Helpful Criteria When Implementing NGS Panels in Childhood Lymphoblastic Leukemia

 , , , , ,
, , , , ,  , , , , , ,
, , , , , ,
Abstract
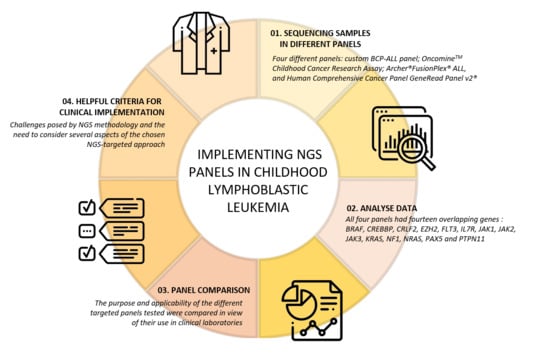
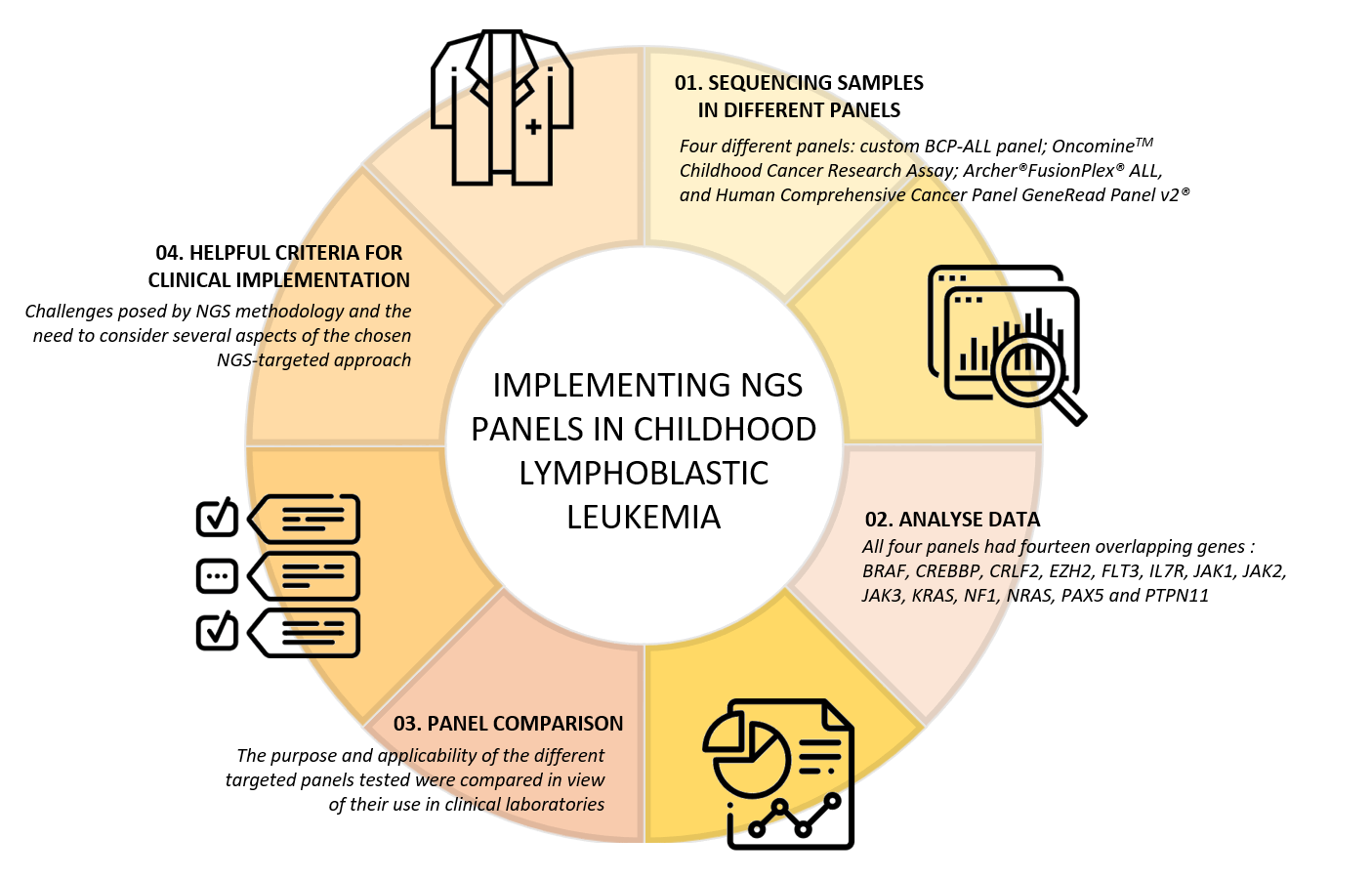
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethical Issues
2.2. Design of the Study
2.3. NGS-Targeted Panels Included in the Study
2.3.1. BCP-ALL Custom Panel
2.3.2. Archer® FusionPlex® ALL Kit
2.3.3. OncomineTM Childhood Cancer Research Assay (OCCRA)
2.3.4. Human Comprehensive Cancer GeneRead Panel v2®
2.4. Sequencing and Variant Data Analysis
3. Results
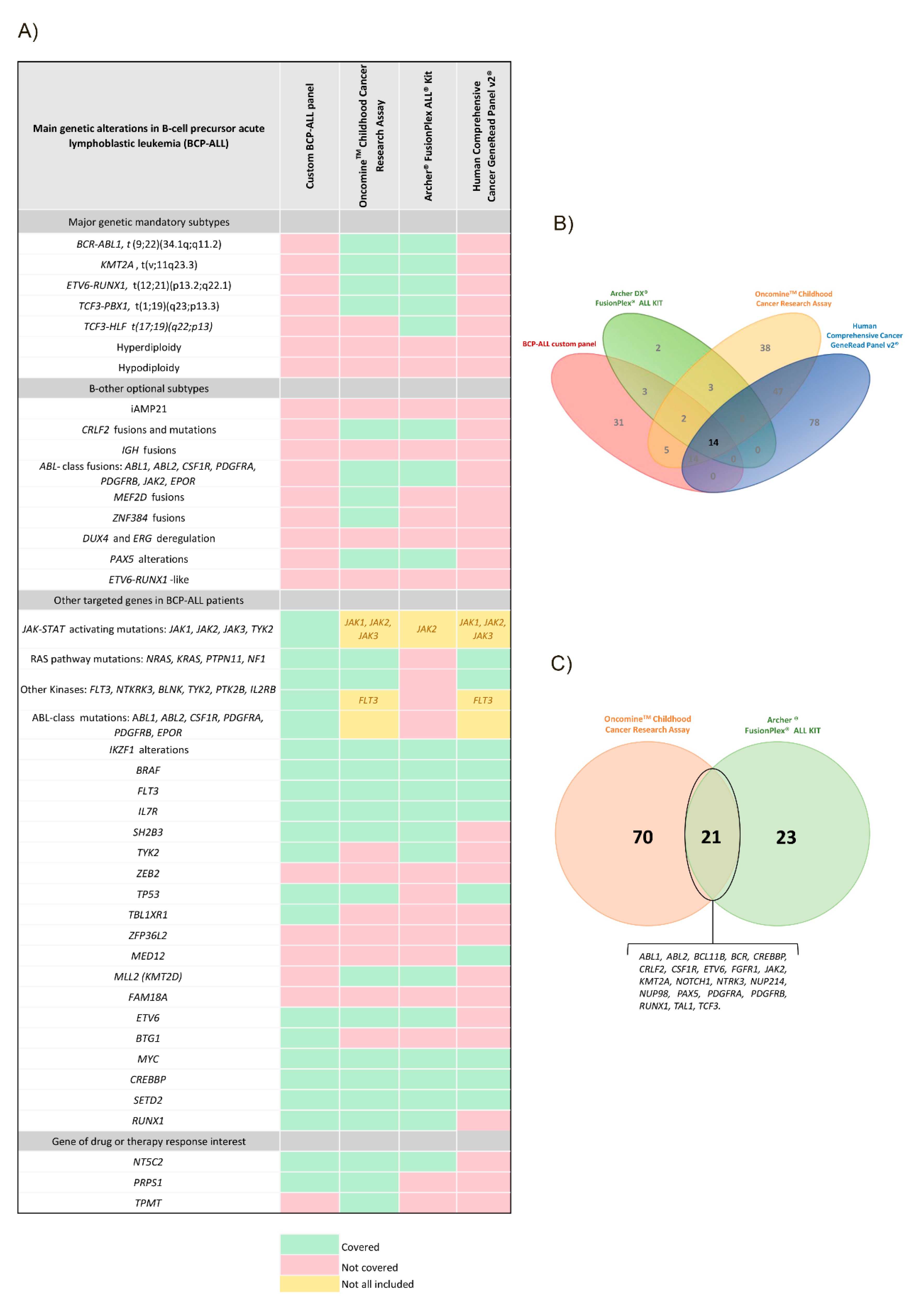
3.1. Dissection of the NGS Panels
3.1.1. BCP-ALL Custom Panel
3.1.2. Archer® FusionPlex® ALL Kit
3.1.3. OncomineTM Childhood Cancer Research Assay (OCCRA)
3.1.4. Human Comprehensive Cancer GeneRead Panel v2®
3.2. Panel Dissection
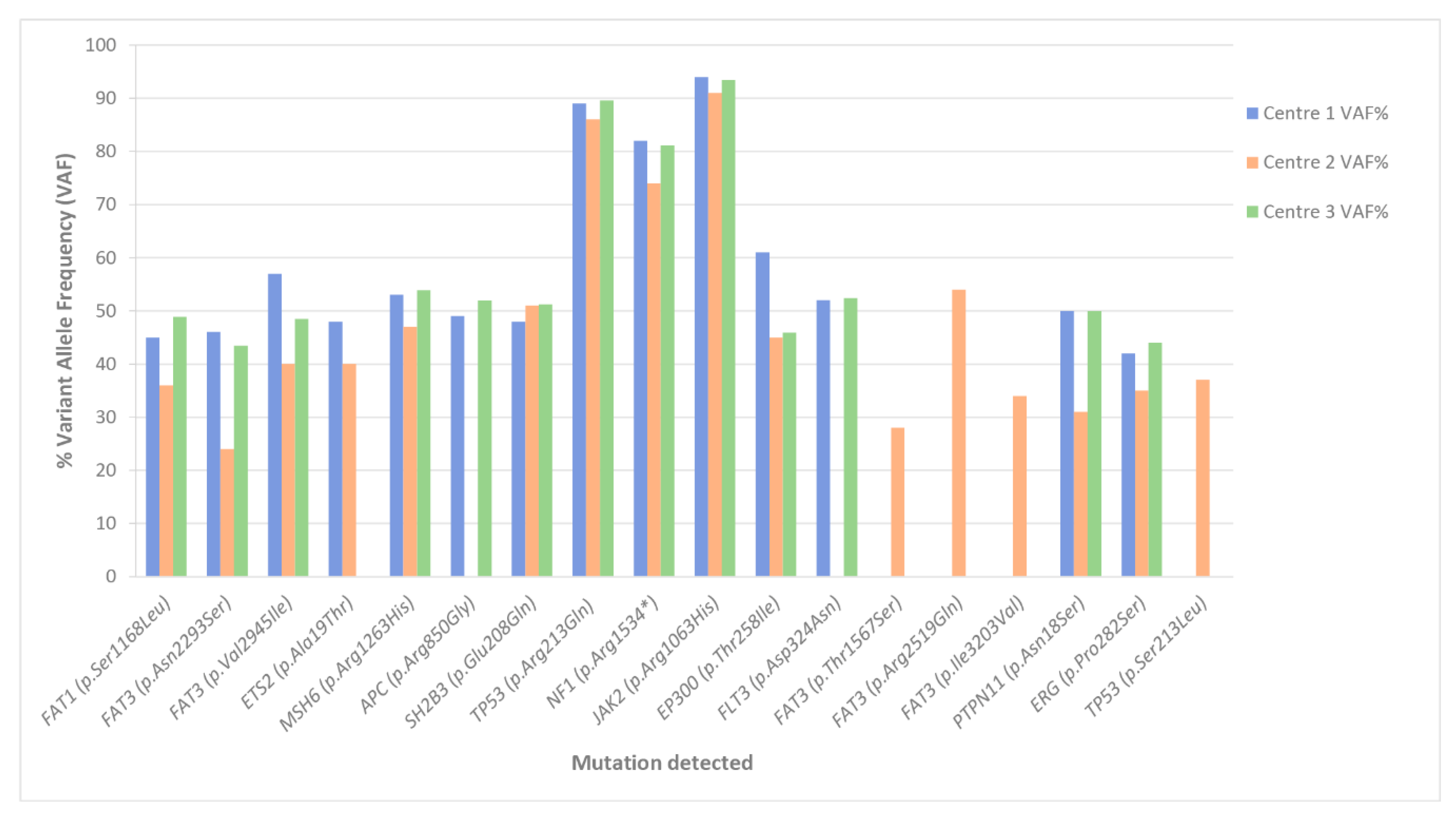
3.2.1. SNV Detection
3.2.2. CNV Detection
3.2.3. RNA Fusion Detection
4. Discussion
4.1. Panel Discussion
4.2. Implementing NGS in Pediatric BCP-ALL
4.2.1. The Panel Choice
4.2.2. Technical Challenges of Each Analyzed Panel for BCP-ALL Diagnosis
4.2.3. Data Analysis for Variant Detection
4.2.4. Variant Prioritization and Reporting
5. Conclusions: Lessons Learned and the Future Ahead
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pui, C.-H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat. Rev. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Gaynon, P.S.; Boyett, J.M.; Chessells, J.M.; Baruchel, A.; Kamps, W.; Silverman, L.B.; Biondi, A.; Harms, D.O.; Vilmer, E.; et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 2002, 359, 1909–1915. [Google Scholar] [CrossRef]
- Gatta, G.; Rossi, S.; Foschi, R.; Trama, A.; Marcos-Gragera, R.; Pastore, G.; Peris-Bonet, R.; Stiller, C.; Capocaccia, R. Survival and cure trends for European children, adolescents and young adults diagnosed with acute lymphoblastic leukemia from 1982 to 2002. Haematologica 2013, 98, 744–752. [Google Scholar] [CrossRef]
- Haslam, K.; Catherwood, M.A.; Dobbin, E.; Sproul, A.; Langabeer, S.E.; Mills, K.I. Inter-Laboratory Evaluation of a Next-Generation Sequencing Panel for Acute Myeloid Leukemia. Mol. Diagn. Ther. 2016, 20, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burghel, G.J.; Hurst, C.D.; Watson, C.M.; Chambers, P.A.; Dickinson, H.; Roberts, P.; Knowles, M.A. Towards a Next-Generation Sequencing Diagnostic Service for Tumour Genotyping: A Comparison of Panels and Platforms. BioMed Res. Int. 2015, 2015, 478017. [Google Scholar] [CrossRef] [Green Version]
- Pathology, M.; Area, H.; Service, P.; Dna, N.; Meldrum, C.; Doyle, M.A.; Tothill, R.W. Next-Generation Sequencing for Cancer Diagnostics: A Practical Perspective. Clin. Biochem. Rev. 2011, 32, 177–195. [Google Scholar]
- Bastida, J.M.; Lozano, M.L.; Benito, R.; Janusz, K.; Palma-Barqueros, V.; Del Rey, M.; Hernández-Sánchez, J.M.; Riesco, S.; Bermejo, N.; González-García, H.; et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica 2018, 103, 148–162. [Google Scholar] [CrossRef]
- Knoppers, B.M.; Nguyen, M.T.; Sénécal, K.; Tassé, A.M.; Zawati, M.H. Next-generation sequencing and the return of results. Cold Spring Harb. Perspect. Med. 2016, 6, a026724. [Google Scholar] [CrossRef] [Green Version]
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Harrison, C.J. Advances in B-cell Precursor Acute Lymphoblastic Leukemia Genomics. HemaSphere 2018, 2, e53. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Fioretos, T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Bacher, U.; Shumilov, E.; Flach, J.; Porret, N.; Joncourt, R.; Wiedemann, G.; Fiedler, M.; Novak, U.; Amstutz, U.; Pabst, T. Challenges in the introduction of next-generation sequencing (NGS) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer J. 2018, 8, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sujobert, P.; Le Bris, Y.; de Leval, L.; Gros, A.; Merlio, J.P.; Pastoret, C.; Huet, S.; Sarkozy, C.; Davi, F.; Callanan, M.; et al. The Need for a Consensus Next-generation Sequencing Panel for Mature Lymphoid Malignancies. HemaSphere 2019, 3, e169. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Humburg, P.; Kanapin, A.; Rivas, M.A.; Gaulton, K.; Cazier, J.-B.; Donnelly, P. Choice of transcripts and software has a large effect on variant annotation. Genome Med. 2014, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- De Leng, W.W.J.; Gadellaa-Van Hooijdonk, C.G.; Barendregt-Smouter, F.A.S.; Koudijs, M.J.; Nijman, I.; Hinrichs, J.W.J.; Cuppen, E.; Van Lieshout, S.; Loberg, R.D.; De Jonge, M.; et al. Targeted next generation sequencing as a reliable diagnostic assay for the detection of somatic mutations in tumours using minimal DNA amounts from formalin fixed paraffin embedded material. PLoS ONE 2016, 11, e0149405. [Google Scholar] [CrossRef]
- García-García, G.; Baux, D.; Faugère, V.; Moclyn, M.; Koenig, M.; Claustres, M.; Roux, A.F. Assessment of the latest NGS enrichment capture methods in clinical context. Sci. Rep. 2016, 6, 20948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, C.M.; Llop, M.; Sargas, C.; Pedrola, L.; Panadero, J.; Hervás, D.; Cervera, J.; Such, E.; Ibáñez, M.; Ayala, R.; et al. Clinical Utility of a Next-Generation Sequencing Panel for Acute Myeloid Leukemia Diagnostics. J. Mol. Diagn. 2019, 21, 228–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Press, R.D.; Eickelberg, G.; Froman, A.; Yang, F.; Stentz, A.; Flatley, E.M.; Fan, G.; Lim, J.Y.; Meyers, G.; Maziarz, R.T.; et al. NGS-Defined Minimal Residual Disease Before Stem Cell Transplantation Predicts Acute Myeloid Leukemia Relapse. Am. J. Hematol. 2019, 94, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Kotrova, M.; Trka, J.; Kneba, M.; Brüggemann, M. Is Next-Generation Sequencing the way to go for Residual Disease Monitoring in Acute Lymphoblastic Leukemia? Mol. Diagn. Ther. 2017, 21, 481–492. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, M.-Y.; Liu, W.-J. Application of minimal residual disease monitoring in pediatric patients with acute lymphoblastic leukemia. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6885–6895. [Google Scholar] [CrossRef]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing–Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vega-Garcia, N.; Benito, R.; Esperanza-Cebollada, E.; Llop, M.; Robledo, C.; Vicente-Garcés, C.; Alonso, J.; Barragán, E.; Fernández, G.; Hernández-Sánchez, J.M.; et al. Helpful Criteria When Implementing NGS Panels in Childhood Lymphoblastic Leukemia. J. Pers. Med. 2020, 10, 244. https://doi.org/10.3390/jpm10040244
Vega-Garcia N, Benito R, Esperanza-Cebollada E, Llop M, Robledo C, Vicente-Garcés C, Alonso J, Barragán E, Fernández G, Hernández-Sánchez JM, et al. Helpful Criteria When Implementing NGS Panels in Childhood Lymphoblastic Leukemia. Journal of Personalized Medicine. 2020; 10(4):244. https://doi.org/10.3390/jpm10040244
Chicago/Turabian StyleVega-Garcia, Nerea, Rocío Benito, Elena Esperanza-Cebollada, Marta Llop, Cristina Robledo, Clara Vicente-Garcés, Javier Alonso, Eva Barragán, Guerau Fernández, Jesús M. Hernández-Sánchez, and et al. 2020. "Helpful Criteria When Implementing NGS Panels in Childhood Lymphoblastic Leukemia" Journal of Personalized Medicine 10, no. 4: 244. https://doi.org/10.3390/jpm10040244