
Transcriptional Responses of Fusarium graminearum Interacted with Soybean to Cause Root Rot

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Preparation and Growth Conditions
2.2. Preparation and Inoculation of the Pathogen
2.3. Extraction of Total RNA
2.4. cDNA Library Construction and Illumina RNA-Sequencing
2.5. RNA-Seq Data Analysis
2.6. qRT-PCR Verification
3. Results
3.1. Transcriptional Data Analysis of F. graminearum Infection on Soybean
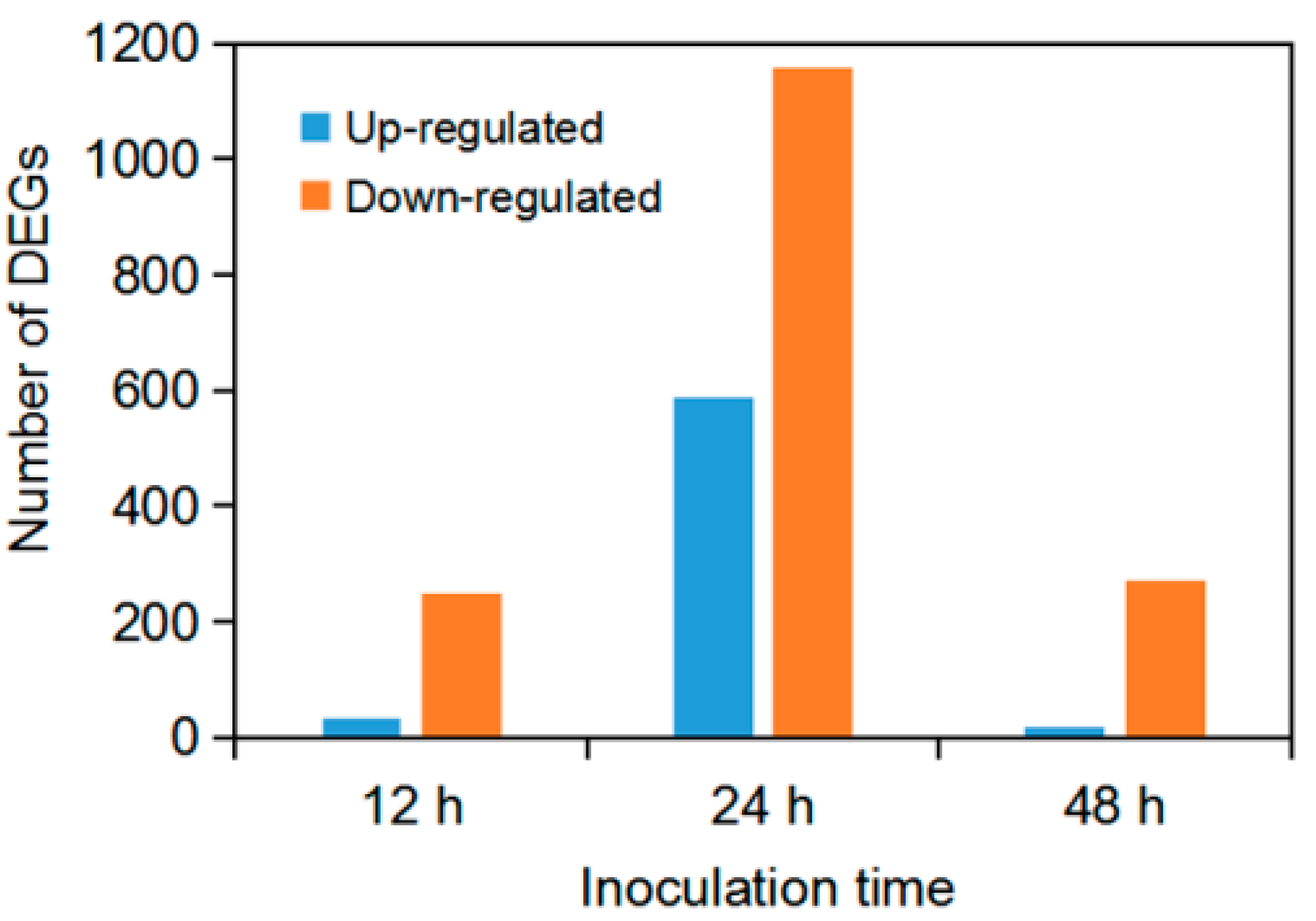
3.2. Differentially Expressed Genes (DEGs) of F. graminearum Infection on Soybean
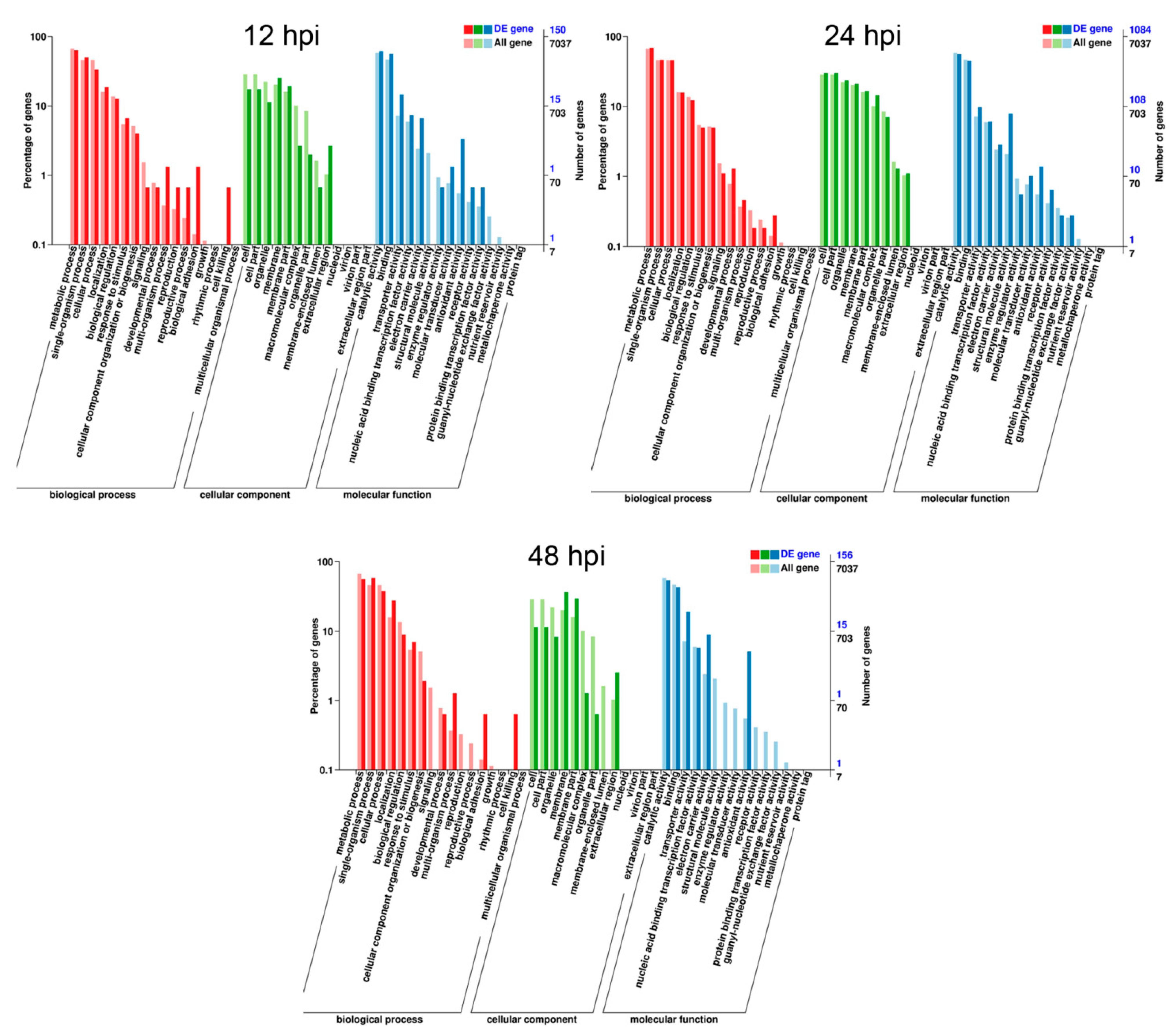
3.3. DEGs Annotation of F. graminearum
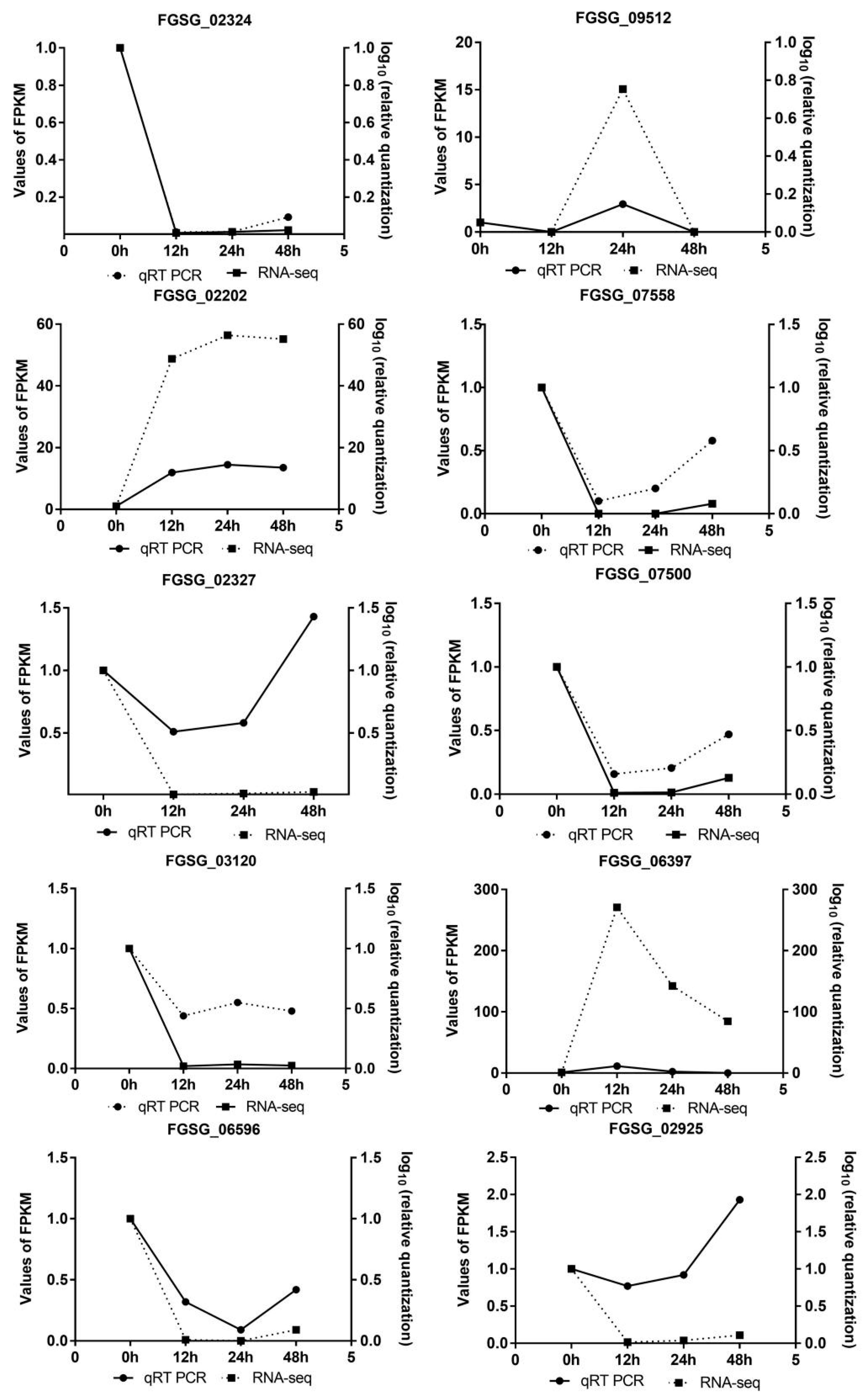
3.4. Validation of Randomly Selected Gene and Their Expression by qRT-PCR
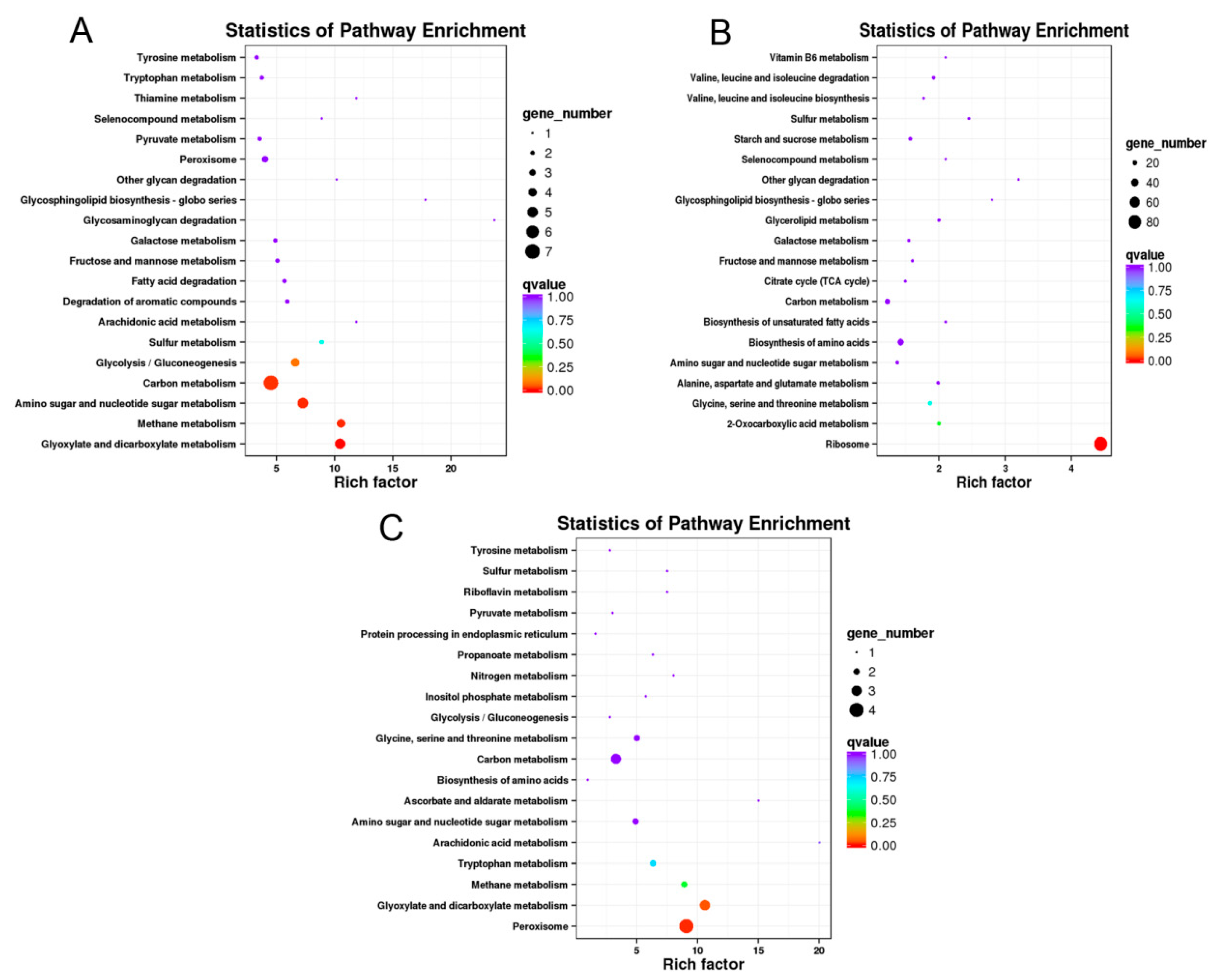
3.5. Expression Profile of DEGs Involving in Carbon Metabolism, Ribosome and Peroxisomes Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Wang, C.; Li, B.; Li, L.; Lin, D.; Chen, H.; Liu, Y.; Li, S.; Qin, W.; Liu, J. Research progress in tofu processing: From raw materials to processing conditions. Crit. Rev. Food Sci. Nutr. 2018, 58, 1448–1467. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, X.; Liao, D.; Lu, F.; Gao, R.; Liu, W.; Yong, T.; Wu, X.; Du, J.; Liu, J. Yield response to different planting geometries in maize–soybean relay strip intercropping systems. Agron. J. 2015, 107, 296–304. [Google Scholar] [CrossRef]
- Du, J.; Han, T.; Gai, J.; Yong, T.; Sun, X.; Wang, X.; Yang, F.; Liu, J.; Shu, K.; Liu, W. Maize-soybean strip intercropping: Achieved a balance between high productivity and sustainability. J. Integr. Agric. 2018, 17, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Rahman, T.; Song, C.; Su, B.; Yang, F.; Yong, T.; Wu, Y.; Zhang, C.; Yang, W. Changes in light environment, morphology, growth and yield of soybean in maize-soybean intercropping systems. Field Crop. Res. 2017, 200, 38–46. [Google Scholar] [CrossRef]
- Chang, X.; Dai, H.; Wang, D.; Zhou, H.; He, W.; Fu, Y.; Ibrahim, F.; Zhou, Y.; Gong, G.; Shang, J. Identification of Fusarium species associated with soybean root rot in Sichuan Province, China. Eur. J. Plant Pathol. 2018, 151, 563–577. [Google Scholar] [CrossRef]
- Dorrance, A.E.; Lipps, P.; Mills, D. Phytophthora damping off and root rot of soybean. In Columbus: Ohio State University Extension Fact Sheet; The Ohio State University: Columbus, OH, USA, 2000. [Google Scholar]
- Yang, X.B.; Hartman, G.L. Pythium damping-off and root rot. In Compendium of Soybean Diseases, 4th ed.; Hartman, G.L., Sinclair, J.B., Rupe, J.C., Eds.; American Phytopathological Society: St. Paul, MN, USA, 1999; pp. 42–44. [Google Scholar]
- Chang, X.; Yan, L.; Naeem, M.; Khaskheli, M.I.; Zhang, H.; Gong, G.; Zhang, M.; Song, C.; Yang, W.; Liu, T. Maize/soybean relay strip intercropping reduces the occurrence of Fusarium root rot and changes the diversity of the pathogenic Fusarium Species. Pathogens 2020, 9, 211. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Naeem, M.; Li, H.; Yan, L.; Liu, T.; Liu, B.; Zhang, H.; Khaskheli, M.; Gong, G.; Zhang, M. First report of Fusarium asiaticum as a causal agent for seed decay of soybean (Glycine max) in Sichuan, China. Plant Dis. 2020, 104, 1542. [Google Scholar] [CrossRef]
- Sella, L.; Gazzetti, K.; Castiglioni, C.; Schäfer, W.; Favaron, F. Fusarium graminearum possesses virulence factors common to Fusarium head blight of wheat and seedling rot of soybean but differing in their impact on disease severity. Phytopathology 2014, 104, 1201–1207. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Van der Lee, T.; Waalwijk, C.; Chen, W.; Xu, J.; Xu, J.; Zhang, Y.; Feng, J. Population analysis of the Fusarium graminearum species complex from wheat in China show a shift to more aggressive isolates. PLoS ONE 2012, 7, e31722. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; He, J.; Jia, L.; Yuan, T.; Zhang, D.; Guo, Y.; Wang, Y.; Tang, W.H. Cellular tracking and gene profiling of Fusarium graminearum during maize stalk rot disease development elucidates its strategies in confronting phosphorus limitation in the host apoplast. PLoS Pathog. 2016, 12, e1005485. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Jacobsen, S.; Jørgensen, H.J.; Collinge, D.B.; Svensson, B.; Finnie, C. Fusarium graminearum and its interactions with cereal heads: Studies in the proteomics era. Front. Plant Sci. 2013, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xia, M.; Xue, B.; Goodwin, P.; Sun, R.; Quan, X.; Lu, W.; Yang, L. First report of Fusarium pseudograminearum causing root rot on soybean (Glycine max) in Henan, China. Plant Dis. 2018, 102, 1454. [Google Scholar] [CrossRef]
- Barros, G.; Zanon, M.A.; Abod, A.; Oviedo, M.; Ramirez, M.; Reynoso, M.; Torres, A.; Chulze, S. Natural deoxynivalenol occurrence and genotype and chemotype determination of a field population of the Fusarium graminearum complex associated with soybean in Argentina. Food Addit. Contam. Part A 2012, 29, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Li, H.; Naeem, M.; Wu, X.; Yong, T.; Song, C.; Liu, T.; Chen, W.; Yang, W. Diversity of the seedborne fungi and pathogenicity of Fusarium species associated with intercropped soybean. Pathogens 2020, 9, 531. [Google Scholar] [CrossRef]
- Baird, R.; Mullinix, B.; Peery, A.; Lang, M. Diversity and longevity of the soybean debris mycobiota in a no-tillage system. Plant Dis. 1997, 81, 530–534. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, A.; Proctor, R. Molecular biology of Fusarium mycotoxins. Int. J. Food Microbiol. 2007, 119, 47–50. [Google Scholar] [CrossRef]
- Ellis, M.; Munkvold, G. Trichothecene genotype of Fusarium graminearum isolates from soybean (Glycine max) seedling and root diseases in the United States. Plant Dis. 2014, 98, 1012. [Google Scholar] [CrossRef] [PubMed]
- Shah, L.; Ali, A.; Yahya, M.; Zhu, Y.; Wang, S.; Si, H.; Rahman, H.; Ma, C. Integrated control of fusarium head blight and deoxynivalenol mycotoxin in wheat. Plant Pathol. 2018, 67, 532–548. [Google Scholar] [CrossRef]
- Cuomo, C.A.; Güldener, U.; Xu, J.R.; Trail, F.; Turgeon, B.G.; Di Pietro, A.; Walton, J.D.; Ma, L.J.; Baker, S.E.; Rep, M. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 2007, 317, 1400–1402. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.J.; Van Der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef]
- Park, J.; Park, J.; Jang, S.; Kim, S.; Kong, S.; Choi, J.; Ahn, K.; Kim, J.; Lee, S.; Kim, S. FTFD: An informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 2008, 24, 1024–1025. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.W.; Jia, L.J.; Zhang, Y.; Jiang, G.; Li, X.; Zhang, D.; Tang, W.-H. In planta stage-specific fungal gene profiling elucidates the molecular strategies of Fusarium graminearum growing inside wheat coleoptiles. Plant Cell 2012, 24, 5159–5176. [Google Scholar] [CrossRef] [Green Version]
- Kazan, K.; Gardiner, D.M.; Manners, J.M. On the trail of a cereal killer: Recent advances in Fusarium graminearum pathogenomics and host resistance. Mol. Plant Pathol. 2012, 13, 399–413. [Google Scholar] [CrossRef]
- Hynes, M.J.; Murray, S.L.; Khew, G.S.; Davis, M.A. Genetic analysis of the role of peroxisomes in the utilization of acetate and fatty acids in Aspergillus nidulans. Genetics 2008, 178, 1355–1369. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-Y.; Soanes, D.M.; Kershaw, M.J.; Talbot, N.J. Functional analysis of lipid metabolism in Magnaporthe grisea reveals a requirement for peroxisomal fatty acid β-oxidation during appressorium-mediated plant infection. Mol. Plant-Microbe Interact. 2007, 20, 475–491. [Google Scholar] [CrossRef] [Green Version]
- Goh, J.; Jeon, J.; Kim, K.S.; Park, J.; Park, S.-Y.; Lee, Y.-H. The PEX7-mediated peroxisomal import system is required for fungal development and pathogenicity in Magnaporthe oryzae. PLoS ONE 2011, 6, e28220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, S.; Yang, L.; Sun, Y.; Zhang, Q.; Li, B.; Wang, B.; Li, L.; Wang, D.; Wang, Z. Reference genes for qRT-PCR normalisation in different tissues, developmental stages, and stress conditions of Hypericum perforatum. PeerJ 2019, 7, e7133. [Google Scholar] [CrossRef]
- Abdullah, A.S.; Moffat, C.S.; Lopez-Ruiz, F.J.; Gibberd, M.R.; Hamblin, J.; Zerihun, A. Host–multi-pathogen warfare: Pathogen interactions in co-infected plants. Front. Plant Sci. 2017, 8, 1806. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Stam, R.; Cano, L.M.; Song, J.; Sklenar, J.; Yoshida, K.; Bozkurt, T.O.; Oliva, R.; Liu, Z.; Tian, M. Effector specialization in a lineage of the Irish potato famine pathogen. Science 2014, 343, 552–555. [Google Scholar] [CrossRef]
- Naeem, M.; Li, H.; Yan, L.; Raza, M.A.; Gong, G.; Chen, H.; Yang, C.; Zhang, M.; Shang, J.; Liu, T. Characterization and pathogenicity of Fusarium species associated with soybean pods in maize/soybean strip intercropping. Pathogens 2019, 8, 245. [Google Scholar] [CrossRef] [Green Version]
- Lysøe, E.; Seong, K.Y.; Kistler, H.C. The transcriptome of Fusarium graminearum during the infection of wheat. Mol. Plant-Microbe Interact. 2011, 24, 995–1000. [Google Scholar] [CrossRef] [Green Version]
- Barna, B.; Fodor, J.; Harrach, B.; Pogány, M.; Király, Z. The Janus face of reactive oxygen species in resistance and susceptibility of plants to necrotrophic and biotrophic pathogens. Plant. Physiol. Biochem. 2012, 59, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–227. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Schäfer, W.; Bormann, J. The stress-activated protein kinase FgOS-2 is a key regulator in the life cycle of the cereal pathogen Fusarium graminearum. Mol. Plant-Microbe Interact. 2012, 25, 1142–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Son, H.; Shin, J.Y.; Choi, G.J.; Lee, Y.W. Genome-wide functional characterization of putative peroxidases in the head blight fungus Fusarium graminearum. Mol. Plant Pathol. 2018, 19, 715–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gessler, N.; Aver’Yanov, A.; Belozerskaya, T. Reactive oxygen species in regulation of fungal development. Biochemistry 2007, 72, 1091–1109. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, H.; Ring, J.; Mayer, T.; Schauer, A.; Büttner, S.; Eisenberg, T.; Carmona-Gutierrez, D.; Kuchler, K.; Madeo, F. Loss of peroxisome function triggers necrosis. FEBS Lett. 2008, 582, 2882–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, K.; Son, H.; Lee, J.; Choi, G.J.; Kim, J.C.; Lee, Y.W. Peroxisome function is required for virulence and survival of Fusarium graminearum. Mol. Plant-Microbe Interact. 2012, 25, 1617–1627. [Google Scholar] [CrossRef] [Green Version]
- Robbertse, B.; Yoder, O.; Nguyen, A.; Schoch, C.L.; Turgeon, B.G. Deletion of all Cochliobolus heterostrophus monofunctional catalase-encoding genes reveals a role for one in sensitivity to oxidative stress but none with a role in virulence. Mol. Plant-Microbe Interact. 2003, 16, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skamnioti, P.; Henderson, C.; Zhang, Z.; Robinson, Z.; Gurr, S.J. A novel role for catalase B in the maintenance of fungal cell-wall integrity during host invasion in the rice blast fungus Magnaporthe grisea. Mol. Plant-Microbe Interact. 2007, 20, 568–580. [Google Scholar] [CrossRef]
- Tanabe, S.; Ishii-Minami, N.; Saitoh, K.-I.; Otake, Y.; Kaku, H.; Shibuya, N.; Nishizawa, Y.; Minami, E. The role of catalase-peroxidase secreted by Magnaporthe oryzae during early infection of rice cells. Mol. Plant-Microbe Interact. 2011, 24, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.-H.; Guo, Y.; Wang, Y.Z.; Zhang, D.; Xu, L.; Tang, W.H. A cytoplasmic Cu-Zn superoxide dismutase SOD1 contributes to hyphal growth and virulence of Fusarium graminearum. Fungal Genet. Biol. 2016, 91, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Yao, S.; Yuan, T.; Wang, Y.; Zhang, D.; Tang, W. The spatiotemporal control of KatG2 catalase-peroxidase contributes to the invasiveness of Fusarium graminearum in host plants. Mol. Plant Pathol. 2019, 20, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, T.; Yoshinari, T.; Sakuda, S. Intracellular superoxide level controlled by manganese superoxide dismutases affects trichothecene production in Fusarium graminearum. FEMS Microbiol. Lett. 2017, 364, fnx213. [Google Scholar] [CrossRef]
- Xu, C.; Li, M.; Zhou, Z.; Li, J.; Chen, D.; Duan, Y.; Zhou, M. Impact of five succinate dehydrogenase inhibitors on DON biosynthesis of Fusarium asiaticum, causing fusarium head blight in wheat. Toxins 2019, 11, 272. [Google Scholar] [CrossRef] [Green Version]
- Liberti, D.; Rollins, J.; Dobinson, K. Peroxysomal carnitine acetyl transferase influences host colonization capacity in Sclerotinia sclerotiorum. Mol. Plant-Microbe Interact. 2013, 26, 768–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Yang, Q.; Yin, C.; Liu, L.; Liang, W. Systematic analysis of the lysine acetylome in Fusarium graminearum. BMC Genom. 2016, 17, 1019. [Google Scholar] [CrossRef] [Green Version]
- Son, H.; Seo, Y.-S.; Min, K.; Park, A.R.; Lee, J.; Jin, J.M.; Lin, Y.; Cao, P.; Hong, S.Y.; Kim, E.K. A phenome-based functional analysis of transcription factors in the cereal head blight fungus, Fusarium graminearum. PLoS Pathog 2011, 7, e1002310. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, S.; Hou, R.; Zhao, Z.; Zheng, Q.; Xu, Q.; Zheng, D.; Wang, G.; Liu, H.; Gao, X. Functional analysis of the kinome of the wheat scab fungus Fusarium graminearum. PLoS Pathog 2011, 7, e1002460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, Y.; Liu, Z.; Yin, Y.; Jiang, J.; Chen, Y.; Xu, J.R.; Ma, Z. Functional analysis of the Fusarium graminearum phosphatome. New Phytol. 2015, 207, 119–134. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Total Reads | Clean Reads | Mapped Reads | Unique Mapped Reads |
|---|---|---|---|---|
| 12 h | 53,979,100 | 26,989,550 | 283,660 (0.53%) | 231,631 (0.43%) |
| 12 h | 82,859,646 | 41,429,823 | 393,276 (0.47%) | 334,586 (0.40%) |
| 12 h | 100,333,768 | 50,166,884 | 399,024 (0.40%) | 333,526 (0.33%) |
| 24 h | 79,795,198 | 39,897,599 | 3,202,980 (4.01%) | 2,987,228 (3.74%) |
| 24 h | 72,608,382 | 36,304,191 | 3,455,298 (4.76%) | 3,104,447 (4.28%) |
| 48 h | 88,036,166 | 44,018,083 | 6,783,005 (7.70%) | 6,214,145 (7.06%) |
| 48 h | 72,301,850 | 36,150,925 | 6,423,924 (8.88%) | 5,767,693 (7.98%) |
| 48 h | 74,999,418 | 37,499,709 | 5,774,290 (7.70%) | 5,243,878 (6.99%) |
| F. graminearum | 102,697,726 | 51,348,863 | 66,062,446 (64.33%) | 58,398,509 (56.86%) |
| F. graminearum | 101,416,397 | 50,176,223 | 64,173,223 (63.28%) | 57,247,119 (56.48%) |
| F. graminearum | 102,385,647 | 51,267,432 | 66,001,101 (64.46%) | 58,266,178 (56.91%) |
| Inoculation time | Gene ID | Description | log2FC |
|---|---|---|---|
| 12 hpi | FGSG_02881 | Catalase, CAT | Infinity |
| FGSG_06596 | Catalase, CAT | −5.82 | |
| FGSG_00840 | carnitine O-acetyltransferase | −3.18 | |
| 24 hpi | FGSG_00724 | peroxin-2, PEX2 | −2.08 |
| FGSG_00666 | peroxin-13, PEX13 | −1.22 | |
| FGSG_02881 | Catalase, CAT | −7.33 | |
| FGSG_06596 | Catalase, CAT | Infinity | |
| FGSG_02051 | superoxide dismutase (SOD), Fe-Mn family | −3.92 | |
| FGSG_02287 | acyl-CoA oxidase | 2.46 | |
| FGSG_10347 | isocitrate dehydrogenase | 2.06 | |
| FGSG_00840 | carnitine O-acetyltransferase | −2.580 | |
| 48 hpi | FGSG_02881 | Catalase, CAT | −2.28 |
| FGSG_06596 | Catalase, CAT | −3.32 | |
| FGSG_02051 | superoxide dismutase (SOD), Fe-Mn family | −2.71 | |
| FGSG_00840 | carnitine O-acetyltransferase | −3.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naeem, M.; Munir, M.; Li, H.; Raza, M.A.; Song, C.; Wu, X.; Irshad, G.; Khalid, M.H.B.; Yang, W.; Chang, X. Transcriptional Responses of Fusarium graminearum Interacted with Soybean to Cause Root Rot. J. Fungi 2021, 7, 422. https://doi.org/10.3390/jof7060422
Naeem M, Munir M, Li H, Raza MA, Song C, Wu X, Irshad G, Khalid MHB, Yang W, Chang X. Transcriptional Responses of Fusarium graminearum Interacted with Soybean to Cause Root Rot. Journal of Fungi. 2021; 7(6):422. https://doi.org/10.3390/jof7060422
Chicago/Turabian StyleNaeem, Muhammd, Maira Munir, Hongju Li, Muhammad Ali Raza, Chun Song, Xiaoling Wu, Gulshan Irshad, Muhammad Hyder Bin Khalid, Wenyu Yang, and Xiaoli Chang. 2021. "Transcriptional Responses of Fusarium graminearum Interacted with Soybean to Cause Root Rot" Journal of Fungi 7, no. 6: 422. https://doi.org/10.3390/jof7060422