A Holistic Perspective: Exosomes Shuttle between Nerves and Immune Cells in the Tumor Microenvironment

 , , ,
, , ,  ,
,  ,
,
Abstract
:1. Introduction
1.1. A New Perspective on the Tumor Microenvironment
1.2. Exosomes and microRNAs Represent a Systemic, and Dynamic Communication Channel between the Components of the Tumor Microenvironment
2. Communication between Nerve Cells and Cancer Cells
2.1. Nerves within the Tumor Microenvironment Can Either Promote or Inhibit Tumorigenesis
2.2. Exosomes Are Key Components of the Communication between Nerve and Cancer Cells
3. Communication between Immune Cells and Cancer Cells
3.1. The Immunological Landscape of Tumors Dictates the Outcome of Cancer Patients
3.2. Communication between Tumor Cells and Adaptive Immunity Is both Direct and Exosome-Mediated
3.3. Exosomes Are Key Components of the Communication between Innate Immunity and Cancer Cells
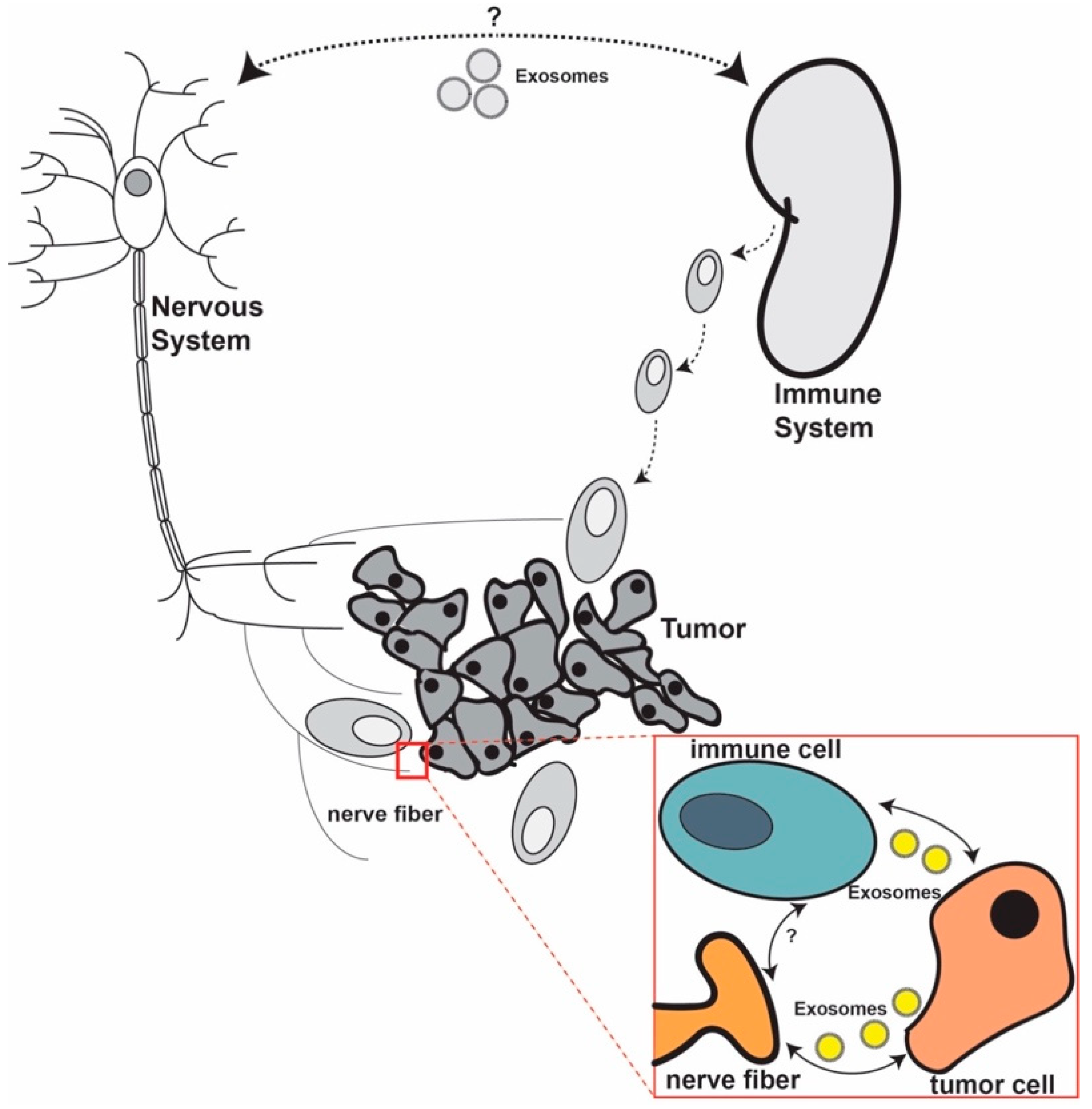
4. Is There any Communication between Nerves and Immune Cells in the Tumor Microenvironment?
5. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Dagenais, G.R.; Leong, D.P.; Rangarajan, S.; Lanas, F.; Lopez-Jaramillo, P.; Gupta, R.; Diaz, R.; Avezum, A.; Oliveira, G.B.F.; Wielgosz, A.; et al. Variations in common diseases, hospital admissions, and deaths in middle-aged adults in 21 countries from five continents (PURE): A prospective cohort study. Lancet 2020, 395, 785–794. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Amit, M.; Na’ara, S.; Gil, Z. Mechanisms of cancer dissemination along nerves. Nat. Rev. Cancer 2016, 16, 399–408. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Lewis, C.E.; Naldini, L.; Brown, J.M.; Ferrara, N.; De Palma, M. Elusive identities and overlapping phenotypes of proangiogenic myeloid cells in tumors. Am. J. Pathol. 2010, 176, 1564–1576. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Demir, I.E.; Friess, H.; Ceyhan, G.O. Nerve-cancer interactions in the stromal biology of pancreatic cancer. Front. Physiol. 2012, 3, 97. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Lyden, D.; Wang, T.C. The evolution of the cancer niche during multistage carcinogenesis. Nat. Rev. Cancer 2013, 13, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Litwack, G. Hormones and transport systems. Preface. Vitam. Horm. 2015, 98, xvii–xviii. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Harding, C.; Heuser, J.; Stahl, P. Endocytosis and intracellular processing of transferrin and colloidal gold-transferrin in rat reticulocytes: Demonstration of a pathway for receptor shedding. Eur. J. Cell Biol. 1984, 35, 256–263. [Google Scholar]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the isolation, characterization, biological function, and multifarious therapeutic approaches of exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Wubbolts, R.; Leckie, R.S.; Veenhuizen, P.T.; Schwarzmann, G.; Mobius, W.; Hoernschemeyer, J.; Slot, J.W.; Geuze, H.J.; Stoorvogel, W. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J. Biol. Chem. 2003, 278, 10963–10972. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Jenjaroenpun, P.; Kremenska, Y.; Nair, V.M.; Kremenskoy, M.; Joseph, B.; Kurochkin, I.V. Characterization of RNA in exosomes secreted by human breast cancer cell lines using next-generation sequencing. PeerJ 2013, 1, e201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, M.A.; Bueso-Ramos, C.; Ferdin, J.; Lopez-Berestein, G.; Sood, A.K.; Calin, G.A. MicroRNAs in body fluids--the mix of hormones and biomarkers. Nat. Rev. Clin. Oncol. 2011, 8, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Pardini, B.; Calin, G.A. MicroRNAs and long non-coding RNAs and their hormone-like activities in cancer. Cancers 2019, 11, 378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Los Santos, M.C.; Dragomir, M.P.; Calin, G.A. The role of exosomal long non-coding RNAs in cancer drug resistance. Cancer Drug Resist. 2019, 2, 1178–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragomir, M.; Chen, B.; Calin, G.A. Exosomal lncRNAs as new players in cell-to-cell communication. Transl. Cancer Res. 2018, 7, S243–S252. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Vasilescu, C.; Tanase, M.; Giza, D.; Procopiuc, L.; Dragomir, M.P.; Calin, A.G.A. How does a tumor get its shape? MicroRNAs act as morphogens at the cancer invasion front. Noncoding RNA 2020, 6, 23. [Google Scholar] [CrossRef]
- Nishida-Aoki, N.; Ochiya, T. Interactions between cancer cells and normal cells via miRNAs in extracellular vesicles. Cell. Mol. Life Sci. 2015, 72, 1849–1861. [Google Scholar] [CrossRef] [Green Version]
- Tkach, M.; Thery, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Josson, S.; Gururajan, M.; Sung, S.Y.; Hu, P.; Shao, C.; Zhau, H.E.; Liu, C.; Lichterman, J.; Duan, P.; Li, Q.; et al. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 2015, 34, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Wehrwein, E.A.; Orer, H.S.; Barman, S.M. Overview of the anatomy, physiology, and pharmacology of the autonomic nervous system. Compr. Physiol. 2016, 6, 1239–1278. [Google Scholar] [CrossRef] [PubMed]
- Saloman, J.L.; Albers, K.M.; Li, D.; Hartman, D.J.; Crawford, H.C.; Muha, E.A.; Rhim, A.D.; Davis, B.M. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 3078–3083. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Smith, M.; Lutgendorf, S.K.; Sood, A.K. Impact of stress on cancer metastasis. Future Oncol. 2010, 6, 1863–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanczkowski, W.; Sue, M.; Bornstein, S.R. Adrenal gland microenvironment and its involvement in the regulation of stress-induced hormone secretion during sepsis. Front. Endocrinol. (Lausanne) 2016, 7, 156. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor neurobiology and the war of nerves in cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Liebig, C.; Ayala, G.; Wilks, J.A.; Berger, D.H.; Albo, D. Perineural invasion in cancer: A review of the literature. Cancer 2009, 115, 3379–3391. [Google Scholar] [CrossRef]
- Aurello, P.; Berardi, G.; Tierno, S.M.; Rampioni Vinciguerra, G.L.; Socciarelli, F.; Laracca, G.G.; Giulitti, D.; Pilozzi, E.; Ramacciato, G. Influence of perineural invasion in predicting overall survival and disease-free survival in patients With locally advanced gastric cancer. Am. J. Surg. 2017, 213, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Skancke, M.; Arnott, S.M.; Amdur, R.L.; Siegel, R.S.; Obias, V.J.; Umapathi, B.A. Lymphovascular invasion and perineural invasion negatively impact overall survival for stage II adenocarcinoma of the colon. Dis. Colon Rectum 2019, 62, 181–188. [Google Scholar] [CrossRef]
- Ayala, G.E.; Dai, H.; Ittmann, M.; Li, R.; Powell, M.; Frolov, A.; Wheeler, T.M.; Thompson, T.C.; Rowley, D. Growth and survival mechanisms associated with perineural invasion in prostate cancer. Cancer Res. 2004, 64, 6082–6090. [Google Scholar] [CrossRef] [Green Version]
- Sheng, L.; Ji, Y.; Du, X. Perineural invasion correlates with postoperative distant metastasis and poor overall survival in patients with PT1-3N0M0 esophageal squamous cell carcinoma. Onco Targets Ther. 2015, 8, 3153–3157. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, S.; Matsuda, K.; Watanabe, T.; Mitani, Y.; Ieda, J.; Iwamoto, H.; Hotta, T.; Takifuji, K.; Yamaue, H. Perineural invasion is associated with poor survival after preoperative chemoradiation therapy for advanced lower rectal cancer. Dig. Surg. 2017, 34, 387–393. [Google Scholar] [CrossRef]
- Murakami, Y.; Uemura, K.; Sudo, T.; Hashimoto, Y.; Kondo, N.; Nakagawa, N.; Muto, T.; Sasaki, H.; Urabe, K.; Sueda, T. Perineural invasion in extrahepatic cholangiocarcinoma: Prognostic impact and treatment strategies. J. Gastrointest. Surg. 2013, 17, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Bakst, R.L.; Glastonbury, C.M.; Parvathaneni, U.; Katabi, N.; Hu, K.S.; Yom, S.S. Perineural invasion and perineural tumor spread in head and neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 1109–1124. [Google Scholar] [CrossRef] [Green Version]
- Partecke, L.I.; Kading, A.; Trung, D.N.; Diedrich, S.; Sendler, M.; Weiss, F.; Kuhn, J.P.; Mayerle, J.; Beyer, K.; von Bernstorff, W.; et al. Subdiaphragmatic vagotomy promotes tumor growth and reduces survival via TNFalpha in a murine pancreatic cancer model. Oncotarget 2017, 8, 22501–22512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renz, B.W.; Tanaka, T.; Sunagawa, M.; Takahashi, R.; Jiang, Z.; Macchini, M.; Dantes, Z.; Valenti, G.; White, R.A.; Middelhoff, M.A.; et al. Cholinergic signaling via muscarinic receptors directly and indirectly suppresses pancreatic tumorigenesis and cancer stemness. Cancer Discov. 2018, 8, 1458–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.M.; Hayakawa, Y.; Kodama, Y.; Muthupalani, S.; Westphalen, C.B.; Andersen, G.T.; Flatberg, A.; Johannessen, H.; Friedman, R.A.; Renz, B.W.; et al. Denervation suppresses gastric tumorigenesis. Sci. Transl. Med. 2014, 6, 250ra115. [Google Scholar] [CrossRef] [Green Version]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Fu, Y.Y.; Grimont, A.; Ketcham, M.; Lafaro, K.; Saglimbeni, J.A.; Askan, G.; Bailey, J.M.; Melchor, J.P.; Zhong, Y.; et al. PanIN neuroendocrine cells promote tumorigenesis via neuronal cross-talk. Cancer Res. 2017, 77, 1868–1879. [Google Scholar] [CrossRef]
- Bai, H.; Li, H.; Zhang, W.; Matkowskyj, K.A.; Liao, J.; Srivastava, S.K.; Yang, G.Y. Inhibition of chronic pancreatitis and pancreatic intraepithelial neoplasia (PanIN) by capsaicin in LSL-KrasG12D/Pdx1-Cre mice. Carcinogenesis 2011, 32, 1689–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, M.B.; Le, X.; Heymach, J.V. Beta-adrenergic signaling in lung cancer: A potential role for beta-blockers. J. Neuroimmune Pharmacol. 2020, 15, 27–36. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Sun, H.; Diao, L.; Tong, P.; Liu, D.; Li, L.; Fan, Y.; Poteete, A.; Lim, S.O.; Howells, K.; et al. Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with beta-blockers. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Richbart, S.D.; Merritt, J.C.; Brown, K.C.; Nolan, N.A.; Akers, A.T.; Lau, J.K.; Robateau, Z.R.; Miles, S.L.; Dasgupta, P. Acetylcholine signaling system in progression of lung cancers. Pharmacol. Ther. 2019, 194, 222–254. [Google Scholar] [CrossRef]
- Shao, J.X.; Wang, B.; Yao, Y.N.; Pan, Z.J.; Shen, Q.; Zhou, J.Y. Autonomic nervous infiltration positively correlates with pathological risk grading and poor prognosis in patients with lung adenocarcinoma. Thorac. Cancer 2016, 7, 588–598. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; He, Z.; Yin, K.; Li, B.; Zhang, L.; Xu, Z. Chronic stress promotes gastric cancer progression and metastasis: An essential role for ADRB2. Cell Death Dis. 2019, 10, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhi, X.; Li, B.; Li, Z.; Zhang, J.; Yu, J.; Zhang, L.; Xu, Z. Adrenergic modulation of AMPKdependent autophagy by chronic stress enhances cell proliferation and survival in gastric cancer. Int. J. Oncol. 2019, 54, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Yang, Z.; Hu, M.; Liu, D.; Hu, Y.; Qian, L.; Zhang, W.; Chen, H.; Guo, L.; Yu, M.; et al. Catecholamine-Induced beta2-adrenergic receptor activation mediates desensitization of gastric cancer cells to trastuzumab by upregulating MUC4 expression. J. Immunol. 2013, 190, 5600–5608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, Y.; Sakitani, K.; Konishi, M.; Asfaha, S.; Niikura, R.; Tomita, H.; Renz, B.W.; Tailor, Y.; Macchini, M.; Middelhoff, M.; et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell 2017, 31, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.C.; Li, J.M.; Lee, K.F.; Huang, Y.C.; Wang, K.C.; Lai, H.C.; Cheng, C.C.; Kuo, Y.H.; Shi, C.S. Selective beta2-AR blockage suppresses colorectal cancer growth through regulation of EGFR-Akt/ERK1/2 signaling, G1-phase arrest, and apoptosis. J. Cell Physiol. 2016, 231, 459–472. [Google Scholar] [CrossRef]
- Han, J.; Jiang, Q.; Ma, R.; Zhang, H.; Tong, D.; Tang, K.; Wang, X.; Ni, L.; Miao, J.; Duan, B.; et al. Norepinephrine-CREB1-miR-373 axis promotes progression of colon cancer. Mol. Oncol. 2020, 14, 1059–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Raufman, J.P. Muscarinic receptor signaling and colon cancer progression. J. Cancer Metastasis Treat. 2016, 2, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Chen, Y.; Cheng, K.; Compadre, C.; Compadre, L.; Zimniak, P. Selective interaction of bile acids with muscarinic receptors: A case of molecular mimicry. Eur. J. Pharmacol. 2002, 457, 77–84. [Google Scholar] [CrossRef]
- Xiao, M.B.; Jin, D.D.; Jiao, Y.J.; Ni, W.K.; Liu, J.X.; Qu, L.S.; Lu, C.H.; Ni, R.Z.; Jiang, F.; Chen, W.C. beta2-AR regulates the expression of AKR1B1 in human pancreatic cancer cells and promotes their proliferation via the ERK1/2 pathway. Mol. Biol. Rep. 2018, 45, 1863–1871. [Google Scholar] [CrossRef]
- Zhang, P.; He, X.; Tan, J.; Zhou, X.; Zou, L. beta-arrestin2 mediates beta-2 adrenergic receptor signaling inducing prostate cancer cell progression. Oncol. Rep. 2011, 26, 1471–1477. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.Q.; Xu, L.H.; Zhang, M.; Xu, C. Muscarinic acetylcholine receptor M1 mediates prostate cancer cell migration and invasion through hedgehog signaling. Asian J. Androl. 2018, 20, 608–614. [Google Scholar] [CrossRef]
- Guo, L.; Liu, Y.; Ding, Z.; Sun, W.; Yuan, M. Signal transduction by M3 muscarinic acetylcholine receptor in prostate cancer. Oncol. Lett. 2016, 11, 385–392. [Google Scholar] [CrossRef]
- Goto, Y.; Ando, T.; Izumi, H.; Feng, X.; Arang, N.; Gilardi, M.; Wang, Z.; Ando, K.; Gutkind, J.S. Muscarinic receptors promote castration-resistant growth of prostate cancer through a FAK-YAP signaling axis. Oncogene 2020, 39, 4014–4027. [Google Scholar] [CrossRef]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; Arevalo, J.M.; Morizono, K.; Karanikolas, B.D.; Wu, L.; et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–7052. [Google Scholar] [CrossRef] [Green Version]
- Erin, N.; Zhao, W.; Bylander, J.; Chase, G.; Clawson, G. Capsaicin-induced inactivation of sensory neurons promotes a more aggressive gene expression phenotype in breast cancer cells. Breast Cancer Res. Treat. 2006, 99, 351–364. [Google Scholar] [CrossRef]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Duhrsen, U.; Frenette, P.S. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, D.; Scheiermann, C.; Chow, A.; Kunisaki, Y.; Bruns, I.; Barrick, C.; Tessarollo, L.; Frenette, P.S. Chemotherapy-induced bone marrow nerve injury impairs hematopoietic regeneration. Nat. Med. 2013, 19, 695–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, B.; Cabadak, H.; Goren, M.Z. Investigation of the roles of non-neuronal acetylcholine in chronic myeloid leukemic cells and their erythroid or megakaryocytic differentiated lines. Anticancer Agents Med. Chem. 2018, 18, 1440–1447. [Google Scholar] [CrossRef]
- Cabadak, H.; Aydin, B.; Kan, B. Regulation of M2, M3, and M4 muscarinic receptor expression in K562 chronic myelogenous leukemic cells by carbachol. J. Recept. Signal Transduct. Res. 2011, 31, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Pufall, M.A. Glucocorticoids and Cancer. Adv. Exp. Med. Biol. 2015, 872, 315–333. [Google Scholar] [CrossRef] [Green Version]
- Mattingly, A.; Finley, J.K.; Knox, S.M. Salivary gland development and disease. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 573–590. [Google Scholar] [CrossRef]
- Aloe, L.; Rocco, M.L.; Balzamino, B.O.; Micera, A. Nerve growth factor: Role in growth, differentiation and controlling cancer cell development. J. Exp. Clin. Cancer Res. 2016, 35, 116. [Google Scholar] [CrossRef] [Green Version]
- Fielder, G.C.; Yang, T.W.; Razdan, M.; Li, Y.; Lu, J.; Perry, J.K.; Lobie, P.E.; Liu, D.X. The GDNF family: A role in cancer? Neoplasia 2018, 20, 99–117. [Google Scholar] [CrossRef]
- Knox, S.M.; Lombaert, I.M.; Haddox, C.L.; Abrams, S.R.; Cotrim, A.; Wilson, A.J.; Hoffman, M.P. Parasympathetic stimulation improves epithelial organ regeneration. Nat. Commun. 2013, 4, 1494. [Google Scholar] [CrossRef] [Green Version]
- Emmerson, E.; May, A.J.; Nathan, S.; Cruz-Pacheco, N.; Lizama, C.O.; Maliskova, L.; Zovein, A.C.; Shen, Y.; Muench, M.O.; Knox, S.M. SOX2 regulates acinar cell development in the salivary gland. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Chang, W.W. Effect of neonatal sympathectomy on the postnatal differentiation of the submandibular gland of the rat. Cell Tissue Res. 1977, 180, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.D.; Carlsoo, B.; Danielsson, A.; Hellstrom, S.; Henriksson, R. Trophic effect of the sympathetic nervous system on the early development of the rat parotid gland: A quantitative ultrastructural study. Anat. Rec. 1981, 201, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, E.F.; Bothwell, M. Spatiotemporal patterns of expression of NGF and the low-affinity NGF receptor in rat embryos suggest functional roles in tissue morphogenesis and myogenesis. J. Neurosci. 1992, 12, 930–945. [Google Scholar] [CrossRef] [PubMed]
- Borden, P.; Houtz, J.; Leach, S.D.; Kuruvilla, R. Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep. 2013, 4, 287–301. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Wang, K.; Demir, I.E.; D’Haese, J.G.; Tieftrunk, E.; Kujundzic, K.; Schorn, S.; Xing, B.; Kehl, T.; Friess, H.; Ceyhan, G.O. The neurotrophic factor neurturin contributes toward an aggressive cancer cell phenotype, neuropathic pain and neuronal plasticity in pancreatic cancer. Carcinogenesis 2014, 35, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Ceyhan, G.O.; Schafer, K.H.; Kerscher, A.G.; Rauch, U.; Demir, I.E.; Kadihasanoglu, M.; Bohm, C.; Muller, M.W.; Buchler, M.W.; Giese, N.A.; et al. Nerve growth factor and artemin are paracrine mediators of pancreatic neuropathy in pancreatic adenocarcinoma. Ann. Surg. 2010, 251, 923–931. [Google Scholar] [CrossRef]
- Pundavela, J.; Demont, Y.; Jobling, P.; Lincz, L.F.; Roselli, S.; Thorne, R.F.; Bond, D.; Bradshaw, R.A.; Walker, M.M.; Hondermarck, H. ProNGF correlates with Gleason score and is a potential driver of nerve infiltration in prostate cancer. Am. J. Pathol. 2014, 184, 3156–3162. [Google Scholar] [CrossRef]
- Dobrenis, K.; Gauthier, L.R.; Barroca, V.; Magnon, C. Granulocyte colony-stimulating factor off-target effect on nerve outgrowth promotes prostate cancer development. Int. J. Cancer 2015, 136, 982–988. [Google Scholar] [CrossRef]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.P.; Firlej, V.; Allory, Y.; Romeo, P.H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019, 569, 672–678. [Google Scholar] [CrossRef]
- Madeo, M.; Colbert, P.L.; Vermeer, D.W.; Lucido, C.T.; Cain, J.T.; Vichaya, E.G.; Grossberg, A.J.; Muirhead, D.; Rickel, A.P.; Hong, Z.; et al. Cancer exosomes induce tumor innervation. Nat. Commun. 2018, 9, 4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucido, C.T.; Wynja, E.; Madeo, M.; Williamson, C.S.; Schwartz, L.E.; Imblum, B.A.; Drapkin, R.; Vermeer, P.D. Innervation of cervical carcinoma is mediated by cancer-derived exosomes. Gynecol. Oncol. 2019, 154, 228–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ching, R.C.; Wiberg, M.; Kingham, P.J. Schwann cell-like differentiated adipose stem cells promote neurite outgrowth via secreted exosomes and RNA transfer. Stem Cell Res. Ther. 2018, 9, 266. [Google Scholar] [CrossRef]
- Amit, M.; Takahashi, H.; Dragomir, M.P.; Lindemann, A.; Gleber-Netto, F.O.; Pickering, C.R.; Anfossi, S.; Osman, A.A.; Cai, Y.; Wang, R.; et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 2020, 578, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Soyano, A.E.; Dholaria, B.; Knutson, K.L.; Lou, Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 8. [Google Scholar] [CrossRef]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of melanoma: Facts and hopes. Clin. Cancer Res. 2019, 25, 5191–5201. [Google Scholar] [CrossRef] [Green Version]
- Massarelli, E.; Papadimitrakopoulou, V.; Welsh, J.; Tang, C.; Tsao, A.S. Immunotherapy in lung cancer. Transl. Lung Cancer Res. 2014, 3, 53–63. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Restifo, N.P.; Antony, P.A.; Finkelstein, S.E.; Leitner, W.W.; Surman, D.P.; Theoret, M.R.; Touloukian, C.E. Assumptions of the tumor ‘escape’ hypothesis. Semin. Cancer Biol. 2002, 12, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccin. Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Anassi, E.; Ndefo, U.A. Sipuleucel-T (provenge) injection: The first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. Pharm. Ther. 2011, 36, 197–202. [Google Scholar]
- Pehlivan, K.C.; Duncan, B.B.; Lee, D.W. CAR-T cell therapy for acute lymphoblastic leukemia: Transforming the treatment of relapsed and refractory disease. Curr. Hematol. Malig. Rep. 2018, 13, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraille, E.; Leo, O. Revisiting the Th1/Th2 paradigm. Scand. J. Immunol. 1998, 47, 1–9. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Walunas, T.L.; Bluestone, J.A. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 1996, 14, 233–258. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Kane, L.P.; Andres, P.G.; Howland, K.C.; Abbas, A.K.; Weiss, A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not TH2 cytokines. Nat. Immunol. 2001, 2, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Pages, F.; Ragueneau, M.; Rottapel, R.; Truneh, A.; Nunes, J.; Imbert, J.; Olive, D. Binding of phosphatidylinositol-3-OH kinase to CD28 is required for T-cell signalling. Nature 1994, 369, 327–329. [Google Scholar] [CrossRef]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Maybruck, B.T.; Pfannenstiel, L.W.; Diaz-Montero, M.; Gastman, B.R. Tumor-derived exosomes induce CD8(+) T cell suppressors. J. Immunother. Cancer 2017, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Bland, C.L.; Byrne-Hoffman, C.N.; Fernandez, A.; Rellick, S.L.; Deng, W.; Klinke, D.J., 2nd. Exosomes derived from B16F0 melanoma cells alter the transcriptome of cytotoxic T cells that impacts mitochondrial respiration. FEBS J. 2018, 285, 1033–1050. [Google Scholar] [CrossRef] [Green Version]
- Olingy, C.E.; Dinh, H.Q.; Hedrick, C.C. Monocyte heterogeneity and functions in cancer. J. Leukoc. Biol. 2019, 106, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.N.; Cekic, C.; Sag, D.; Tacke, R.; Thomas, G.D.; Nowyhed, H.; Herrley, E.; Rasquinha, N.; McArdle, S.; Wu, R.; et al. Patrolling monocytes control tumor metastasis to the lung. Science 2015, 350, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Zhao, X.; Qu, J.; Sun, Y.; Wang, J.; Liu, X.; Wang, F.; Zhang, H.; Wang, W.; Ma, X.; Gao, X.; et al. Prognostic significance of tumor-associated macrophages in breast cancer: A meta-analysis of the literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.W.; Joyce, J.A. Alternative activation of tumor-associated macrophages by IL-4: Priming for protumoral functions. Cell Cycle 2010, 9, 4824–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Turkowski, K.; Mora, J.; Brune, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plebanek, M.P.; Angeloni, N.L.; Vinokour, E.; Li, J.; Henkin, A.; Martinez-Marin, D.; Filleur, S.; Bhowmick, R.; Henkin, J.; Miller, S.D.; et al. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat. Commun. 2017, 8, 1319. [Google Scholar] [CrossRef]
- Kanlikilicer, P.; Bayraktar, R.; Denizli, M.; Rashed, M.H.; Ivan, C.; Aslan, B.; Mitra, R.; Karagoz, K.; Bayraktar, E.; Zhang, X.; et al. Exosomal miRNA confers chemo resistance via targeting Cav1/p-gp/M2-type macrophage axis in ovarian cancer. EBioMedicine 2018, 38, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef] [Green Version]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Fridman, E.; Yaari, Z.; Milman, N.; Schroeder, A.; Ben David, G.; Shlomi, T.; Gil, Z. Transfer of miRNA in macrophage-derived exosomes induces drug resistance in pancreatic adenocarcinoma. Cancer Res. 2018, 78, 5287–5299. [Google Scholar] [CrossRef] [Green Version]
- Challagundla, K.B.; Wise, P.M.; Neviani, P.; Chava, H.; Murtadha, M.; Xu, T.; Kennedy, R.; Ivan, C.; Zhang, X.; Vannini, I.; et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]
- Frank, A.C.; Ebersberger, S.; Fink, A.F.; Lampe, S.; Weigert, A.; Schmid, T.; Ebersberger, I.; Syed, S.N.; Brune, B. Apoptotic tumor cell-derived microRNA-375 uses CD36 to alter the tumor-associated macrophage phenotype. Nat. Commun. 2019, 10, 1135. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ying, X.; Wang, X.; Wu, X.; Zhu, Q.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol. Rep. 2017, 38, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Qiu, W.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Xue, H.; Li, G. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten Pathways. Oncogene 2018, 37, 4239–4259. [Google Scholar] [CrossRef]
- Guo, X.; Qiu, W.; Wang, J.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Guo, Q.; et al. Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int. J. Cancer 2019, 144, 3111–3126. [Google Scholar] [CrossRef]
- Langers, I.; Renoux, V.M.; Thiry, M.; Delvenne, P.; Jacobs, N. Natural killer cells: Role in local tumor growth and metastasis. Biologics 2012, 6, 73–82. [Google Scholar] [CrossRef]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Maurer, S.; Kropp, K.N.; Klein, G.; Steinle, A.; Haen, S.P.; Walz, J.S.; Hinterleitner, C.; Marklin, M.; Kopp, H.G.; Salih, H.R. Platelet-mediated shedding of NKG2D ligands impairs NK cell immune-surveillance of tumor cells. Oncoimmunology 2018, 7, e1364827. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Wise, P.M.; Murtadha, M.; Liu, C.W.; Wu, C.H.; Jong, A.Y.; Seeger, R.C.; Fabbri, M. Natural killer-derived exosomal miR-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. 2019, 79, 1151–1164. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Conforti, R.; Viaud, S.; Spatz, A.; Zitvogel, L. The Janus face of dendritic cells in cancer. Oncogene 2008, 27, 5920–5931. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Pedroza-Gonzalez, A.; Gallegos, M.; Tindle, S.; Burton, E.C.; Su, D.; Marches, F.; Banchereau, J.; Palucka, A.K. Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J. Exp. Med. 2007, 204, 1037–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Chen, J.; Zhou, L.; Chen, W.; Ding, G.; Cao, L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol. 2014, 292, 65–69. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Arnal-Estape, A.; Maryanovich, M.; Nakahara, F.; Cruz, C.D.; Finley, L.W.S.; Frenette, P.S. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017, 358, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Mo, R.J.; Han, Z.D.; Liang, Y.K.; Ye, J.H.; Wu, S.L.; Lin, S.X.; Zhang, Y.Q.; Song, S.D.; Jiang, F.N.; Zhong, W.D.; et al. Expression of PD-L1 in tumor-associated nerves correlates with reduced CD8(+) tumor-associated lymphocytes and poor prognosis in prostate cancer. Int. J. Cancer 2019, 144, 3099–3110. [Google Scholar] [CrossRef]
- Cavel, O.; Shomron, O.; Shabtay, A.; Vital, J.; Trejo-Leider, L.; Weizman, N.; Krelin, Y.; Fong, Y.; Wong, R.J.; Amit, M.; et al. Endoneurial macrophages induce perineural invasion of pancreatic cancer cells by secretion of GDNF and activation of RET tyrosine kinase receptor. Cancer Res. 2012, 72, 5733–5743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.W.; Tao, L.Y.; Jiang, Y.S.; Yang, J.Y.; Huo, Y.M.; Liu, D.J.; Li, J.; Fu, X.L.; He, R.; Lin, C.; et al. Perineural invasion reprograms the immune microenvironment through cholinergic signaling in pancreatic ductal adenocarcinoma. Cancer Res. 2020, 80, 1991–2003. [Google Scholar] [CrossRef] [Green Version]
- Amit, M.; Na’ara, S.; Leider-Trejo, L.; Binenbaum, Y.; Kulish, N.; Fridman, E.; Shabtai-Orbach, A.; Wong, R.J.; Gil, Z. Upregulation of RET induces perineurial invasion of pancreatic adenocarcinoma. Oncogene 2017, 36, 3232–3239. [Google Scholar] [CrossRef]
- Simeoli, R.; Montague, K.; Jones, H.R.; Castaldi, L.; Chambers, D.; Kelleher, J.H.; Vacca, V.; Pitcher, T.; Grist, J.; Al-Ahdal, H.; et al. Exosomal cargo including microRNA regulates sensory neuron to macrophage communication after nerve trauma. Nat. Commun. 2017, 8, 1778. [Google Scholar] [CrossRef] [Green Version]
- Frick, L.R.; Arcos, M.L.; Rapanelli, M.; Zappia, M.P.; Brocco, M.; Mongini, C.; Genaro, A.M.; Cremaschi, G.A. Chronic restraint stress impairs T-cell immunity and promotes tumor progression in mice. Stress 2009, 12, 134–143. [Google Scholar] [CrossRef]
- Estrada, L.D.; Agac, D.; Farrar, J.D. Sympathetic neural signaling via the beta2-adrenergic receptor suppresses T-cell receptor-mediated human and mouse CD8(+) T-cell effector function. Eur. J. Immunol. 2016, 46, 1948–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daher, C.; Vimeux, L.; Stoeva, R.; Peranzoni, E.; Bismuth, G.; Wieduwild, E.; Lucas, B.; Donnadieu, E.; Bercovici, N.; Trautmann, A.; et al. Blockade of beta-adrenergic receptors improves CD8(+) T-cell priming and cancer vaccine efficacy. Cancer Immunol. Res. 2019, 7, 1849–1863. [Google Scholar] [CrossRef]
- Mohammadpour, H.; MacDonald, C.R.; Qiao, G.; Chen, M.; Dong, B.; Hylander, B.L.; McCarthy, P.L.; Abrams, S.I.; Repasky, E.A. beta2 adrenergic receptor-mediated signaling regulates the immunosuppressive potential of myeloid-derived suppressor cells. J. Clin. Investig. 2019, 129, 5537–5552. [Google Scholar] [CrossRef] [PubMed]
- Araujo, L.P.; Maricato, J.T.; Guereschi, M.G.; Takenaka, M.C.; Nascimento, V.M.; de Melo, F.M.; Quintana, F.J.; Brum, P.C.; Basso, A.S. The sympathetic nervous system mitigates CNS autoimmunity via beta2-adrenergic receptor signaling in immune cells. Cell Rep. 2019, 28, 3120–3130.e3125. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Tang, X.Y.; Li, Y.X.; Zhao, D.D.; Cao, Q.H.; Wu, H.X.; Yang, H.B.; Hao, K.; Yang, Y. Depression-Induced Neuropeptide Y Secretion promotes prostate cancer growth by recruiting myeloid cells. Clin. Cancer Res. 2019, 25, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Chida, Y.; Hamer, M.; Wardle, J.; Steptoe, A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat. Clin. Pract. Oncol. 2008, 5, 466–475. [Google Scholar] [CrossRef]
- Dragomir, M.; Chen, B.; Fu, X.; Calin, G.A. Key questions about the checkpoint blockade-are microRNAs an answer? Cancer Biol. Med. 2018, 15, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Bapat, A.A.; Munoz, R.M.; Von Hoff, D.D.; Han, H. Blocking nerve growth factor signaling reduces the neural invasion potential of pancreatic cancer cells. PLoS ONE 2016, 11, e0165586. [Google Scholar] [CrossRef] [Green Version]
- Kokolus, K.M.; Zhang, Y.; Sivik, J.M.; Schmeck, C.; Zhu, J.; Repasky, E.A.; Drabick, J.J.; Schell, T.D. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology 2018, 7, e1405205. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Receptor | Subtype | Effector | Ligand |
|---|---|---|---|
| Adrenergic receptors (AR) | alpha 1 | Gq: ↑PLC, ↑PIP3, ↑DAG, ↑Ca2+ | adrenaline, noradrenaline |
| alpha 2 | Gi: ↓AC, ↓cAMP | adrenaline, noradrenaline | |
| beta 1 | Gs: ↑AC, ↑cAMP | adrenaline, noradrenaline | |
| beta 2 | Gs: ↑AC, ↑cAMP | adrenaline, noradrenaline | |
| beta 3 | Gs: ↑AC, ↑cAMP | adrenaline, noradrenaline | |
| Cholinergic receptors | Nicotinic | ↑Na+, ↑K+ | acetylcholine, nicotine |
| Muscarinic M1 | Gq: ↑ PLC, ↑PIP3, ↑DAG, ↑Ca2+ | acetylcholine, muscarine | |
| Muscarinic M2 | Gi: ↓AC, ↓cAMP | acetylcholine, muscarine | |
| Muscarinic M3 | Gq: ↑ PLC, ↑PIP3, ↑DAG, ↑Ca2+ | acetylcholine, muscarine | |
| Muscarinic M4 | Gi: ↓AC, ↓cAMP | acetylcholine, muscarine | |
| Muscarinic M5 | Gq: ↑ PLC, ↑PIP3, ↑DAG, ↑Ca2+ | acetylcholine, muscarine |
| Cancer Type | Sympathetic Innervation | Parasympathetic Innervation | References |
|---|---|---|---|
| Lung Cancer | Pro-tumorigenic | Pro-tumorigenic | [52,53,54,55] |
| Gastric cancer | Pro-tumorigenic | Pro-tumorigenic | [47,56,57,58,59] |
| Colorectal Cancer | Pro-tumorigenic | Pro-tumorigenic | [60,61,62,63] |
| Pancreatic ductal adenocarcinoma | Pro-tumorigenic | Anti-tumorigenic | [46,64] |
| Prostate cancer | Pro-tumorigenic | Pro-tumorigenic | [65,66,67,68] |
| Breast cancer | Pro-tumorigenic | Anti-tumorigenic | [69,70] |
| Hematological malignancies | Anti-tumorigenic | Anti-tumorigenic | [71,72,73,74] |
| Exosome Origin | Target Cells | Key Molecules Involved | Effect | Ref |
|---|---|---|---|---|
| Head and neck squamous cell carcinoma cell lines (other cancers: CRC, BC, melanoma) | PC12 cells (rat pheochromocytoma cell line) | EphrinB1 protein | Neurite outgrowth (in vitro) and tumor innervation (in vivo). | [92] |
| dADSC, primary Schwann cells | NG108–15 neurons | miRNAs: miR-18a, miR-21, miR-182, miR-222, miR-1, and mRNAs: GAP43 and Tau | Neurite outgrowth. | [94] |
| p53 null head and neck cancer cells | Peritumoral nerve fibers, DRGs and TGs | Low levels of miR-34a and high levels of miR-21 and miR-324 | Neurite outgrowth and transdifferentiation of sensory neurons in adrenergic neurons. | [95] |
| Head and neck cancer cells | CD8+ T cells | Galectin-1 (immunoregulatory protein) | Stimulation of CD8+ T-cell suppressor phenotype. | [115] |
| Melanoma cell lines | CTLL2 Cytotoxic T cell lines | miR-709, miR-2137, miR-2861, miR-1195, miR-762 (the five most highly abundant miRNAs) | Transcriptome signature changes resulting in mitochondrial respiration alteration. | [116] |
| Poorly metastatic melanoma cells | Patrolling monocytes (PMo) | Nr4a transcription factor and pigment epithelium-derived factor | PMo conditioned innate immune response with cancer cell clearance at the metastatic niche. | [124] |
| Neuroblastoma cell lines | Monocytes | miR-21 | Protumoral activity of monocytes through miR-21/TLR8-NF-кB/exosomic miR-155/TERF1 signaling pathway. | [129] |
| Ovarian cancer cell lines | Macrophages | miR-1246 | Transfer of oncogenic miR-1246 to M2-type macrophages, but not M0-type macrophages. | [125] |
| p53 mutant CRC cells | Macrophages | miR-1246 | Macrophage miR-1246-dependent reprogramming into a cancer promoting state with increased TGF-beta activity. | [126] |
| Melanoma cell lines | Macrophages | let-7a | Macrophage increased oxidative phosphorylation activity and M2-like polarization. | [127] |
| Glioma cell lines under hypoxic conditions | MDSC | miR-10a and miR-21 | Hypoxia-inducible expression of miR-10a and miR-21 mediates MDSC expansion and activation by targeting RORA and PTEN. | [134] |
| Glioma cell lines | MDSC | miR-29a and miR-92a | MDSC expansion and function activation through miRNA-29a/Hbp1 and miRNA-92a/Prkar1a pathways. | [135] |
| Pancreatic adenocarcinoma cell lines | DC | miR-203 | Downregulation of TLR4 and downstream cytokines. | [143] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dragomir, M.P.; Moisoiu, V.; Manaila, R.; Pardini, B.; Knutsen, E.; Anfossi, S.; Amit, M.; Calin, G.A. A Holistic Perspective: Exosomes Shuttle between Nerves and Immune Cells in the Tumor Microenvironment. J. Clin. Med. 2020, 9, 3529. https://doi.org/10.3390/jcm9113529
Dragomir MP, Moisoiu V, Manaila R, Pardini B, Knutsen E, Anfossi S, Amit M, Calin GA. A Holistic Perspective: Exosomes Shuttle between Nerves and Immune Cells in the Tumor Microenvironment. Journal of Clinical Medicine. 2020; 9(11):3529. https://doi.org/10.3390/jcm9113529
Chicago/Turabian StyleDragomir, Mihnea P., Vlad Moisoiu, Roxana Manaila, Barbara Pardini, Erik Knutsen, Simone Anfossi, Moran Amit, and George A. Calin. 2020. "A Holistic Perspective: Exosomes Shuttle between Nerves and Immune Cells in the Tumor Microenvironment" Journal of Clinical Medicine 9, no. 11: 3529. https://doi.org/10.3390/jcm9113529