A Novel Kindred with Familial Gastrointestinal Stromal Tumors Caused by a Rare KIT Germline Mutation (N655K): Clinico-Pathological Presentation and TKI Sensitivity

 , , , , , and
, , , , , and
Abstract
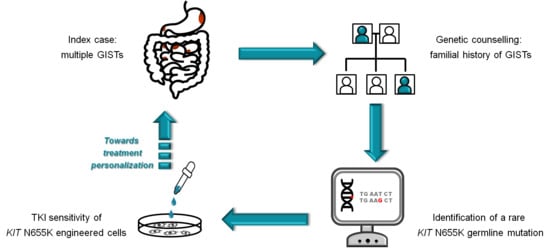
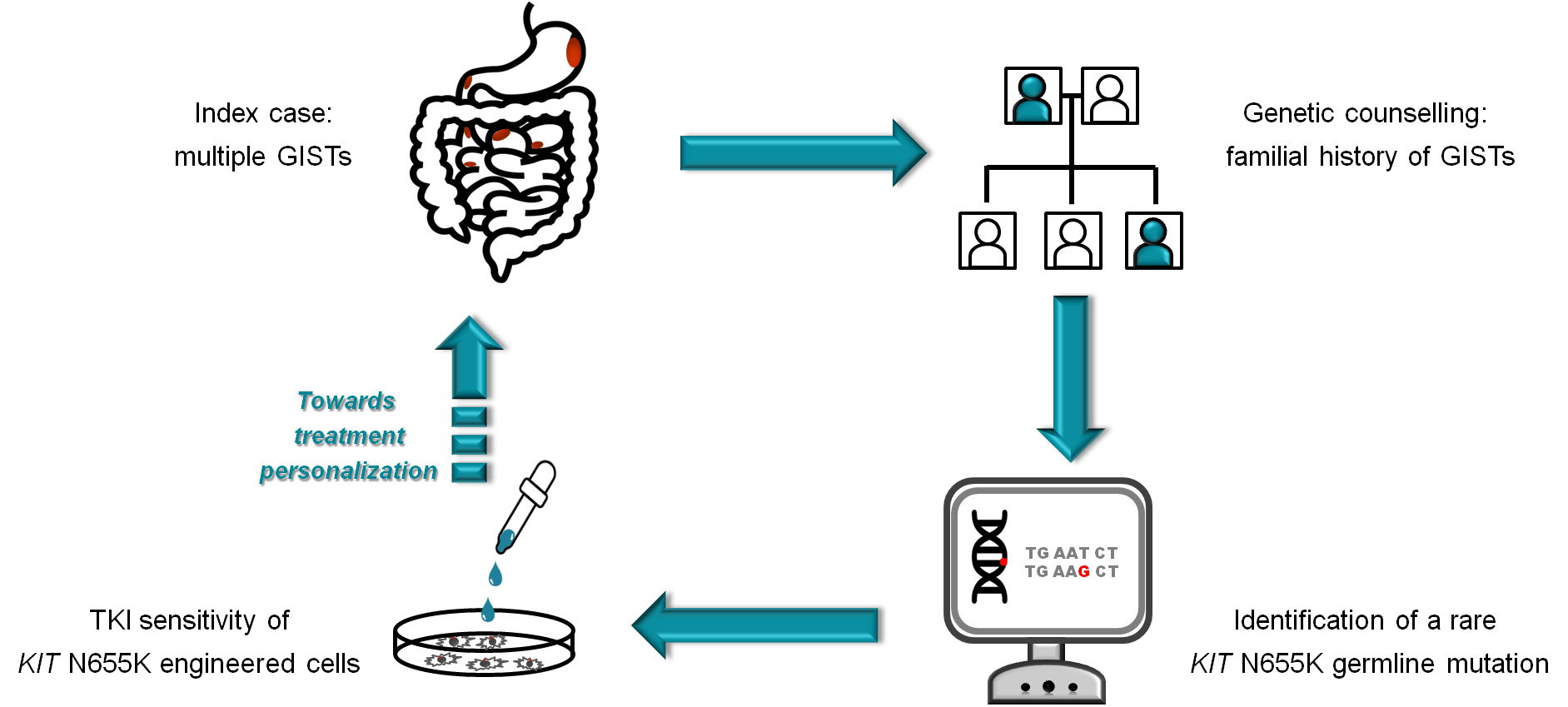
:
1. Introduction
2. Materials and Methods
2.1. Histology and Immunohistochemistry
2.2. Molecular Analysis
2.3. Engineering of Ba/F3 Cells for the Expression of KIT Mutants
2.4. TKI Cell Viability Assay
2.5. Ethical Approval
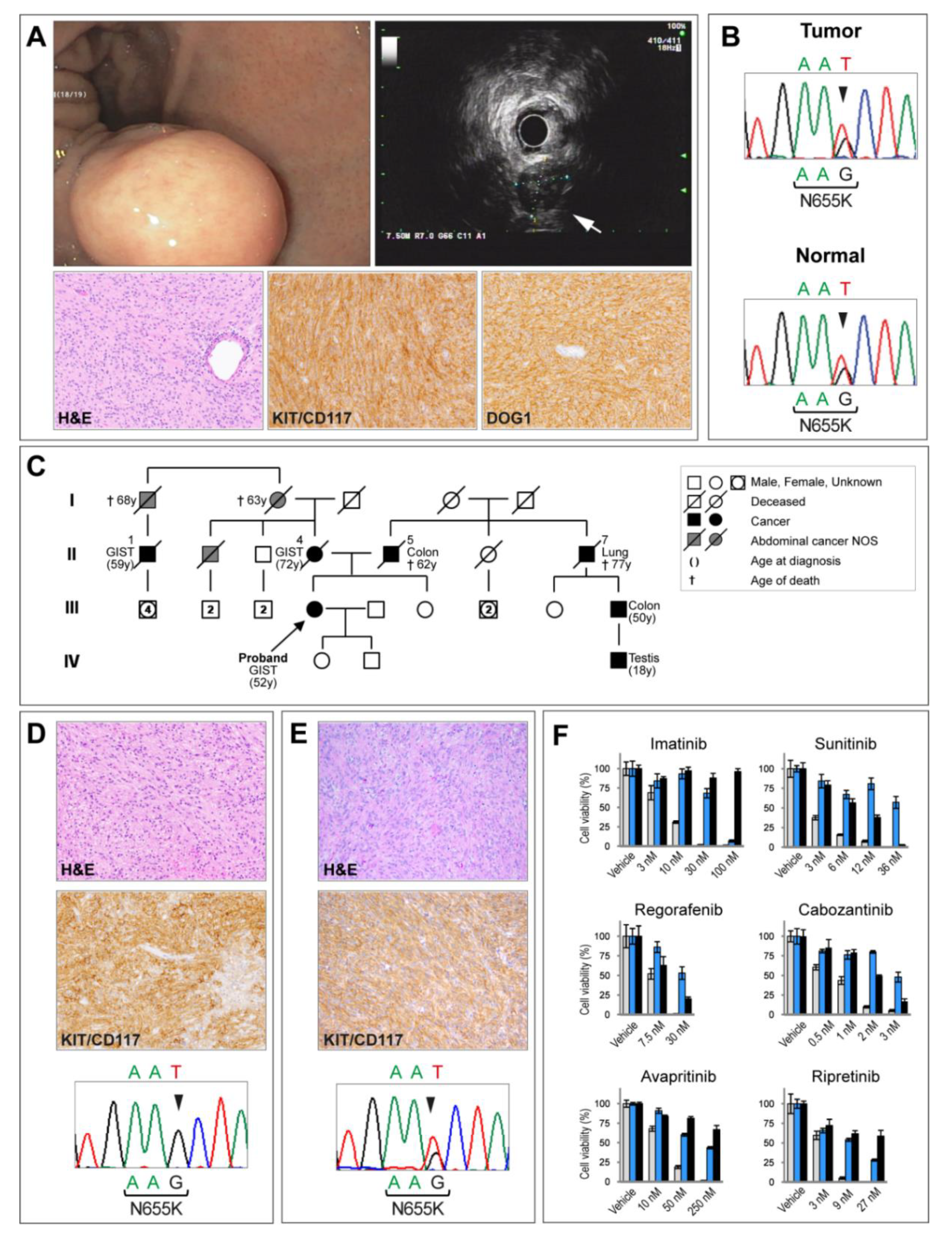
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Corless, C.L. Gastrointestinal stromal tumors: What do we know now? Mod. Pathol. 2014, 27, S1–S16. [Google Scholar] [CrossRef] [Green Version]
- Mei, L.; Smith, S.C.; Faber, A.C.; Trent, J.; Grossman, S.R.; Stratakis, C.A.; Boikos, S.A. Gastrointestinal Stromal Tumors: The GIST of Precision Medicine. Trends Cancer 2018, 4, 74–91. [Google Scholar] [CrossRef]
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Dei Tos, A.P.; Maestro, R. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J. Pathol. 2016, 238, 543–549. [Google Scholar] [CrossRef]
- Gasparotto, D.; Rossi, S.; Polano, M.; Tamborini, E.; Lorenzetto, E.; Sbaraglia, M.; Mondello, A.; Massani, M.; Lamon, S.; Bracci, R.; et al. Quadruple-Negative GIST Is a Sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin. Cancer Res. 2017, 23, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Ricci, R. Syndromic gastrointestinal stromal tumors. Hered. Cancer Clin. Pract. 2016, 14, 15. [Google Scholar] [CrossRef]
- Agarwal, R.; Robson, M. Inherited predisposition to gastrointestinal stromal tumor. Hematol. Oncol. Clin. N. Am. 2009, 23, 1–13. [Google Scholar] [CrossRef]
- Garner, A.P.; Gozgit, J.M.; Anjum, R.; Vodala, S.; Schrock, A.; Zhou, T.; Serrano, C.; Eilers, G.; Zhu, M.; Ketzer, J.; et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin. Cancer Res. 2014, 20, 5745–5755. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, K.; Hirota, S.; Isozaki, K.; Nishitani, A.; Tsutsui, S.; Watabe, K.; Tamura, S.; Ishikawa, T.; Kanda, T.; Nishida, T.; et al. Characterization of tyrosine kinase I domain c-kit gene mutation Asn655Lys newly found in primary jejunal gastrointestinal stromal tumor. Am. J. Gastroenterol. 2007, 102, 1134–1136. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors: Pathology and prognosis at different sites. Semin. Diagn. Pathol. 2006, 23, 70–83. [Google Scholar] [CrossRef]
- Farag, S.; Smith, M.J.; Fotiadis, N.; Constantinidou, A.; Jones, R.L. Revolutions in treatment options in gastrointestinal stromal tumours (GISTs): The latest updates. Curr. Treat. Options Oncol. 2020, 21, 55. [Google Scholar] [CrossRef]
- Hartmann, K.; Wardelmann, E.; Ma, Y.; Merkelbach-Bruse, S.; Preussner, L.M.; Woolery, C.; Baldus, S.E.; Heinicke, T.; Thiele, J.; Buettner, R.; et al. Novel germline mutation of KIT associated with familial gastrointestinal stromal tumors and mastocytosis. Gastroenterology 2005, 129, 1042–1046. [Google Scholar] [CrossRef]
- Speight, R.A.; Nicolle, A.; Needham, S.J.; Verrill, M.W.; Bryon, J.; Panter, S. Rare, germline mutation of KIT with imatinib-resistant multiple GI stromal tumors and mastocytosis. J. Clin. Oncol. 2013, 31, e245–e247. [Google Scholar] [CrossRef]
- Halpern, A.L.; Torphy, R.J.; McCarter, M.D.; Sciotto, C.G.; Glode, L.M.; Robinson, W.A. A familial germline mutation in KIT associated with achalasia, mastocytosis and gastrointestinal stromal tumors shows response to kinase inhibitors. Cancer Genet. 2019, 233–234, 1–6. [Google Scholar] [CrossRef]
- Nakai, M.; Hashikura, Y.; Ohkouchi, M.; Yamamura, M.; Akiyama, T.; Shiba, K.; Kajimoto, N.; Tsukamoto, Y.; Hao, H.; Isozaki, K.; et al. Characterization of novel germline c-kit gene mutation, KIT-Tyr553Cys, observed in a family with multiple gastrointestinal stromal tumors. Lab. Investig. 2012, 92, 451–457. [Google Scholar] [CrossRef]
- Robson, M.E.; Glogowski, E.; Sommer, G.; Antonescu, C.R.; Nafa, K.; Maki, R.G.; Ellis, N.; Besmer, P.; Brennan, M.; Offit, K. Pleomorphic characteristics of a germ-line KIT mutation in a large kindred with gastrointestinal stromal tumors, hyperpigmentation, and dysphagia. Clin. Cancer Res. 2004, 10, 1250–1254. [Google Scholar] [CrossRef] [Green Version]
- Hirota, S.; Okazaki, T.; Kitamura, Y.; O’Brien, P.; Kapusta, L.; Dardick, I. Cause of familial and multiple gastrointestinal autonomic nerve tumors with hyperplasia of interstitial cells of Cajal is germline mutation of the c-kit gene. Am. J. Surg. Pathol. 2000, 24, 326–327. [Google Scholar] [CrossRef]
- Farag, S.; van der Kolk, L.E.; van Boven, H.H.; van Akkooi, A.C.J.; Beets, G.L.; Wilmink, J.W.; Steeghs, N. Remarkable effects of imatinib in a family with young onset gastrointestinal stromal tumors and cutaneous hyperpigmentation associated with a germline KIT-Trp557Arg mutation: Case report and literature overview. Fam. Cancer 2018, 17, 247–253. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Viale, A.; Sarran, L.; Tschernyavsky, S.J.; Gonen, M.; Segal, N.H.; Maki, R.G.; Socci, N.D.; DeMatteo, R.P.; Besmer, P. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin. Cancer Res. 2004, 10, 3282–3290. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Shimizu, A.; Ieta, K.; Shibusawa, K.; Ishikawa, O.; Ishida-Yamamoto, A.; Tamura, A. Generalized lentigines associated with familial gastrointestinal stromal tumors dramatically improved by imatinib treatment. J. Dermatol. 2020, 47, e241–e242. [Google Scholar] [CrossRef]
- Sekido, Y.; Ohigashi, S.; Takahashi, T.; Hayashi, N.; Suzuki, K.; Hirota, S. Familial Gastrointestinal Stromal Tumor with Germline KIT Mutations Accompanying Hereditary Breast and Ovarian Cancer Syndrome. Anticancer Res. 2017, 37, 1425–1431. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Chandrashekar, L.; Larizza, L.; Colombo, E.A.; Fontana, L.; Gervasini, C.; Thappa, D.M.; Rajappa, M.; Rajendiran, K.S.; Sreenath, G.S.; et al. Familial gastrointestinal stromal tumors, lentigines, and café-au-lait macules associated with germline c-kit mutation treated with imatinib. Int. J. Dermatol. 2017, 56, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Maeyama, H.; Hidaka, E.; Ota, H.; Minami, S.; Kajiyama, M.; Kuraishi, A.; Mori, H.; Matsuda, Y.; Wada, S.; Sodeyama, H.; et al. Familial gastrointestinal stromal tumor with hyperpigmentation: Association with a germline mutation of the c-kit gene. Gastroenterology 2001, 120, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Adela Avila, S.; Peñaloza, J.; González, F.; Abdo, I.; Rainville, I.; Root, E.; Carrero Valenzuela, R.D.; Garber, J. Dysphagia, melanosis, gastrointestinal stromal tumors and a germinal mutation of the KIT gene in an Argentine family. Acta Gastroenterol. Latinoam. 2014, 44, 9–15. [Google Scholar] [PubMed]
- Beghini, A.; Tibiletti, M.G.; Roversi, G.; Chiaravalli, A.M.; Serio, G.; Capella, C.; Larizza, L. Germline mutation in the juxtamembrane domain of the kit gene in a family with gastrointestinal stromal tumors and urticaria pigmentosa. Cancer 2001, 92, 657–662. [Google Scholar] [CrossRef]
- Kuroda, N.; Tanida, N.; Hirota, S.; Daum, O.; Hes, O.; Michal, M.; Lee, G.H. Familial gastrointestinal stromal tumor with germ line mutation of the juxtamembrane domain of the KIT gene observed in relatively young women. Ann. Diagn. Pathol. 2011, 15, 358–361. [Google Scholar] [CrossRef]
- Kim, H.J.; Lim, S.J.; Park, K.; Yuh, Y.J.; Jang, S.J.; Choi, J. Multiple gastrointestinal stromal tumors with a germline c-kit mutation. Pathol. Int. 2005, 55, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Hirota, S.; Taniguchi, M.; Hashimoto, K.; Isozaki, K.; Nakamura, H.; Kanakura, Y.; Tanaka, T.; Takabayashi, A.; Matsuda, H.; et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat. Genet. 1998, 19, 323–324. [Google Scholar] [CrossRef]
- Bamba, S.; Hirota, S.; Inatomi, O.; Ban, H.; Nishimura, T.; Shioya, M.; Imaeda, H.; Nishida, A.; Sasaki, M.; Murata, S.; et al. Familial and multiple gastrointestinal stromal tumors with fair response to a half-dose of imatinib. Intern. Med. 2015, 54, 759–764. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.Y.; Park, C.K.; Choi, J.S.; Jin, S.Y.; Kim, H.J.; Joo, M.; Kang, M.S.; Moon, W.S.; Yun, K.J.; Yu, E.S.; et al. Multiple gastrointestinal stromal tumors: Clinicopathologic and genetic analysis of 12 patients. Am. J. Surg. Pathol. 2007, 31, 224–232. [Google Scholar] [CrossRef]
- Woźniak, A.; Rutkowski, P.; Sciot, R.; Ruka, W.; Michej, W.; Debiec-Rychter, M. Rectal gastrointestinal stromal tumors associated with a novel germline KIT mutation. Int. J. Cancer 2008, 122, 2160–2164. [Google Scholar] [CrossRef]
- Neuhann, T.M.; Mansmann, V.; Merkelbach-Bruse, S.; Klink, B.; Hellinger, A.; Höffkes, H.G.; Wardelmann, E.; Schildhaus, H.U.; Tinschert, S. A novel germline KIT mutation (p.L576P) in a family presenting with juvenile onset of multiple gastrointestinal stromal tumors, skin hyperpigmentations, and esophageal stenosis. Am. J. Surg. Pathol. 2013, 37, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Vale Rodrigues, R.; Santos, F.; Pereira da Silva, J.; Francisco, I.; Claro, I.; Albuquerque, C.; Lemos, M.M.; Limbert, M.; Dias Pereira, A. A case of multiple gastrointestinal stromal tumors caused by a germline KIT gene mutation (p.Leu576Pro). Fam. Cancer 2017, 16, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Carballo, M.; Roig, I.; Aguilar, F.; Pol, M.A.; Gamundi, M.J.; Hernan, I.; Martinez-Gimeno, M. Novel c-KIT germline mutation in a family with gastrointestinal stromal tumors and cutaneous hyperpigmentation. Am. J. Med. Genet. A 2005, 132, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Forde, P.M.; Cochran, R.L.; Boikos, S.A.; Zabransky, D.J.; Beaver, J.A.; Meyer, C.F.; Thornton, K.A.; Montgomery, E.A.; Lidor, A.O.; Donehower, R.C.; et al. Familial GI Stromal Tumor With Loss of Heterozygosity and Amplification of Mutant KIT. J. Clin. Oncol. 2016, 34, e13–e16. [Google Scholar] [CrossRef]
- Tarn, C.; Merkel, E.; Canutescu, A.A.; Shen, W.; Skorobogatko, Y.; Heslin, M.J.; Eisenberg, B.; Birbe, R.; Patchefsky, A.; Dunbrack, R.; et al. Analysis of KIT mutations in sporadic and familial gastrointestinal stromal tumors: Therapeutic implications through protein modeling. Clin. Cancer Res. 2005, 11, 3668–3677. [Google Scholar] [CrossRef] [Green Version]
- Wali, G.N.; Halliday, D.; Dua, J.; Ieremia, E.; McPherson, T.; Matin, R.N. Cutaneous hyperpigmentation and familial gastrointestinal stromal tumour associated with KIT mutation. Clin. Exp. Dermatol. 2019, 44, 418–421. [Google Scholar] [CrossRef]
- Jones, D.H.; Caracciolo, J.T.; Hodul, P.J.; Strosberg, J.R.; Coppola, D.; Bui, M.M. Familial gastrointestinal stromal tumor syndrome: Report of 2 cases with KIT exon 11 mutation. Cancer Control 2015, 22, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Lasota, J.; Miettinen, M. A new familial GIST identified. Am. J. Surg. Pathol. 2006, 30, 1342. [Google Scholar] [CrossRef]
- Kleinbaum, E.P.; Lazar, A.J.F.; Tamborini, E.; Mcauliffe, J.C.; Sylvestre, P.B.; Sunnenberg, T.D.; Strong, L.; Chen, L.L.; Choi, H.; Benjamin, R.S.; et al. Clinical, histopathologic, molecular and therapeutic findings in a large kindred with gastrointestinal stromal tumor. Int. J. Cancer 2008, 122, 711–718. [Google Scholar] [CrossRef]
- Yamanoi, K.; Higuchi, K.; Kishimoto, H.; Nishida, Y.; Nakamura, M.; Sudoh, M.; Hirota, S. Multiple gastrointestinal stromal tumors with novel germline c-kit gene mutation, K642T, at exon 13. Hum. Pathol. 2014, 45, 884–888. [Google Scholar] [CrossRef]
- Wadt, K.; Andersen, M.K.; Hansen, T.V.O.; Gerdes, A.M. A new genetic diagnosis of familiar gastrointestinal stromal tumour. Ugeskr. Laeg. 2012, 174, 1462–1464. [Google Scholar] [PubMed]
- Bachet, J.B.; Landi, B.; Laurent-Puig, P.; Italiano, A.; Le Cesne, A.; Lévy, P.; Safar, V.; Duffaud, F.; Blay, J.Y.; Emile, J.F. Diagnosis, prognosis and treatment of patients with gastrointestinal stromal tumour (GIST) and germline mutation of KIT exon 13. Eur. J. Cancer 2013, 49, 2531–2541. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, K.; Terris, B.; Belghiti, J.; Schiffmann, S.; Hirota, S.; Vanderwinden, J.M. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am. J. Pathol. 2000, 157, 1581–1585. [Google Scholar] [CrossRef] [Green Version]
- Graham, J.; Debiec-Rychter, M.; Corless, C.L.; Reid, R.; Davidson, R.; White, J.D. Imatinib in the management of multiple gastrointestinal stromal tumors associated with a germline KIT K642E mutation. Arch. Pathol. Lab. Med. 2007, 131, 1393–1396. [Google Scholar] [CrossRef]
- Vilain, R.E.; Dudding, T.; Braye, S.G.; Groombridge, C.; Meldrum, C.; Spigelman, A.D.; Ackland, S.; Ashman, L.; Scott, R.J. Can a familial gastrointestinal tumour syndrome be allelic with Waardenburg syndrome? Clin. Genet. 2011, 79, 554–560. [Google Scholar] [CrossRef]
- Engin, G.; Eraslan, S.; Kayserili, H.; Kapran, Y.; Akman, H.; Akyuz, A.; Aykan, N.F. Imatinib response of gastrointestinal stromal tumor patients with germline mutation on KIT exon 13: A family report. World J. Radiol. 2017, 9, 365–370. [Google Scholar] [CrossRef]
- Hirota, S.; Nishida, T.; Isozaki, K.; Taniguchi, M.; Nishikawa, K.; Ohashi, A.; Takabayashi, A.; Obayashi, T.; Okuno, T.; Kinoshita, K.; et al. Familial gastrointestinal stromal tumors associated with dysphagia and novel type germline mutation of KIT gene. Gastroenterology 2002, 122, 1493–1499. [Google Scholar] [CrossRef]
- O’Riain, C.; Corless, C.L.; Heinrich, M.C.; Keegan, D.; Vioreanu, M.; Maguire, D.; Sheahan, K. Gastrointestinal stromal tumors: Insights from a new familial GIST kindred with unusual genetic and pathologic features. Am. J. Surg. Pathol. 2005, 29, 1680–1683. [Google Scholar] [CrossRef]
- Veiga, I.; Silva, M.; Vieira, J.; Pinto, C.; Pinheiro, M.; Torres, L.; Soares, M.; Santos, L.; Duarte, H.; Bastos, A.L.; et al. Hereditary gastrointestinal stromal tumors sharing the KIT Exon 17 germline mutation p.Asp820Tyr develop through different cytogenetic progression pathways. Genes Chromosomes Cancer 2010, 49, 91–98. [Google Scholar] [CrossRef]
- Arima, J.; Hiramatsu, M.; Taniguchi, K.; Kobayashi, T.; Tsunematsu, I.; Kagota, S.; Sakane, J.; Suzuki, Y.; Hirota, S. Multiple gastrointestinal stromal tumors caused by a novel germline KIT gene mutation (Asp820Gly): A case report and literature review. Gastric Cancer 2020, 23, 760–764. [Google Scholar] [CrossRef]
- Thalheimer, A.; Schlemmer, M.; Bueter, M.; Merkelbach-Bruse, S.; Schildhaus, H.U.; Buettner, R.; Hartung, E.; Thiede, A.; Meyer, D.; Fein, M.; et al. Familial gastrointestinal stromal tumors caused by the novel KIT exon 17 germline mutation N822Y. Am. J. Surg. Pathol. 2008, 32, 1560–1565. [Google Scholar] [CrossRef] [PubMed]
- de Raedt, T.; Cools, J.; Debiec-Rychter, M.; Brems, H.; Mentens, N.; Sciot, R.; Himpens, J.; de Wever, I.; Schöffski, P.; Marynen, P.; et al. Intestinal neurofibromatosis is a subtype of familial GIST and results from a dominant activating mutation in PDGFRA. Gastroenterology 2006, 131, 1907–1912. [Google Scholar] [CrossRef] [Green Version]
- Pasini, B.; Matyakhina, L.; Bei, T.; Muchow, M.; Boikos, S.; Ferrando, B.; Carney, J.A.; Stratakis, C.A. Multiple gastrointestinal stromal and other tumors caused by platelet-derived growth factor receptor alpha gene mutations: A case associated with a germline V561D defect. J. Clin. Endocrinol. Metab. 2007, 92, 3728–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carney, J.A.; Stratakis, C.A. Stromal, fibrous, and fatty gastrointestinal tumors in a patient with a PDGFRA gene mutation. Am. J. Surg. Pathol. 2008, 32, 1412–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, R.; Martini, M.; Cenci, T.; Carbone, A.; Lanza, P.; Biondi, A.; Rindi, G.; Cassano, A.; Larghi, A.; Persiani, R.; et al. PDGFRA-mutant syndrome. Mod. Pathol. 2015, 28, 954–964. [Google Scholar] [CrossRef] [Green Version]
- Ricci, R.; Martini, M.; Cenci, T.; Riccioni, M.E.; Maria, G.; Cassano, A.; Larocca, L.M. Divergent gastrointestinal stromal tumors in syndromic settings. Cancer Genet. 2016, 209, 354–358. [Google Scholar] [CrossRef]
- Chompret, A.; Kannengiesser, C.; Barrois, M.; Terrier, P.; Dahan, P.; Tursz, T.; Lenoir, G.M.; Bressac-De Paillerets, B. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 2004, 126, 318–321. [Google Scholar] [CrossRef]
- Manley, P.N.; Abu-Abed, S.; Kirsch, R.; Hawrysh, A.; Perrier, N.; Feilotter, H.; Pollett, A.; Riddell, R.H.; Hookey, L.; Walia, J.S. Familial PDGFRA-mutation syndrome: Somatic and gastrointestinal phenotype. Hum. Pathol. 2018, 76, 52–57. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Gene | Exon | Mutation | No. of Kindreds | Main Clinical Features [Reference] |
|---|---|---|---|---|
| KIT | 8 | D419del | 1 | systemic mastocytosis, multiple GISTs, dysphagia [11] |
| 9 | K509I | 2 | systemic mastocytosis, multiple GISTs [12]; achalasia; mastocytosis, multiple GISTs [13] | |
| 11 | Y533C | 1 | multiple GISTs [14] | |
| 11 | W557R | 4 | multiple GISTs, skin hyperpigmentation, dysphagia [15]; multiple gastrointestinal autonomic nerve tumor [16]; multiple GISTs; skin hyperpigmentation [17]; multiple GISTs [18] | |
| 11 | W557S | 1 | multiple GISTs; lentigines [19] | |
| 11 | W557L K558E | 1 | multiple GISTs, hereditary breast cancer [20] | |
| 11 | V559A | 7 | multiple GISTs; lentigines, cafe-au-lait macules [21]; multiple GIST, cutaneous hyperpigmentation [22]; multiple GISTs, melanosis, lentiginosis, hyperpigmentation, dysphagia [23]; multiple GISTs, hyperpigmentation, urticaria pigmentosa [24]; multiple GISTs, cutaneous hyperpigmentation [25]; multiple GISTs [26] | |
| 11 | V559_V560del | 1 | multiple GISTs, cutaneous hyperpigmentation [27] | |
| 11 | V560del | 1 | multiple GISTs [28] | |
| 11 | V560G | 1 | multiple GISTs, cutaneous hyperpigmentation [29] | |
| 11 | V560A | 1 | multiple GISTs [29] | |
| 11 | Q575_P577delinsH | 1 | rectal GIST [30] | |
| 11 | L576P | 2 | multiple GISTs; skin hyperpigmentation, achalasia-like stenosis [31]; multiple GISTs [32] | |
| 11 | L576_P577insQL | 1 | multiple GISTs, cutaneous hyperpigmentation [33] | |
| 11 | D579del | 7 | multiple GISTs, cutaneous hyperpigmentation, dysphagia [34]; GIST [35]; GIST, cutaneous hyperpigmentation [36]; multiple GISTs [37]; multiple GISTs [38]; multiple GIST, nevi, hyperpigmentation [39] | |
| 13 | K642T | 1 | multiple GISTs, dysphagia [40]; | |
| 13 | K642E | 7 | multiple GISTs, breast cancer [41]; multiple GISTs, dysphagia, multiple nevi and lentigines [42]; multiple GISTs [43]; multiple GISTs including rectal GIST [44]; multiple GISTs, dysphagia, pigmentary defects (hyper- and hypopigmentation) [45]; multiple GISTs including rectal GIST, pigmentary defects (hyper- and hypopigmentation) [46] | |
| 13 | N655K | 1 | multiple GISTs, lentigines, atypical junctional nevus, breast and thyroid cancer [present report] | |
| 17 | D820Y | 3 | multiple GISTs, dysphagia [47]; multiple GISTs [48]; multiple GISTs including rectal GIST [49] | |
| 17 | D820G | 1 | multiple GISTs [50] | |
| 17 | N822Y | 1 | multiple GISTs [51] | |
| PDGFRA | 12 | Y555C | 1 | multiple GISTs, intestinal neurofibromatosis, glaucoma, coarse facies, broad hands [52] |
| 12 | V561D | 1 | multiple GISTs, fibrous tumors, lipoma [53,54] | |
| 14 | P653L | 2 | multiple GISTs, fibrous tumors, inflammatory fibroid polyps [55,56] | |
| 18 | D846Y | 1 | multiple GISTs, broad hands [57] | |
| 18 | D846V | 1 | multiple GISTs, coarse facies/skin, broad extremities [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fornasarig, M.; Gasparotto, D.; Foltran, L.; Campigotto, M.; Lombardi, S.; Del Savio, E.; Buonadonna, A.; Puglisi, F.; Sulfaro, S.; Canzonieri, V.; et al. A Novel Kindred with Familial Gastrointestinal Stromal Tumors Caused by a Rare KIT Germline Mutation (N655K): Clinico-Pathological Presentation and TKI Sensitivity. J. Pers. Med. 2020, 10, 234. https://doi.org/10.3390/jpm10040234
Fornasarig M, Gasparotto D, Foltran L, Campigotto M, Lombardi S, Del Savio E, Buonadonna A, Puglisi F, Sulfaro S, Canzonieri V, et al. A Novel Kindred with Familial Gastrointestinal Stromal Tumors Caused by a Rare KIT Germline Mutation (N655K): Clinico-Pathological Presentation and TKI Sensitivity. Journal of Personalized Medicine. 2020; 10(4):234. https://doi.org/10.3390/jpm10040234
Chicago/Turabian StyleFornasarig, Mara, Daniela Gasparotto, Luisa Foltran, Michele Campigotto, Sara Lombardi, Elisa Del Savio, Angela Buonadonna, Fabio Puglisi, Sandro Sulfaro, Vincenzo Canzonieri, and et al. 2020. "A Novel Kindred with Familial Gastrointestinal Stromal Tumors Caused by a Rare KIT Germline Mutation (N655K): Clinico-Pathological Presentation and TKI Sensitivity" Journal of Personalized Medicine 10, no. 4: 234. https://doi.org/10.3390/jpm10040234