release 2008, 129, 107–116. [Google Scholar] [CrossRef]
- Zuckerman, J.E.; Choi, C.H.; Han, H.; Davis, M.E. Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane. Proc. Natl. Acad. Sci. USA 2012, 109, 3137–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, F.; Minakuchi, Y.; Nagahara, S.; Honma, K.; Sasaki, H.; Hirai, K.; Teratani, T.; Namatame, N.; Yamamoto, Y.; Hanai, K. Efficient delivery of small interfering RNA to bone-metastatic tumors by using atelocollagen in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 12177–12182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudoh, K.; Fukuda, N.; Kasugai, S.; Tachikawa, N.; Koyano, K.; Matsushita, Y.; Ogino, Y.; Ishikawa, K.; Miyamoto, Y. Maxillary Sinus Floor Augmentation Using Low-Crystalline Carbonate Apatite Granules With Simultaneous Implant Installation: First-in-Human Clinical Trial. J. Oral Maxillofac. Surg. 2019, 77, 985.e1–985.e11. [Google Scholar] [CrossRef]
- Nakagawa, T.; Kudoh, K.; Fukuda, N.; Kasugai, S.; Tachikawa, N.; Koyano, K.; Matsushita, Y.; Sasaki, M.; Ishikawa, K.; Miyamoto, Y. Application of low-crystalline carbonate apatite granules in 2-stage sinus floor augmentation: A prospective clinical trial and histomorphometric evaluation. J. Periodontal Implant Sci. 2019, 49, 382–396. [Google Scholar] [CrossRef]
- Campbell, R.B.; Fukumura, D.; Brown, E.B.; Mazzola, L.M.; Izumi, Y.; Jain, R.K.; Torchilin, V.P.; Munn, L.L. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002, 62, 6831–6836. [Google Scholar]
- Li, S.D.; Huang, L. Pharmacokinetics and biodistribution of nanoparticles. Mol. Pharm. 2008, 5, 496–504. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Yokoyama, Y.; Takahashi, H.; Kouda, S.; Yamamoto, H.; Wang, J.; Morimoto, Y.; Minami, K.; Hata, T.; Shamma, A.; et al. Improved In Vivo Delivery of Small RNA Based on the Calcium Phosphate Method. J. Pers. Med. 2021, 11, 1160. https://doi.org/10.3390/jpm11111160
Wu X, Yokoyama Y, Takahashi H, Kouda S, Yamamoto H, Wang J, Morimoto Y, Minami K, Hata T, Shamma A, et al. Improved In Vivo Delivery of Small RNA Based on the Calcium Phosphate Method. Journal of Personalized Medicine. 2021; 11(11):1160. https://doi.org/10.3390/jpm11111160
Chicago/Turabian StyleWu, Xin, Yuhki Yokoyama, Hidekazu Takahashi, Shihori Kouda, Hiroyuki Yamamoto, Jiaqi Wang, Yoshihiro Morimoto, Kazumasa Minami, Tsuyoshi Hata, Awad Shamma, and et al. 2021. "Improved In Vivo Delivery of Small RNA Based on the Calcium Phosphate Method" Journal of Personalized Medicine 11, no. 11: 1160. https://doi.org/10.3390/jpm11111160