A Potential Theragnostic Regulatory Axis for Arthrofibrosis Involving Adiponectin (ADIPOQ) Receptor 1 and 2 (ADIPOR1 and ADIPOR2), TGFβ1, and Smooth Muscle α-Actin (ACTA2)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA-Sequencing Analysis
2.2. Cell Culture
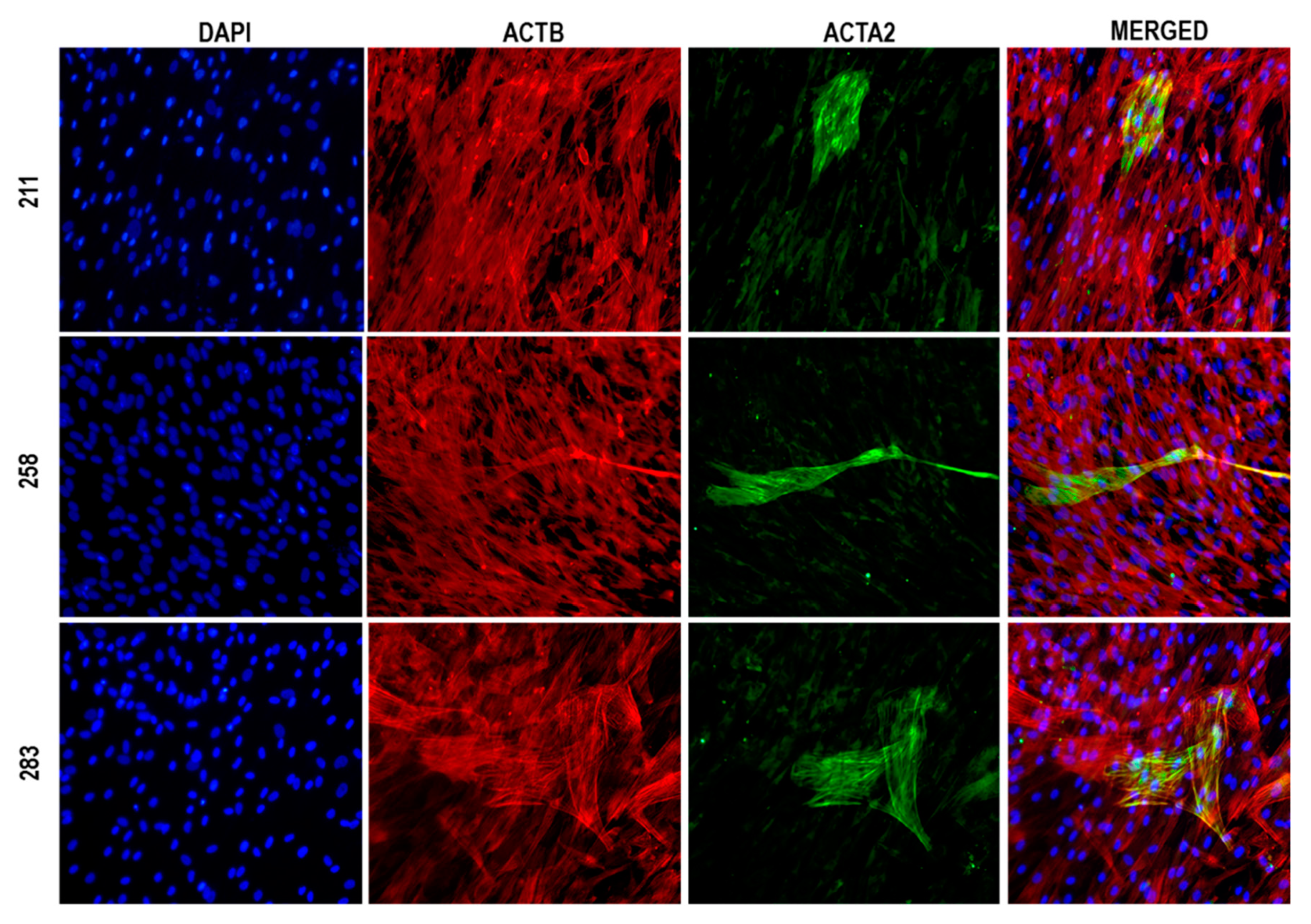
2.3. Immunofluorescence Microscopy
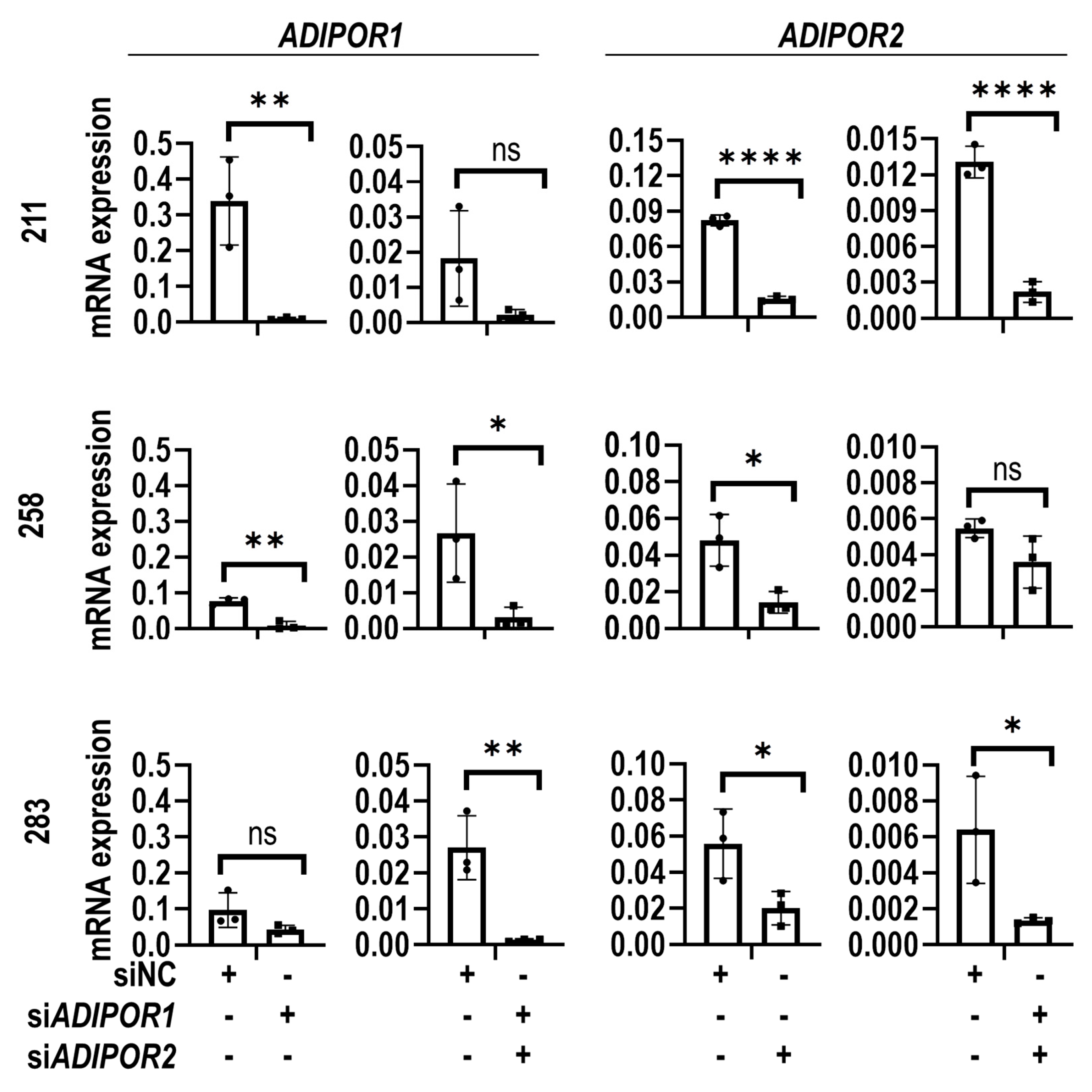
2.4. RNA Interference
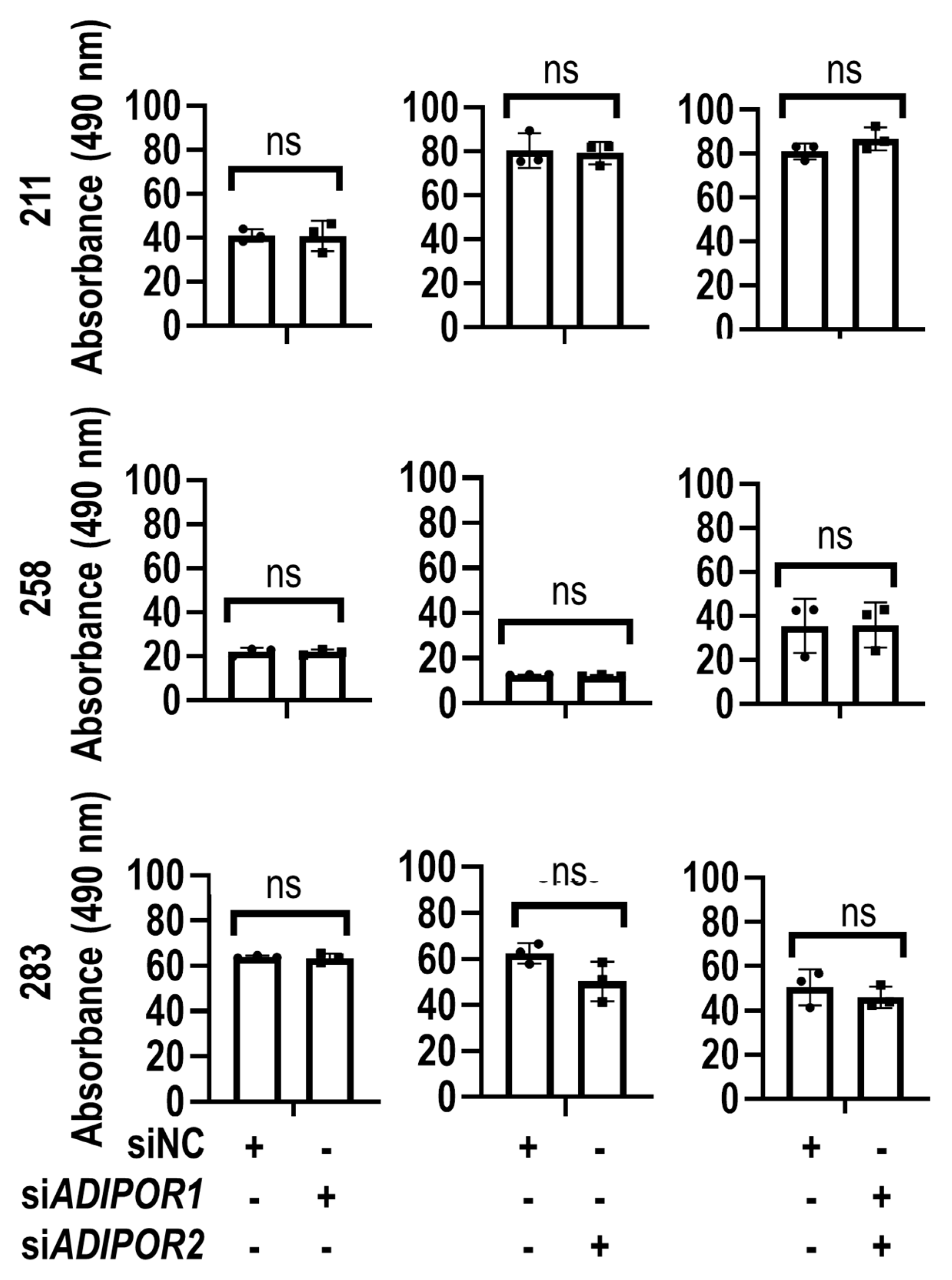
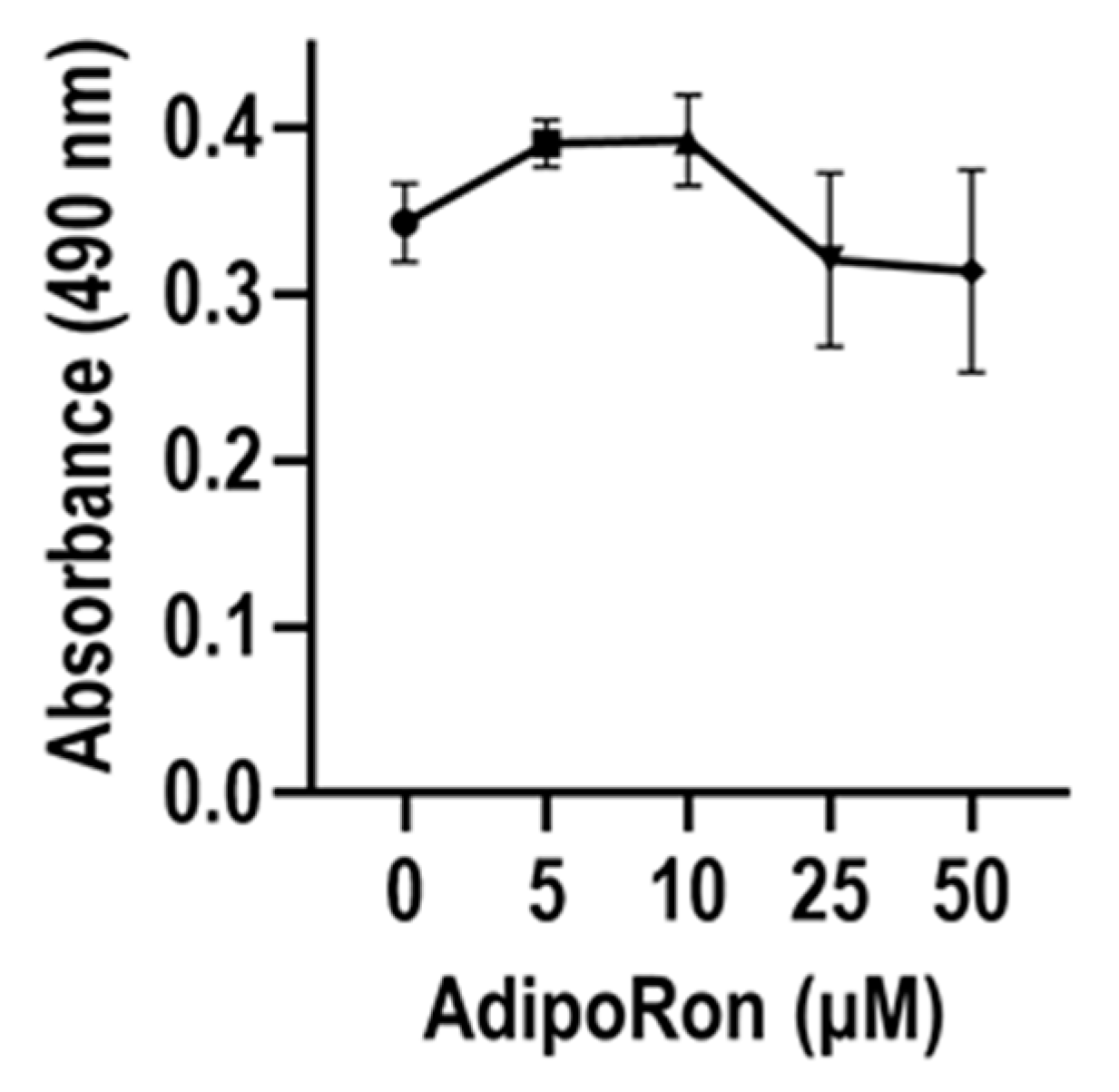
2.5. MTS Assay
2.6. AdipoRon Treatment During TGFβ1 Induction of Myofibroblast Formation
2.7. Analysis of Gene Expression
2.8. Protein Extraction and Immunoblotting
2.9. Statistical Analysis
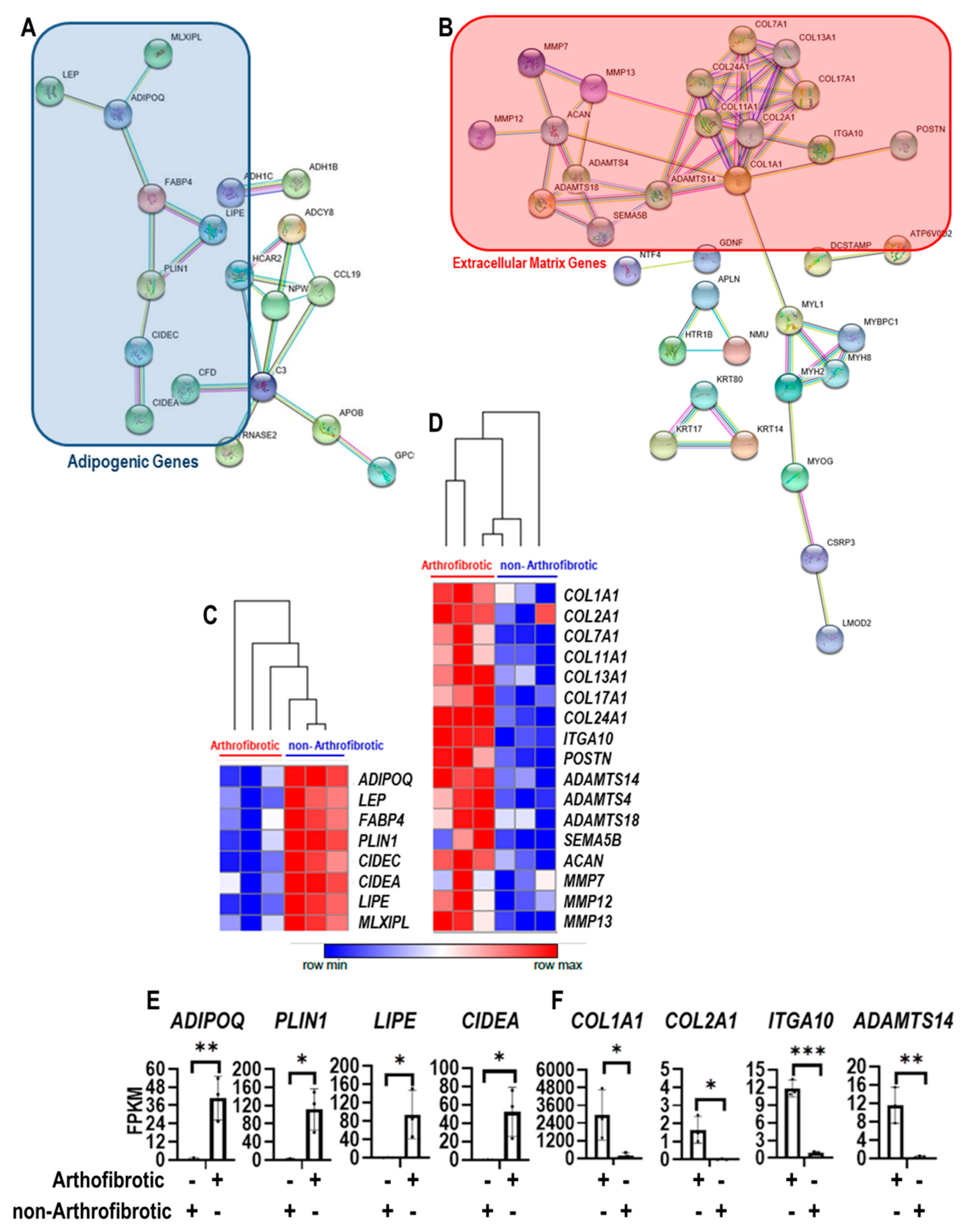
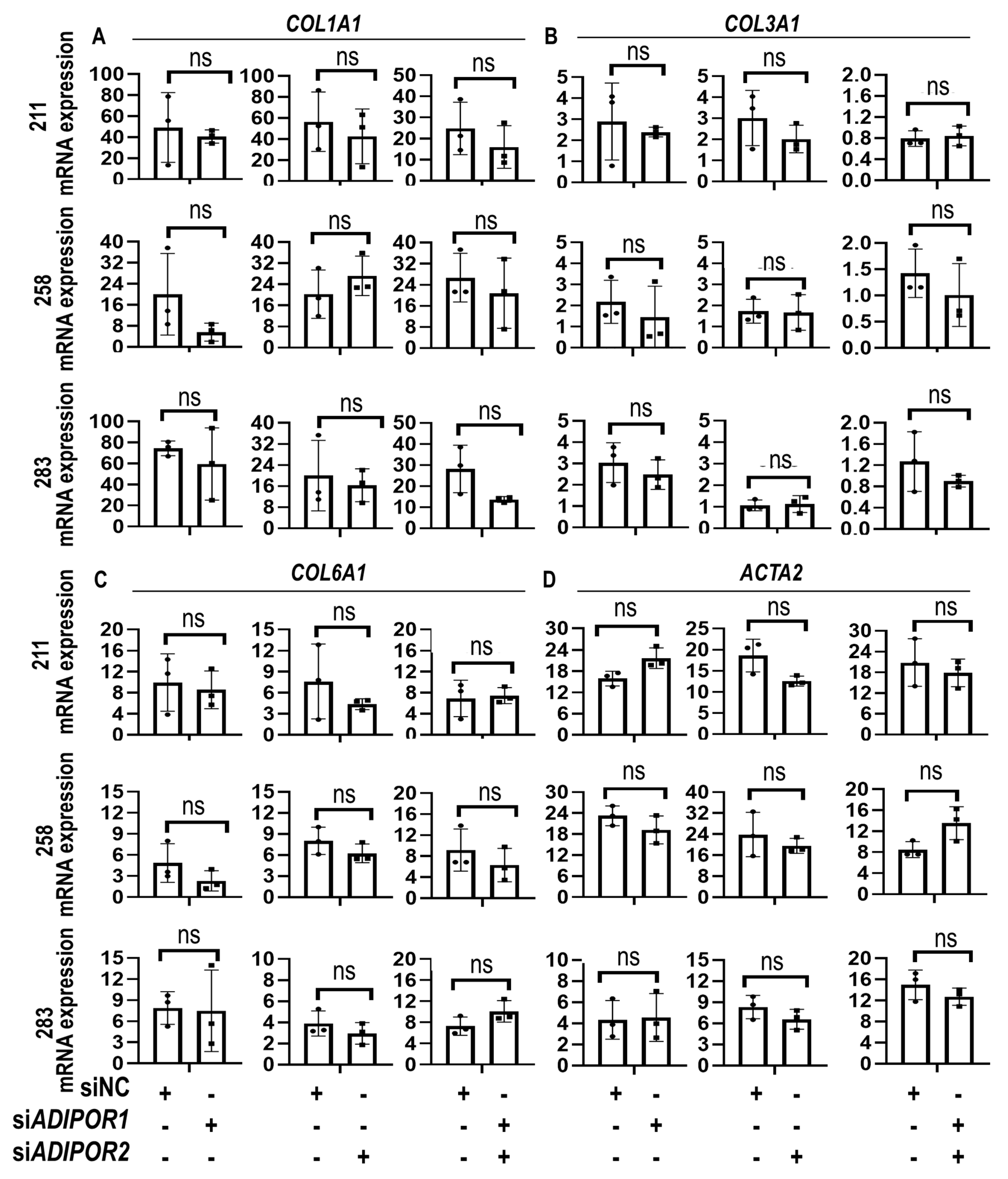
3. Results
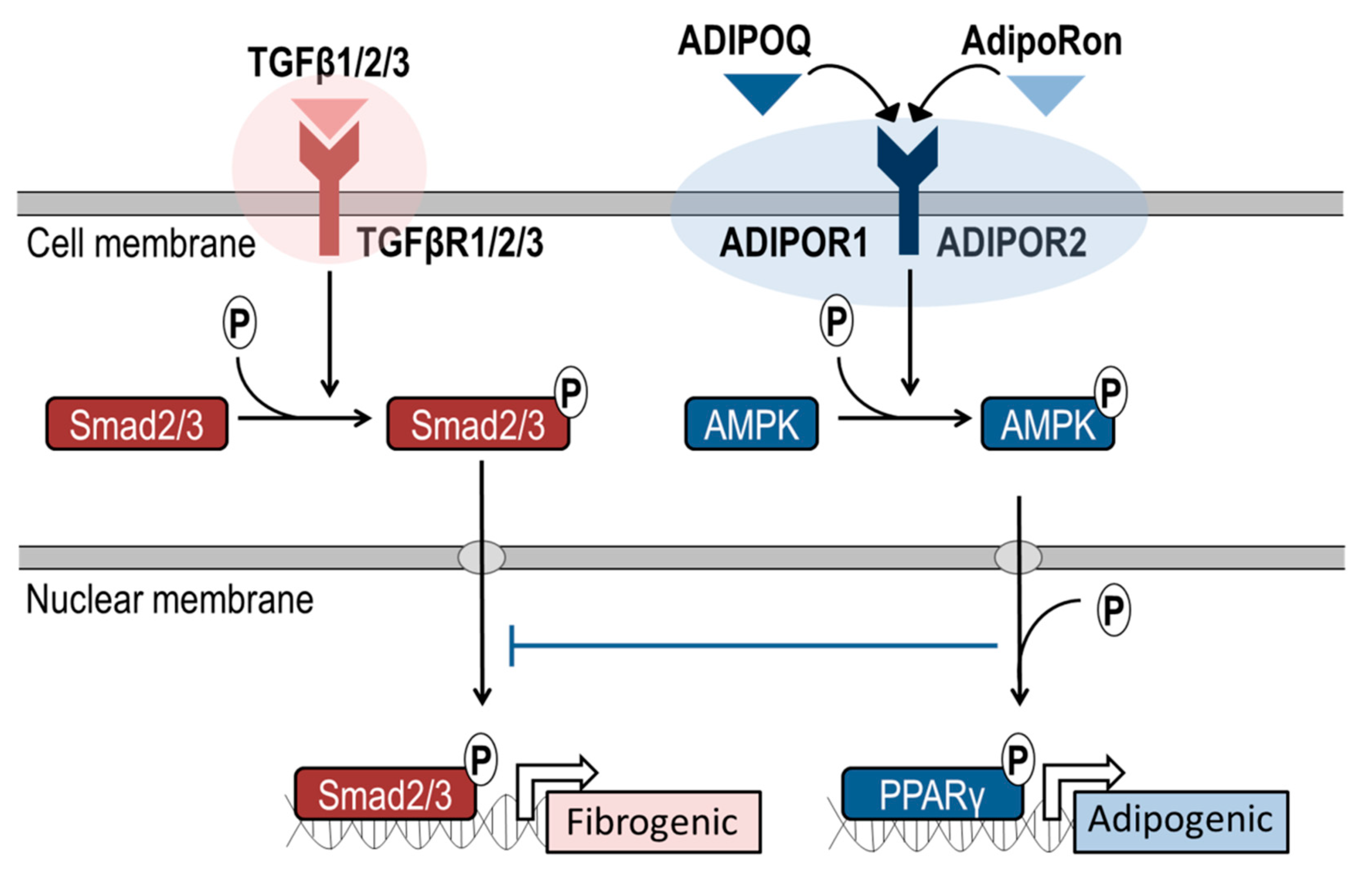
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheuy, V.A.; Foran, J.R.; Paxton, R.J.; Bade, M.J.; Zeni, J.A.; Stevens-Lapsley, J.E. Arthrofibrosis Associated with Total Knee Arthroplasty. J. Arthroplast. 2017, 32, 2604–2611. [Google Scholar] [CrossRef]
- Tibbo, M.E.; Limberg, A.K.; Salib, C.G.; Ahmed, A.T.; Van Wijnen, A.J.; Berry, D.J.; Abdel, M.P. Acquired Idiopathic Stiffness After Total Knee Arthroplasty. J. Bone Jt. Surg. Am. 2019, 101, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Pitta, M.; Esposito, C.I.; Li, Z.; Lee, Y.-Y.; Wright, T.M.; Padgett, D.E. Failure After Modern Total Knee Arthroplasty: A Prospective Study of 18,065 Knees. J. Arthroplast. 2018, 33, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Bingham, J.S.; Bukowski, B.R.; Wyles, C.C.; Pareek, A.; Berry, D.J.; Abdel, M.P. Rotating-Hinge Revision Total Knee Arthroplasty for Treatment of Severe Arthrofibrosis. J. Arthroplast. 2019, 34, S271–S276. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Klingberg, F.; Hinz, B.; White, E.S. The Myofibroblast Matrix: Implications for Tissue Repair and Fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Šumová, B.; Mallano, T.; Chen, C.-W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of STAT3 Integrates Common Profibrotic Pathways to Promote Fibroblast Activation and Tissue Fibrosis. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Bayram, B.; Limberg, A.K.; Salib, C.G.; Bettencourt, J.W.; Trousdale, W.H.; Lewallen, E.A.; Reina, N.; Paradise, C.R.; Thaler, R.; Morrey, M.E.; et al. Molecular Pathology of Human Knee Arthrofibrosis Defined by RNA Sequencing. Genomics 2020, 112, 2703–2712. [Google Scholar] [CrossRef]
- Yamauchi, T.; Iwabu, M.; Okada-Iwabu, M.; Kadowaki, T. Adiponectin Receptors: A Review of Their Structure, Function and How They Work. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 15–23. [Google Scholar] [CrossRef]
- Fang, X.; Sweeney, G. Mechanisms Regulating Energy Metabolism by Adiponectin in Obesity and Diabetes. Biochem. Soc. Trans. 2006, 34, 798–801. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING Database in 2017: Quality-Controlled Protein–Protein Association Networks, Made Broadly Accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.F.; Gundersen, G.W.; Rahman, A.; Grimes, M.L.; Rikova, K.; Hornbeck, P.; Ma’Ayan, A. Cluster Grammer, a Web-Based Heatmap Visualization and Analysis Tool for High-Dimensional Biological Data. Sci. Data 2017, 4, 170151. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, E.T.; Gustafson, M.P.; Dudakovic, A.; Riester, S.M.; Garces, C.G.; Paradise, C.R.; Takai, H.; Karperien, M.; Cool, S.; Sampen, H.-J.I.; et al. Identification and Validation of Multiple Cell Surface Markers of Clinical-Grade Adipose-Derived Mesenchymal Stromal Cells as Novel Release Criteria for Good Manufacturing Practice-Compliant Production. Stem Cell Res. Ther. 2016, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudakovic, A.D.; Camilleri, E.; Riester, S.M.; Lewallen, E.A.; Kvasha, S.; Chen, X.; Radel, D.J.; Anderson, J.M.; Nair, A.A.; Evans, J.M.; et al. High-Resolution Molecular Validation of Self-Renewal and Spontaneous Differentiation in Clinical-Grade Adipose-Tissue Derived Human Mesenchymal Stem Cells. J. Cell. Biochem. 2014, 115, 1816–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galeano-Garces, C.; Camilleri, E.T.; Riester, S.M.; Dudakovic, A.; Larson, D.R.; Qu, W.; Smith, J.; Dietz, A.B.; Im, H.-J.; Krych, A.J.; et al. Molecular Validation of Chondrogenic Differentiation and Hypoxia Responsiveness of Platelet-Lysate Expanded Adipose Tissue–Derived Human Mesenchymal Stromal Cells. Cartilage 2016, 8, 283–299. [Google Scholar] [CrossRef] [Green Version]
- Mader, E.K.; Butler, G.; Dowdy, S.C.; Mariani, A.; Knutson, K.L.; Federspiel, M.J.; Russell, S.J.; Galanis, E.; Dietz, A.B.; Peng, K.W. Optimizing Patient Derived Mesenchymal Stem Cells as Virus Carriers for a Phase I Clinical Trial in Ovarian Cancer. J. Transl. Med. 2013, 11, 20. [Google Scholar] [CrossRef] [Green Version]
- Crespo-Diaz, R.; Behfar, A.; Butler, G.W.; Padley, D.J.; Sarr, M.G.; Bartunek, J.; Dietz, A.B.; Terzic, A. Platelet Lysate Consisting of a Natural Repair Proteome Supports Human Mesenchymal Stem Cell Proliferation and Chromosomal Stability. Cell Transplant. 2011, 20, 797–812. [Google Scholar] [CrossRef] [Green Version]
- Park, P.-H.; Sanz-Garcia, C.; Nagy, L.E. Adiponectin as an Anti-fibrotic and Anti-inflammatory Adipokine in the Liver. Curr. Pathobiol. Rep. 2015, 3, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulou, P.; Fotoulaki, M.; Manolitsas, A.; Pavlitou-Tsiontsi, E.; Tsitouridis, I.; Nousia-Arvanitakis, S. Adiponectin and Body Composition in Cystic Fibrosis. J. Cyst. Fibros. 2008, 7, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lin, S.-C.; Chen, G.; He, L.; Hu, Z.; Chan, L.; Trial, J.; Entman, M.L.; Wang, Y. Adiponectin Promotes Monocyte-to-Fibroblast Transition in Renal Fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1644–1659. [Google Scholar] [CrossRef] [Green Version]
- Marangoni, R.G.; Masui, Y.; Fang, F.; Korman, B.; Lord, G.; Lee, J.; Lakota, K.; Wei, J.; Scherer, P.E.; Otvos, L.; et al. Adiponectin Is an Endogenous Anti-Fibrotic Mediator and Therapeutic Target. Sci. Rep. 2017, 7, 4397. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in Fibrosis: Novel Roles and Mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtman, M.K.; Otero-Viñas, M.; Falanga, V. Transforming Growth Factor Beta (TGF-β) Isoforms in Wound Healing and Fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Scheja, L.; Heeren, J. The Endocrine Function of Adipose Tissues in Health and Cardiometabolic Disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Abdel, M.P.; Morrey, M.E.; Barlow, J.D.; Kreofsky, C.R.; An, K.-N.; Steinmann, S.P.; Morrey, B.F.; Sanchez-Sotelo, J. Myofibroblast Cells Are Preferentially Expressed Early in a Rabbit Model of Joint Contracture. J. Orthop. Res. 2012, 30, 713–719. [Google Scholar] [CrossRef]
- Abdel, M.P.; Morrey, M.E.; Grill, D.E.; Kolbert, C.P.; An, K.-N.; Steinmann, S.P.; Sanchez-Sotelo, J.; Morrey, B.F. Effects of Joint Contracture on the Contralateral Unoperated Limb in a Rabbit Knee Contracture Model: A Biomechanical and Genetic Study. J. Orthop. Res. 2012, 30, 1581–1585. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, K.A.; Zhang, M.; Hart, D.A. Myofibroblast Upregulators are Elevated in Joint Capsules in Posttraumatic Contractures. Clin. Orthop. Relat. Res. 2007, 456, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. Common and Unique Mechanisms Regulate Fibrosis in Various Fibroproliferative Diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Falke, L.L.; Gholizadeh, S.; Goldschmeding, R.; Kok, R.J.; Nguyen, T.Q. Diverse Origins of the Myofibroblast—Implications for Kidney Fibrosis. Nat. Rev. Nephrol. 2015, 11, 233–244. [Google Scholar] [CrossRef]
- Hecker, L.; Kato, K.; Shin, Y.-J.; Thannickal, V. Impaired Myofibroblast Dedifferentiation Contributes to Non-Resolving Fibrosis in Aging. Am. J. Respir. Cell Mol. Biol. 2020, 62, 633–644. [Google Scholar] [CrossRef]
- Kim, Y.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Yoon, H.E.; Shin, S.J.; Choi, B.S.; Kim, Y.-S.; Chang, Y.S.; Park, C.W. The Adiponectin Receptor Agonist AdipoRon Ameliorates Diabetic Nephropathy in a Model of Type 2 Diabetes. J. Am. Soc. Nephrol. 2018, 29, 1108–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ötvös, L. Potential Adiponectin Receptor Response Modifier Therapeutics. Front. Endocrinol. 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted Disruption of AdipoR1 and AdipoR2 Causes Abrogation of Adiponectin Binding and Metabolic Actions. Nat. Med. 2007, 13, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Żółkiewicz, J.; Stochmal, A.; Rudnicka, L. The Role of Adipokines in Systemic Sclerosis: A Missing Link? Arch. Dermatol. Res. 2019, 311, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Tontonoz, P.; Spiegelman, B.M. Fat and Beyond: The Diverse Biology of PPAR Gamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Baker, A.R.; Da Silva, N.F.; Quinn, D.W.; Harte, A.L.; Pagano, D.; Bonser, R.S.; Kumar, S.; McTernan, P.G. Human Epicardial Adipose Tissue Expresses a Pathogenic Profile of Adipocytokines in Patients with Cardiovascular Disease. Cardiovasc. Diabetol. 2006, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Yao, R.; Cao, Y.; He, Y.-R.; Lau, W.B.; Zeng, Z.; Liang, Z.-A. Adiponectin Attenuates Lung Fibroblasts Activation and Pulmonary Fibrosis Induced by Paraquat. PLoS ONE 2015, 10, e0125169. [Google Scholar] [CrossRef] [Green Version]
- Barlow, J.D.; Morrey, M.E.; Hartzler, R.U.; Arsoy, D.; Riester, S.; van Wijnen, A.J.; Morrey, B.F.; Sanchez-Sotelo, J.; Abdel, M.P. Effectiveness of rosiglitazone in reducing flexion contracture in a rabbit model of arthrofibrosis with surgical capsular release: A biomechanical, histological, and genetic analysis. Bone Joint Res. 2016, 5, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Arsoy, D.; Salib, C.G.; Trousdale, W.H.; Tibbo, M.E.; Limberg, A.K.; Viste, A.; Lewallen, E.A.; Reina, N.; Yaszemski, M.J.; Berry, D.J.; et al. Joint contracture is reduced by intra-articular implantation of rosiglitazone-loaded hydrogels in a rabbit model of arthrofibrosis. J. Orthop. Res. 2018, 36, 2949–2955. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; You, L.; Zhao, M. Rosiglitazone Attenuates Paraquat-Induced Lung Fibrosis in Rats in a PPAR Gamma-Dependent Manner. Eur. J. Pharmacol. 2019, 851, 133–143. [Google Scholar] [CrossRef]
- Zha, D.; Wu, X.; Gao, P. Adiponectin and Its Receptors in Diabetic Kidney Disease: Molecular Mechanisms and Clinical Potential. Endocrinology 2017, 158, 2022–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakota, K.; Wei, J.; Carns, M.A.; Hinchcliff, M.; Lee, J.; Whitfield, M.L.; Sodin-Semrl, S.; Varga, J. Levels of Adiponectin, a Marker for PPAR-Gamma Activity, Correlate With Skin Fibrosis in Systemic Sclerosis: Potential Utility as a Biomarker? Arthritis Res. Ther. 2012, 14, R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enomoto, N.; Oyama, Y.; Yasui, H.; Karayama, M.; Hozumi, H.; Suzuki, Y.; Kono, M.; Furuhashi, K.; Fujisawa, T.; Inui, N.; et al. Analysis of Serum Adiponectin and Leptin in Patients With Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Sci. Rep. 2019, 9, 10484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Wei, W.-Y.; Liao, H.-H.; Yang, Z.; Hu, C.; Wang, S.-S.; Deng, W.; Tang, Q. AdipoRon, an Adiponectin Receptor Agonist, Attenuates Cardiac Remodeling Induced by Pressure Overload. J. Mol. Med. 2018, 96, 1345–1357. [Google Scholar] [CrossRef]
- Sha, M.; Gao, Y.; Deng, C.; Wan, Y.; Zhuang, Y.; Hu, X.; Wang, Y. Therapeutic Effects of AdipoRon on Liver Inflammation and Fibrosis Induced by CCl4 in Mice. Int. Immunopharmacol. 2020, 79, 106157. [Google Scholar] [CrossRef]
- Yamashita, T.; Lakota, K.; Taniguchi, T.; Yoshizaki, A.; Sato, S.; Hong, W.; Zhou, X.; Sodin-Semrl, S.; Fang, F.; Asano, Y.; et al. An Orally-Active Adiponectin Receptor Agonist Mitigates Cutaneous Fibrosis, Inflammation and Microvascular Pathology in a Murine Model of Systemic Sclerosis. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayram, B.; Owen, A.R.; Dudakovic, A.; Dagneaux, L.; Turner, T.W.; Bettencourt, J.W.; Limberg, A.K.; Tibbo, M.E.; Morrey, M.E.; Sanchez-Sotelo, J.; et al. A Potential Theragnostic Regulatory Axis for Arthrofibrosis Involving Adiponectin (ADIPOQ) Receptor 1 and 2 (ADIPOR1 and ADIPOR2), TGFβ1, and Smooth Muscle α-Actin (ACTA2). J. Clin. Med. 2020, 9, 3690. https://doi.org/10.3390/jcm9113690
Bayram B, Owen AR, Dudakovic A, Dagneaux L, Turner TW, Bettencourt JW, Limberg AK, Tibbo ME, Morrey ME, Sanchez-Sotelo J, et al. A Potential Theragnostic Regulatory Axis for Arthrofibrosis Involving Adiponectin (ADIPOQ) Receptor 1 and 2 (ADIPOR1 and ADIPOR2), TGFβ1, and Smooth Muscle α-Actin (ACTA2). Journal of Clinical Medicine. 2020; 9(11):3690. https://doi.org/10.3390/jcm9113690
Chicago/Turabian StyleBayram, Banu, Aaron R. Owen, Amel Dudakovic, Louis Dagneaux, Travis W. Turner, Jacob W. Bettencourt, Afton K. Limberg, Meagan E. Tibbo, Mark E. Morrey, Joaquin Sanchez-Sotelo, and et al. 2020. "A Potential Theragnostic Regulatory Axis for Arthrofibrosis Involving Adiponectin (ADIPOQ) Receptor 1 and 2 (ADIPOR1 and ADIPOR2), TGFβ1, and Smooth Muscle α-Actin (ACTA2)" Journal of Clinical Medicine 9, no. 11: 3690. https://doi.org/10.3390/jcm9113690