
Functional Characterization of 21 Rare Allelic CYP1A2 Variants Identified in a Population of 4773 Japanese Individuals by Assessing Phenacetin O-Deethylation

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Sanger Sequencing Analysis for the Detection of CYP1A2 Sequence Alterations
2.3. Expression of CYP1A2 Variants in 293FT Cells
2.4. Western Blotting
2.5. Determination of Cytochrome (CYP), Cytochrome P450 Oxidoreductase (CPR), and Cytochrome b5 Content
2.6. Phenacetin O-Deethylation
2.7. 3D Structural Modeling of CYP1A2
2.8. Data Analysis
3. Results
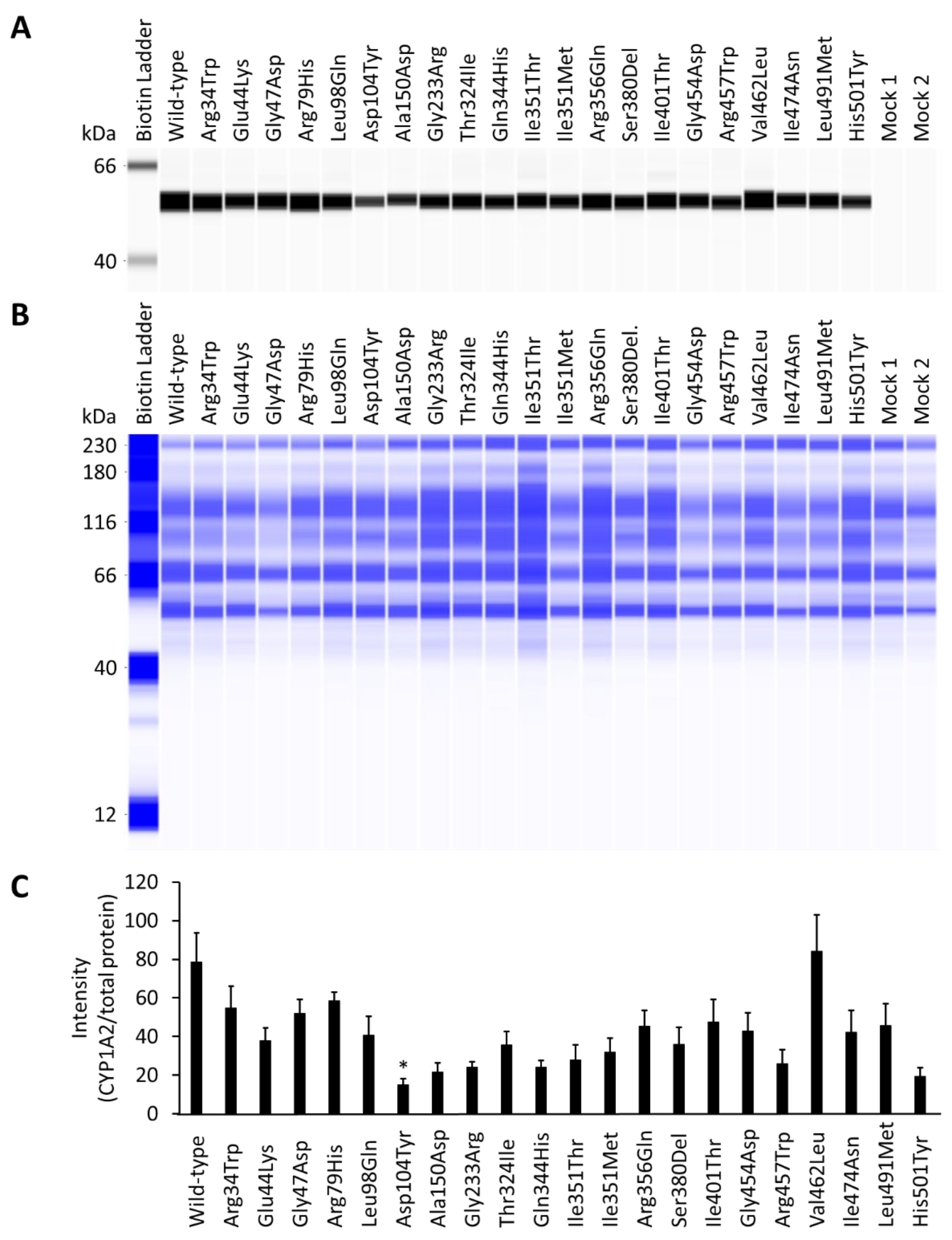
3.1. Effect of CYP1A2 Variants on the Expression of CPR and Cytochrome b5
3.2. Holoprotein Content
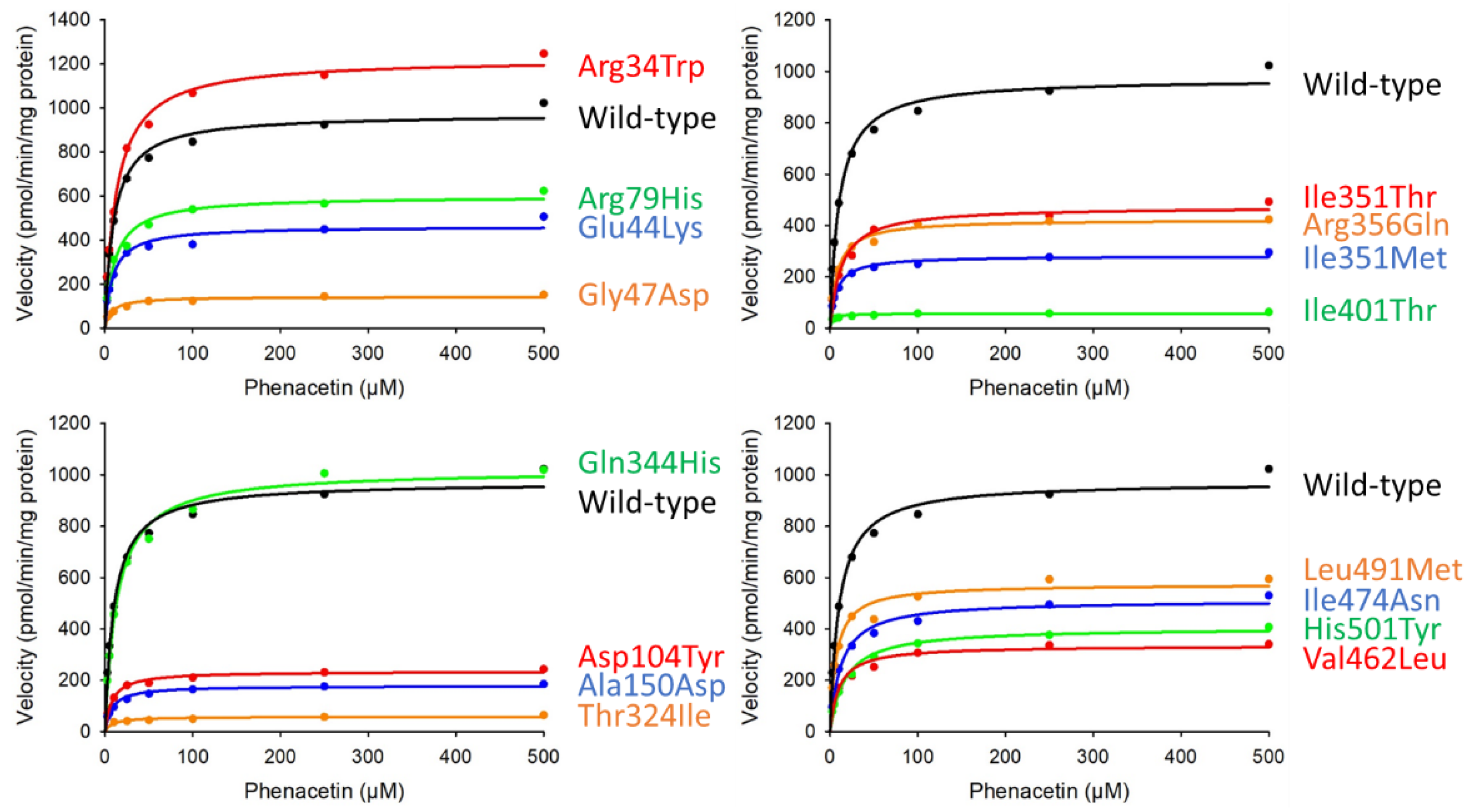
3.3. O-Deethylation of Phenacetin
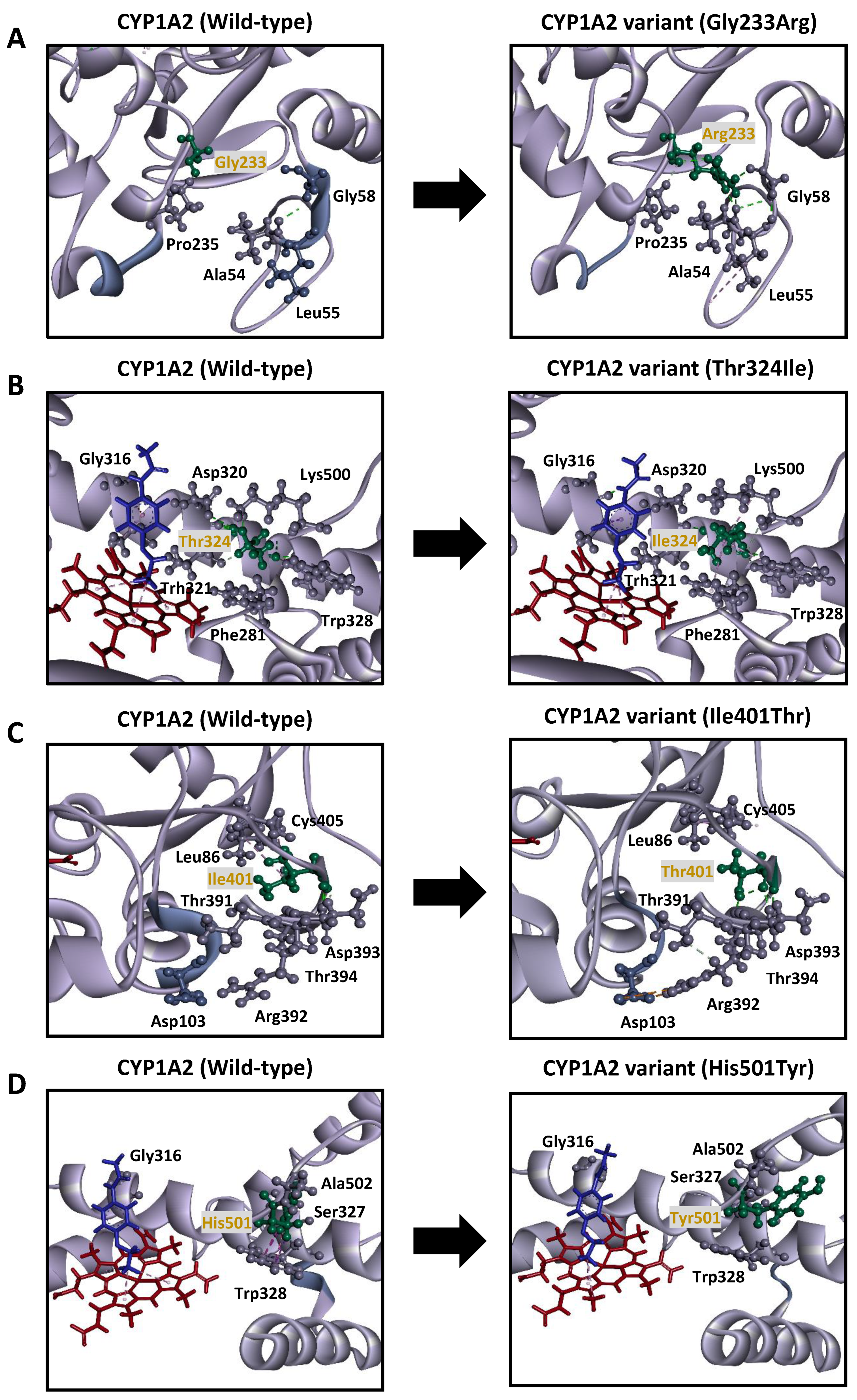
3.4. 3D Molecular Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Belle, D.J.; Singh, H. Genetic factors in drug metabolism. Am. Fam. Physician 2008, 77, 1553–1560. [Google Scholar]
- Zhou, S.F.; Liu, J.P.; Chowbay, B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 2009, 41, 89–295. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, M. In vitro assessment of the allelic variants of cytochrome P450. Drug Metab. Pharm. 2012, 27, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, C.; Liu, G.; Liu, R.; Chen, Y.; Li, W.; Cao, Z.; Zhao, B.; Lu, C.; Liu, Y. Drug-Metabolizing Cytochrome P450 Enzymes Have Multifarious Influences on Treatment Outcomes. Clin. Pharm. 2021, 60, 585–601. [Google Scholar] [CrossRef]
- Solus, J.F.; Arietta, B.J.; Harris, J.R.; Sexton, D.P.; Steward, J.Q.; McMunn, C.; Ihrie, P.; Mehall, J.M.; Edwards, T.L.; Dawson, E.P. Genetic variation in eleven phase I drug metabolism genes in an ethnically diverse population. Pharmacogenomics 2004, 5, 895–931. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharm. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Ahmed, S.; Zhou, Z.; Zhou, J.; Chen, S.Q. Corrigendum to “Pharmacogenomics of Drug Metabolizing Enzymes and Transporters: Relevance to Precision Medicine” [Genomics Proteomics Bioinformatics 14 (5) (2016) 298-313]. Genom. Proteom. Bioinform. 2018, 16, 152–153. [Google Scholar] [CrossRef]
- Achour, B.; Barber, J.; Rostami-Hodjegan, A. Expression of hepatic drug-metabolizing cytochrome p450 enzymes and their intercorrelations: A meta-analysis. Drug Metab. Dispos. 2014, 42, 1349–1356. [Google Scholar] [CrossRef] [Green Version]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Inui, Y.; Guengerich, F.P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharm. Exp. Ther. 1994, 270, 414–423. [Google Scholar]
- Klein, K.; Winter, S.; Turpeinen, M.; Schwab, M.; Zanger, U.M. Pathway-Targeted Pharmacogenomics of CYP1A2 in Human Liver. Front. Pharm. 2010, 1, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghodke-Puranik, Y.A.; Lamba, J.K. Chapter 7—Pharmacogenomics. In Innovative Approaches in Drug Discovery; Patwardhan, B., Chaguturu, R., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 195–234. [Google Scholar]
- Zhou, S.F.; Chan, E.; Zhou, Z.W.; Xue, C.C.; Lai, X.; Duan, W. Insights into the structure, function, and regulation of human cytochrome P450 1A2. Curr. Drug Metab. 2009, 10, 713–729. [Google Scholar] [CrossRef]
- Zhou, S.F.; Yang, L.P.; Zhou, Z.W.; Liu, Y.H.; Chan, E. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009, 11, 481–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunes, A.; Dahl, M.L. Variation in CYP1A2 activity and its clinical implications: Influence of environmental factors and genetic polymorphisms. Pharmacogenomics 2008, 9, 625–637. [Google Scholar] [CrossRef]
- Yim, E.Y.; Kang, H.R.; Jung, J.W.; Sohn, S.W.; Cho, S.H. CYP1A2 polymorphism and theophylline clearance in Korean non-smoking asthmatics. Asia Pac. Allergy 2013, 3, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Laika, B.; Leucht, S.; Heres, S.; Schneider, H.; Steimer, W. Pharmacogenetics and olanzapine treatment: CYP1A2*1F and serotonergic polymorphisms influence therapeutic outcome. Pharm. J. 2010, 10, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Myrand, S.P.; Sekiguchi, K.; Man, M.Z.; Lin, X.; Tzeng, R.Y.; Teng, C.H.; Hee, B.; Garrett, M.; Kikkawa, H.; Lin, C.Y.; et al. Pharmacokinetics/genotype associations for major cytochrome P450 enzymes in native and first- and third-generation Japanese populations: Comparison with Korean, Chinese, and Caucasian populations. Clin. Pharm. Ther. 2008, 84, 347–361. [Google Scholar] [CrossRef]
- Soyama, A.; Saito, Y.; Hanioka, N.; Maekawa, K.; Komamura, K.; Kamakura, S.; Kitakaze, M.; Tomoike, H.; Ueno, K.; Goto, Y.; et al. Single nucleotide polymorphisms and haplotypes of CYP1A2 in a Japanese population. Drug Metab. Pharm. 2005, 20, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Ota, T.; Kamada, Y.; Hayashida, M.; Iwao-Koizumi, K.; Murata, S.; Kinoshita, K. Combination analysis in genetic polymorphisms of drug-metabolizing enzymes CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A5 in the Japanese population. Int. J. Med. Sci. 2015, 12, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Palma, B.B.; Silva, E.S.M.; Urban, P.; Rueff, J.; Kranendonk, M. Functional characterization of eight human CYP1A2 variants: The role of cytochrome b5. Pharm. Genom. 2013, 23, 41–52. [Google Scholar] [CrossRef]
- Ito, M.; Katono, Y.; Oda, A.; Hirasawa, N.; Hiratsuka, M. Functional characterization of 20 allelic variants of CYP1A2. Drug Metab. Pharm. 2015, 30, 247–252. [Google Scholar] [CrossRef]
- Saito, Y.; Hanioka, N.; Maekawa, K.; Isobe, T.; Tsuneto, Y.; Nakamura, R.; Soyama, A.; Ozawa, S.; Tanaka-Kagawa, T.; Jinno, H.; et al. Functional analysis of three CYP1A2 variants found in a Japanese population. Drug Metab. Dispos. 2005, 33, 1905–1910. [Google Scholar] [CrossRef] [Green Version]
- Venkatakrishnan, K.; von Moltke, L.L.; Greenblatt, D.J. Human cytochromes P450 mediating phenacetin O-deethylation in vitro: Validation of the high affinity component as an index of CYP1A2 activity. J. Pharm. Sci. 1998, 87, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Tassaneeyakul, W.; Birkett, D.J.; Veronese, M.E.; McManus, M.E.; Tukey, R.H.; Quattrochi, L.C.; Gelboin, H.V.; Miners, J.O. Specificity of substrate and inhibitor probes for human cytochromes P450 1A1 and 1A2. J. Pharm. Exp. Ther. 1993, 265, 401–407. [Google Scholar]
- Tucker, G.T.; Houston, J.B.; Huang, S.M. Optimizing drug development: Strategies to assess drug metabolism/transporter interaction potential--towards a consensus. Br. J. Clin. Pharm. 2001, 52, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasaki, M.; Yasuda, J.; Katsuoka, F.; Nariai, N.; Kojima, K.; Kawai, Y.; Yamaguchi-Kabata, Y.; Yokozawa, J.; Danjoh, I.; Saito, S.; et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun. 2015, 6, 8018. [Google Scholar] [CrossRef]
- Kumondai, M.; Hishinuma, E.; Gutierrez Rico, E.M.; Ito, A.; Nakanishi, Y.; Saigusa, D.; Hirasawa, N.; Hiratsuka, M. Heterologous expression of high-activity cytochrome P450 in mammalian cells. Sci. Rep. 2020, 10, 14193. [Google Scholar] [CrossRef]
- Simple Western Size-Based Total Protein Assays. Available online: https://www.proteinsimple.com/simple_western_size-based_total_protein_assays.html (accessed on 11 July 2021).
- Guengerich, F.P.; Martin, M.V.; Sohl, C.D.; Cheng, Q. Measurement of cytochrome P450 and NADPH-cytochrome P450 reductase. Nat. Protoc. 2009, 4, 1245–1251. [Google Scholar] [CrossRef] [Green Version]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef] [Green Version]
- Kumondai, M.; Ito, A.; Gutierrez Rico, E.M.; Hishinuma, E.; Ueda, A.; Saito, S.; Nakayoshi, T.; Oda, A.; Tadaka, S.; Kinoshita, K.; et al. Functional Assessment of 12 Rare Allelic CYP2C9 Variants Identified in a Population of 4773 Japanese Individuals. J. Pers. Med. 2021, 11, 94. [Google Scholar] [CrossRef]
- Oda, A.; Yamaotsu, N.; Hirono, S. New AMBER force field parameters of heme iron for cytochrome P450s determined by quantum chemical calculations of simplified models. J. Comput. Chem. 2005, 26, 818–826. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turesky, R.J.; Le Marchand, L. Metabolism and biomarkers of heterocyclic aromatic amines in molecular epidemiology studies: Lessons learned from aromatic amines. Chem. Res. Toxicol. 2011, 24, 1169–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonrungsesomboon, N.; Khatsri, R.; Wongchompoo, P.; Teekachunhatean, S. The impact of genetic polymorphisms on CYP1A2 activity in humans: A systematic review and meta-analysis. Pharm. J. 2018, 18, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef] [Green Version]
- Eaton, D.L.; Gallagher, E.P.; Bammler, T.K.; Kunze, K.L. Role of cytochrome P4501A2 in chemical carcinogenesis: Implications for human variability in expression and enzyme activity. Pharmacogenetics 1995, 5, 259–274. [Google Scholar] [CrossRef]
- Xie, C.; Pogribna, M.; Word, B.; Lyn-Cook, L., Jr.; Lyn-Cook, B.D.; Hammons, G.J. In vitro analysis of factors influencing CYP1A2 expression as potential determinants of interindividual variation. Pharm. Res. Perspect. 2017, 5, e00299. [Google Scholar] [CrossRef] [Green Version]
- Lobo, E.D.; Bergstrom, R.F.; Reddy, S.; Quinlan, T.; Chappell, J.; Hong, Q.; Ring, B.; Knadler, M.P. In vitro and in vivo evaluations of cytochrome P450 1A2 interactions with duloxetine. Clin. Pharm. 2008, 47, 191–202. [Google Scholar] [CrossRef]
- Xiong, S.; Li, L. The effect of CYP1A2 gene polymorphism on the metabolism of theophylline. Exp. Ther. Med. 2018, 15, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Melkersson, K.I.; Scordo, M.G.; Gunes, A.; Dahl, M.L. Impact of CYP1A2 and CYP2D6 polymorphisms on drug metabolism and on insulin and lipid elevations and insulin resistance in clozapine-treated patients. J. Clin. Psychiatry 2007, 68, 697–704. [Google Scholar] [CrossRef]
- Saiz-Rodriguez, M.; Ochoa, D.; Belmonte, C.; Roman, M.; Vieira de Lara, D.; Zubiaur, P.; Koller, D.; Mejia, G.; Abad-Santos, F. Polymorphisms in CYP1A2, CYP2C9 and ABCB1 affect agomelatine pharmacokinetics. J. Psychopharmacol. 2019, 33, 522–531. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.X.; Sun, Y.Q. Glycine residues provide flexibility for enzyme active sites. J. Biol. Chem. 1997, 272, 3190–3194. [Google Scholar] [CrossRef] [Green Version]
- Shalit, Y.; Tuvi-Arad, I. Side chain flexibility and the symmetry of protein homodimers. PLoS ONE 2020, 15, e0235863. [Google Scholar] [CrossRef]
- Zhou, S.F.; Wang, B.; Yang, L.P.; Liu, J.P. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab. Rev. 2010, 42, 268–354. [Google Scholar] [CrossRef]
- Campelo, D.; Esteves, F.; Brito Palma, B.; Costa Gomes, B.; Rueff, J.; Lautier, T.; Urban, P.; Truan, G.; Kranendonk, M. Probing the Role of the Hinge Segment of Cytochrome P450 Oxidoreductase in the Interaction with Cytochrome P450. Int. J. Mol. Sci. 2018, 19, 3914. [Google Scholar] [CrossRef] [Green Version]
- Pochapsky, T.C.; Kazanis, S.; Dang, M. Conformational plasticity and structure/function relationships in cytochromes P450. Antioxid. Redox Signal. 2010, 13, 1273–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, S.C.; Roberts, A.G.; Halpert, J.R. Structural features of cytochromes P450 and ligands that affect drug metabolism as revealed by X-ray crystallography and NMR. Future Med. Chem. 2010, 2, 1451–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, B.; Zhong, Y.; Wang, J. Impact of peripheral mutations on the access channels of human cytochrome P450 1A2. J. Biomol. Struct. Dyn. 2020, 38, 4906–4913. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.H.; Jamakhandi, A.P.; Sullivan, S.Z.; Yun, C.H.; Hollenberg, P.F.; Miller, G.P. Beta sheet 2-alpha helix C loop of cytochrome P450 reductase serves as a docking site for redox partners. Biochim. Biophys. Acta 2010, 1804, 1285–1293. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.F.; Connick, J.P.; Reed, J.R.; Backes, W.L.; Desai, M.C.; Xu, L.; Estrada, D.F.; Laurence, J.S.; Scott, E.E. Correlating structure and function of drug-metabolizing enzymes: Progress and ongoing challenges. Drug Metab. Dispos. 2014, 42, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Kang, S.; Dong, M.S.; Park, J.D.; Park, J.; Rhee, S.; Ryu, D.Y. Characterization of the Ala62Pro polymorphic variant of human cytochrome P450 1A1 using recombinant protein expression. Toxicol. Appl. Pharm. 2015, 285, 159–169. [Google Scholar] [CrossRef]
- Lee, S.H.; Yu, H.J.; Lee, S.; Ryu, D.Y. Characterization of the Gly45Asp variant of human cytochrome P450 1A1 using recombinant expression. Toxicol. Lett. 2015, 239, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Kato, K.; Fukuyoshi, S.; Hiratsuka, M.; Yamaotsu, N.; Hirono, S.; Gouda, H.; Oda, A. Effect of the Arg456His mutation on the three-dimensional structure of cytochrome P450 1A2 predicted by molecular dynamics simulations. J. Phys. Conf. Ser. 2018, 1136, 012023. [Google Scholar] [CrossRef]
- Kemper, B. Structural basis for the role in protein folding of conserved proline-rich regions in cytochromes P450. Toxicol. Appl. Pharm. 2004, 199, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, S.B.; Guengerich, F.P.; Park, Y.I.; Dong, M.S. Effects of N-terminal modification of recombinant human cytochrome P450 1A2 on catalytic activity. Xenobiotica 2007, 37, 356–365. [Google Scholar] [CrossRef]
- Khan, M.K.A.; Akhtar, S.; Arif, J.M. Development of In Silico Protocols to Predict Structural Insights into the Metabolic Activation Pathways of Xenobiotics. Interdiscip. Sci. 2018, 10, 329–345. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Mutations | rs Number | Amino Acid Substitutions | Frequency a (%) | PolyPhen-2 | SIFT |
|---|---|---|---|---|---|
| 100C>T | rs201934979 | Arg34Trp | 0.01 | Benign | Tolerated |
| 130G>A | rs3743482 | Glu44Lys | 0.10 | Benign | Damaging |
| 140G>A | rs777457540 | Gly47Asp | 0.02 | Benign | Damaging |
| 236G>A | rs752037611 | Arg79His | 0.01 | Benign | Tolerated |
| 293T>A | rs773366123 | Leu98Gln | 0.04 | Probably damaging | Damaging |
| 310G>T | Asp104Tyr | 0.01 | Probably damaging | Damaging | |
| 449C>A | rs1477440128 | Ala150Asp | 0.04 | Probably damaging | Tolerated |
| 697G>A | rs201537008 | Gly233Arg | 0.02 | Probably damaging | Damaging |
| 971C>T | rs778097570 | Thr324Ile | 0.03 | Probably damaging | Damaging |
| 1032G>C | Gln344His | 0.03 | Benign | Tolerated | |
| 1052T>C | Ile351Thr | 0.01 | Benign | Damaging | |
| 1053T>G | Ile351Met | 0.01 | Possibly damaging | Damaging | |
| 1067G>A | rs55918015 | Arg356Gln | 0.02 | Benign | Tolerated |
| 1139_1141delCCT | rs757356377 | Ser380del | 0.01 | N.D. | N.D. |
| 1202T>C | Ile401Thr | 0.01 | Probably damaging | Damaging | |
| 1361G>A | Gly454Asp | 0.01 | Probably damaging | Damaging | |
| 1369C>T | rs34151816 | Arg457Trp | 0.01 | Probably damaging | Damaging |
| 1384G>C | rs199528490 | Val462Leu | 0.04 | Benign | Tolerated |
| 1421T>A | rs1300577537 | Ile474Asn | 0.03 | Probably damaging | Damaging |
| 1471C>A | Leu491Met | 0.02 | Benign | Tolerated | |
| 1501C>T | His501Tyr | 0.02 | Benign | Tolerated |
| Variants | CYP Holoenzyme Content (pmol/mg Protein) | CPR Content (pmol/mg Protein) | Cytochrome b5 Content (pmol/mg Protein) | CYP:CPR Ratio | CYP:Cytochrome b5 Ratio |
|---|---|---|---|---|---|
| Wild-type | 145.91 ± 4.11 | 80.52 ± 16.79 | 21.11 ± 2.44 | 1.81 | 6.91 |
| Arg34Trp | 34.50 ± 11.84 * | 135.50 ± 45.33 | 28.34 ± 2.11 # | 0.25 | 1.22 |
| Glu44Lys | 41.69 ± 4.62 *** | 89.69 ± 20.20 | 27.14 ± 3.24 | 0.46 | 1.54 |
| Gly47Asp | 23.21 ± 6.09 *** | 98.12 ± 31.17 | 27.54 ± 2.01 | 0.24 | 0.84 |
| Arg79His | 21.31 ± 3.02 *** | 158.64 ± 24.52 | 27.09 ± 3.18 | 0.13 | 0.79 |
| Leu98Gln | N.D. | 146.98 ± 9.95 | 25.43 ± 1.49 | N.D. | N.D. |
| Asp104Tyr | 15.21 ± 1.52 *** | 150.87 ± 57.19 | 26.46 ± 1.69 | 0.10 | 0.57 |
| Ala150Asp | 20.75 ± 1.41 *** | 127.57 ± 47.56 | 19.28 ± 1.74 | 0.16 | 1.08 |
| Gly233Arg | N.D. | 109.67 ± 52.40 | 24.82 ± 2.06 | N.D. | N.D. |
| Thr324Ile | 21.89 ± 6.31 *** | 103.76 ± 15.09 | 17.46 ± 3.16 | 0.21 | 1.25 |
| Gln344His | 18.83 ± 2.93 *** | 156.04 ± 4.92 | 30.98 ± 2.52 ### | 0.12 | 0.61 |
| Ile351Thr | 36.09 ± 2.85 *** | 133.09 ± 12.25 | 27.72 ± 2.79 # | 0.27 | 1.30 |
| Ile351Met | 43.77 ± 1.61 *** | 129.07 ± 6.34 | 17.71 ± 1.17 | 0.34 | 2.47 |
| Arg356Gln | 88.92 ± 0.88 * | 129.54 ± 1.51 | 25.77 ± 3.52 | 0.69 | 3.45 |
| Ser380del | N.D. | 140.78 ± 25.66 | 19.72 ± 2.48 | N.D. | N.D. |
| Ile401Thr | N.D. | 95.05 ± 33.43 | 18.27 ± 3.62 | N.D. | N.D. |
| Gly454Asp | 12.88 ± 4.98 *** | 120.75 ± 18.54 | 16.95 ± 1.45 | 0.11 | 0.76 |
| Arg457Trp | 14.34 ± 2.24 *** | 126.46 ± 16.37 | 17.00 ± 2.12 | 0.11 | 0.84 |
| Val462Leu | 14.59 ± 5.64 *** | 113.72 ± 62.19 | 23.71 ± 3.57 | 0.13 | 0.62 |
| Ile474Asn | 33.80 ± 4.70 *** | 136.52 ± 28.20 | 26.53 ± 3.32 | 0.25 | 1.27 |
| Leu491Met | 144.17 ± 6.56 | 109.22 ± 35.05 | 23.54 ± 1.60 | 1.32 | 6.13 |
| His501Tyr | 43.22 ± 2.31 *** | 141.73 ± 9.07 | 31.52 ± 4.02 ### | 0.30 | 1.37 |
| Variants | Km (μM) | Vmax (pmol/min/mg Protein) | CLint (µL/min/mg Protein) (% of Wild-Type) |
|---|---|---|---|
| Wild-type | 10.14 ± 0.68 | 972.99 ± 39.96 | 96.10 ± 2.47 (100.00) |
| Arg34Trp | 12.97 ± 0.65 | 1224.92 ± 48.86 | 94.51 ± 1.30 (98.35) |
| Glu44Lys | 8.86 ± 0.27 | 462.54 ± 12.05 * | 52.23 ± 2.04 *** (54.35) |
| Gly47Asp | 7.42 ± 0.32 | 142.46 ± 3.76 ** | 19.22 ± 0.50 *** (20.00) |
| Arg79His | 11.06 ± 0.74 | 599.00 ± 17.84 * | 54.23 ± 1.99 *** (56.43) |
| Leu98Gln | N.D. | N.D. | N.D. |
| Asp104Tyr | 7.83 ± 0.43 | 234.53 ± 5.62 ** | 30.04 ± 2.06 *** (31.26) |
| Ala150Asp | 7.71 ± 0.27 | 178.11 ± 3.53 ** | 23.12 ± 1.13 *** (24.06) |
| Gly233Arg | N.D. | N.D. | N.D. |
| Thr324Ile | 110.07 ± 2.08 | 153.36 ± 0.59 ** | 1.39 ± 0.02 *** (1.45) |
| Gln344His | 13.18 ± 0.30 | 1019.25 ± 28.36 | 77.36 ± 2.97 * (80.50) |
| Ile351Thr | 12.92 ± 1.10 | 473.86 ± 16.59 ** | 36.93 ± 4.50 *** (38.44) |
| Ile351Met | 7.37 ± 0.58 | 285.80 ± 16.34 *** | 38.85 ± 0.84 *** (40.43) |
| Arg356Gln | 8.03 ± 0.21 | 423.55 ± 25.17 *** | 52.74 ± 1.75 *** (54.88) |
| Ser380del | N.D. | N.D. | N.D. |
| Ile401Thr | 2.31 ± 0.26 | 57.05 ± 1.90 ** | 24.88 ± 2.09 *** (25.89) |
| Gly454Asp | N.D. | N.D. | N.D. |
| Arg457Trp | N.D. | N.D. | N.D. |
| Val462Leu | 9.97 ± 1.39 | 334.57 ± 8.54 ** | 34.06 ± 5.64 ** (35.44) |
| Ile474Asn | 12.41 ± 1.45 | 511.48 ± 10.98 * | 41.63 ± 5.48 * (43.31) |
| Leu491Met | 6.97 ± 0.41 | 574.36 ± 9.20 * | 82.58 ± 4.18 (85.93) |
| His501Tyr | 16.83 ± 1.29 | 404.49 ± 14.69 ** | 24.08 ± 1.13 *** (25.06) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumondai, M.; Gutiérrez Rico, E.M.; Hishinuma, E.; Nakanishi, Y.; Yamazaki, S.; Ueda, A.; Saito, S.; Tadaka, S.; Kinoshita, K.; Saigusa, D.; et al. Functional Characterization of 21 Rare Allelic CYP1A2 Variants Identified in a Population of 4773 Japanese Individuals by Assessing Phenacetin O-Deethylation. J. Pers. Med. 2021, 11, 690. https://doi.org/10.3390/jpm11080690
Kumondai M, Gutiérrez Rico EM, Hishinuma E, Nakanishi Y, Yamazaki S, Ueda A, Saito S, Tadaka S, Kinoshita K, Saigusa D, et al. Functional Characterization of 21 Rare Allelic CYP1A2 Variants Identified in a Population of 4773 Japanese Individuals by Assessing Phenacetin O-Deethylation. Journal of Personalized Medicine. 2021; 11(8):690. https://doi.org/10.3390/jpm11080690
Chicago/Turabian StyleKumondai, Masaki, Evelyn Marie Gutiérrez Rico, Eiji Hishinuma, Yuya Nakanishi, Shuki Yamazaki, Akiko Ueda, Sakae Saito, Shu Tadaka, Kengo Kinoshita, Daisuke Saigusa, and et al. 2021. "Functional Characterization of 21 Rare Allelic CYP1A2 Variants Identified in a Population of 4773 Japanese Individuals by Assessing Phenacetin O-Deethylation" Journal of Personalized Medicine 11, no. 8: 690. https://doi.org/10.3390/jpm11080690