The Role of Molecular Chaperones in Virus Infection and Implications for Understanding and Treating COVID-19

 , , , and
, , , and 
Abstract
:1. Introduction
2. Molecular Chaperones and Viral Infection
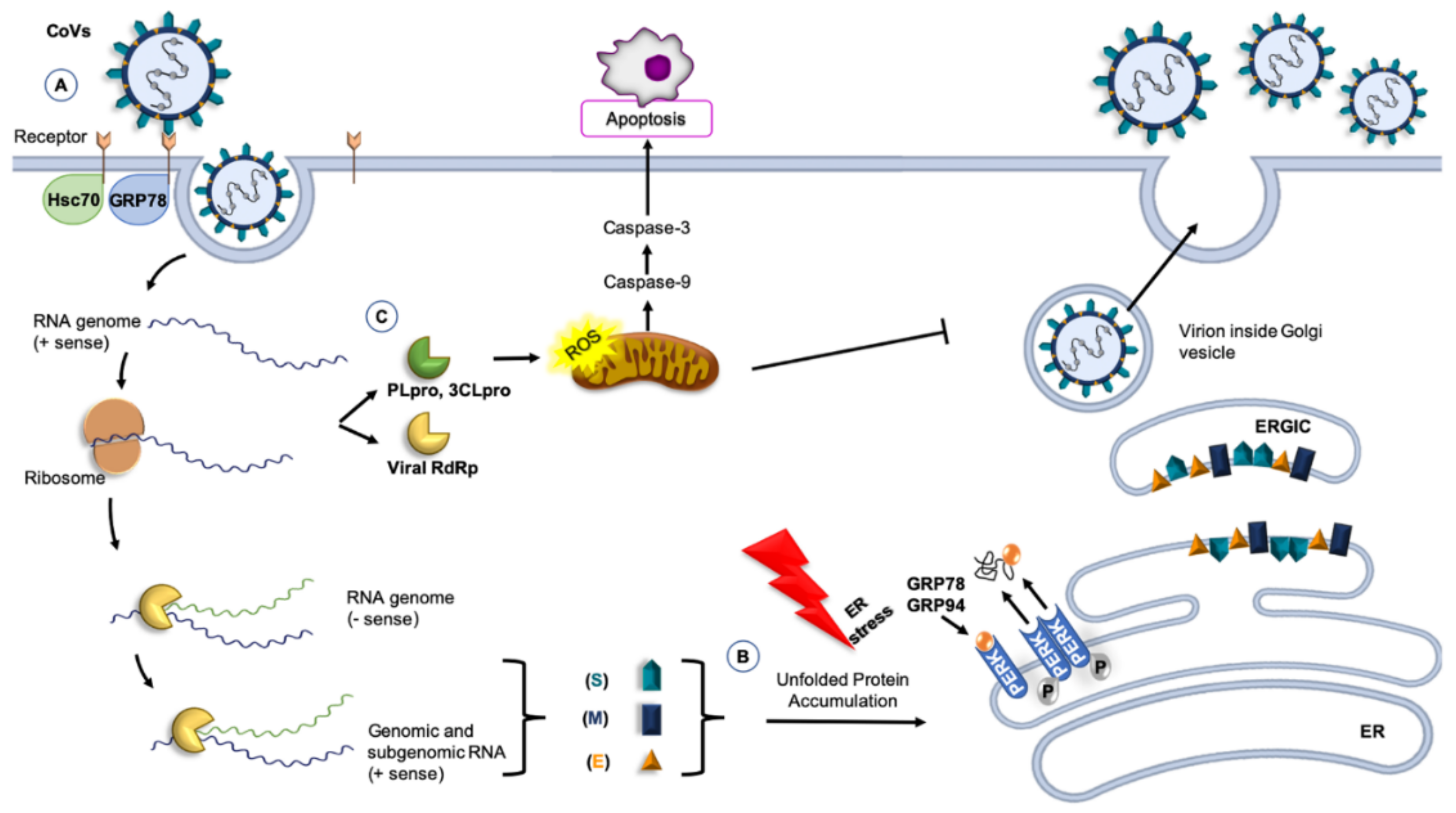
3. Coronavirus and Molecular Chaperones
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tyrrell, D.A.; Almeida, J.D.; Cunningham, C.H.; Dowdle, W.R.; Hofstad, M.S.; McIntosh, K.; Tajima, M.; Zakstelskaya, L.Y.; Easterday, B.C.; Kapikian, A.; et al. Coronaviridae. Intervirology 1975, 5, 76–82. [Google Scholar] [CrossRef]
- Hulswit, R.J.; De Haan, C.A.; Bosch, B.J. Coronavirus spike protein and tropism changes. Adv. Virus Res. 2016, 96, 29–57. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. SARS-CoV-2: An emerging coronavirus that causes a global threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.A.; Macnaughton, M.R. Comparison of the morphology of three coronaviruses. Arch. Virol. 1979, 59, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus. Res. 2006, 66, 193–292. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L.; Siddell, S.G. A contemporary view of coronavirus transcription. J. Virol. 2007, 81, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Heald-Sargent, T.; Gallagher, T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 2012, 4, 557–580. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.; To, K.K.; Tse, H.; Jin, D.Y.; Yuen, K.Y. Interspecies transmission and emergence of novel viruses: Lessons from bats and birds. Trends Microbiol. 2013, 21, 544–555. [Google Scholar] [CrossRef]
- Phan, M.V.T.; Ngo Tri, T.; Hong Anh, P.; Baker, S.; Kellam, P.; Cotton, M. Identification and characterization of Coronaviridae genomes from Vietnamese bats and rats based on conserved protein domains. Virus Evol. 2018, 4, vey035. [Google Scholar] [CrossRef]
- Ammirati, E.; Wang, D.W. SARS-CoV-2 inflames the heart. The importance of awareness of myocardial injury in COVID-19 patients. Int. J. Cardiol. 2020, 311, 122–123. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naicker, S.; Yang, C.W.; Hwang, S.J.; Liu, B.C.; Chen, J.H.; Jha, V. The Novel Coronavirus 2019 epidemic and kidneys. Kidney Int. 2020, 97, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Velavan, T.P.; Meyer, C.G. The COVID-19 epidemic. Trop. Med. Int. Health 2020, 25, 278–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Shi, L.; Wang, F.S. Liver injury in COVID-19: Management and challenges. Lancet Gastroenterol. Hepatol. 2020, 5, 428–430. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Cappello, F. Is COVID-19 a proteiform disease inducing also molecular mimicry phenomena? Cell Stress Chaperones 2020, 25, 381–382. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [Green Version]
- Meyerholz, D.K.; Lambertz, A.M.; McCray, P.B., Jr. Dipeptidyl Peptidase 4 distribution in the human respiratory tract: Implications for the Middle East Respiratory Syndrome. Am. J. Pathol. 2016, 186, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macario, A.J.L.; Conway de Macario, E. Chapter 12—Chaperone proteins and chaperonopathies. In Stress: Physiology, Biochemistry, and Pathology; Fink, G., Ed.; Elsevier/Academic Press: London, UK, 2019; Volume 3, pp. 135–152. [Google Scholar]
- Macario, A.J.L.; Conwey de Macario, E. Molecular mechanisms in chaperonopathies: Clues to understanding the histopathological abnormalities and developing novel therapies. J. Pathol. 2020, 250, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahiya, V.; Buchner, J. Functional principles and regulation of molecular chaperones. Adv. Protein Chem. Struct. Biol. 2019, 114, 1–60. [Google Scholar] [CrossRef]
- Macario, A.J.L. Heat-shock proteins and molecular chaperones: Implications for pathogenesis, diagnostics, and therapeutics. Int. J. Clin. Lab. Res. 1995, 25, 59–70. [Google Scholar] [CrossRef]
- Macario, A.J.L.; Conway de Macario, E. Sick chaperones, cellular stress, and disease. N. Engl. J. Med. 2005, 353, 1489–1501. [Google Scholar] [CrossRef]
- Macario, A.J.L.; Conway de Macario, E. Chaperonopathies by defect, excess, or mistake. Ann. N. Y. Acad. Sci. 2007, 1113, 178–191. [Google Scholar] [CrossRef]
- Panella, S.; Marcocci, M.E.; Celestino, I.; Valente, S.; Zwergel, C.; Li Puma, D.D.; Nencioni, L.; Mai, A.; Palamara, A.T.; Simonetti, G. MC1568 inhibits HDAC6/8 activity and influenza A virus replication in lung epithelial cells: Role of Hsp90 acetylation. Future Med. Chem. 2016, 8, 2017–2031. [Google Scholar] [CrossRef]
- Wan, Q.; Song, D.; Li, H.; He, M.L. Stress proteins: The biological functions in virus infection, present and challenges for target-based antiviral drug development. Signal Transduct. Target. Ther. 2020, 5, 125. [Google Scholar] [CrossRef]
- Hooper, P.L.; Hightower, L.E.; Hooper, P.L. Loss of stress response as a consequence of viral infection: Implications for disease and therapy. Cell Stress Chaperones 2012, 17, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Oglesbee, M.J.; Pratt, M.; Carsillo, T. Role for heat shock proteins in the immune response to measles virus infection. Viral Immunol. 2002, 15, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kanai, F.; Kawakami, T.; Tateishi, K.; Ijichi, H.; Kawabe, T.; Arakawa, Y.; Kawakami, T.; Nishimura, T.; Shirakata, Y.; et al. Interaction of the hepatitis B virus X protein (HBx) with heat shock protein 60 enhances HBx-mediated apoptosis. Biochem. Biophys. Res. Commun. 2004, 318, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Kim, S.J.; Kim, J.H.; Lee, W.; Kim, G.W.; Lee, K.H.; Choi, K.Y.; Oh, J.W. Interaction of hepatitis C virus core protein with Hsp60 triggers the production of reactive oxygen species and enhances TNF-alpha-mediated apoptosis. Cancer Lett. 2009, 279, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, C.A.; Bouyssounade, D.; Zárate, S.; Isa, P.; López, T.; Espinosa, R.; Romero, P.; Méndez, E.; López, S.; Arias, C.F. Heat shock cognate protein 70 is involved in rotavirus cell entry. J. Virol. 2002, 76, 4096–4102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Del Valle, J.; Chávez-Salinas, S.; Medina, F.; Del Angel, R.M. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J. Virol. 2005, 79, 4557–4567. [Google Scholar] [CrossRef] [Green Version]
- Thongtan, T.; Wikan, N.; Wintachai, P.; Rattanarungsan, C.; Srisomsap, C.; Cheepsunthorn, P.; Smith, D.R. Characterization of putative Japanese encephalitis virus receptor molecules on microglial cells. J. Med. Virol. 2012, 84, 615–623. [Google Scholar] [CrossRef]
- Wyżewski, Z.; Gregorczyk, K.P.; Szczepanowska, J.; Szulc-Dąbrowska, L. Functional role of Hsp60 as a positive regulator of human viral infection progression. Acta Virol. 2018, 62, 33–40. [Google Scholar] [CrossRef]
- Pujhari, S.; Brustolin, M.; Macias, V.M.; Nissly, R.H.; Nomura, M.; Kuchipudi, S.V.; Rasgon, J.L. Heat shock protein 70 (Hsp70) mediates Zika virus entry, replication, and egress from host cells. Emerg. Microbes Infect. 2019, 8, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Ivanovic, T.; Agosto, M.A.; Chandran, K.; Nibert, M.L. A role for molecular chaperone Hsc70 in reovirus outer capsid disassembly. J. Biol. Chem. 2007, 282, 12210–12219. [Google Scholar] [CrossRef] [Green Version]
- Park, S.G.; Lee, S.M.; Jung, G. Antisense oligodeoxynucleotides targeted against molecular chaperonin Hsp60 block human hepatitis B virus replication. J. Biol. Chem. 2003, 278, 39851–39857. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Mitra, D. Heat shock protein 40 is necessary for human immunodeficiency virus-1 Nef-mediated enhancement of viral gene expression and replication. J. Biol. Chem. 2005, 280, 40041–40050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, T.; Nishimura, Y.; Ichimura, T.; Suzuki, K.; Miyamura, T.; Suzuki, T.; Moriishi, K.; Matsuura, Y. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006, 25, 5015–5025. [Google Scholar] [CrossRef]
- Naito, T.; Momose, F.; Kawaguchi, A.; Nagata, K. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J. Virol. 2007, 81, 1339–1349. [Google Scholar] [CrossRef] [Green Version]
- Fislová, T.; Thomas, B.; Graef, K.M.; Fodor, E. Association of the influenza virus RNA polymerase subunit PB2 with the host chaperonin CCT. J. Virol. 2010, 84, 8691–8699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Rawat, P.; Khan, S.Z.; Dhamija, N.; Chaudhary, P.; Ravi, D.S.; Mitra, D. Reciprocal regulation of human immunodeficiency virus-1 gene expression and replication by heat shock proteins 40 and 70. J. Mol. Biol. 2011, 410, 944–958. [Google Scholar] [CrossRef]
- Kawashima, D.; Kanda, T.; Murata, T.; Saito, S.; Sugimoto, A.; Narita, Y.; Tsurumi, T. Nuclear transport of Epstein-Barr virus DNA polymerase is dependent on the BMRF1 polymerase processivity factor and molecular chaperone Hsp90. J. Virol. 2013, 87, 6482–6491. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Chen, Z.; Zhang, B.; Miao, H.; Zohaib, A.; Xu, Q.; Chen, H.; Cao, S. Heat shock protein 70 is associated with replicase complex of Japanese encephalitis virus and positively regulates viral genome replication. PLoS ONE 2013, 8, e75188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, X.; Zan, J.; Wu, Y.; Ye, C.; Ruan, X.; Zhou, J. Cellular chaperonin CCTγ contributes to rabies virus replication during infection. J. Virol. 2013, 87, 7608–7621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ye, C.; Ruan, X.; Zan, J.; Xu, Y.; Liao, M.; Zhou, J. The chaperonin CCTα is required for efficient transcription and replication of rabies virus. Microbiol. Immunol. 2014, 58, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Batra, J.; Tripathi, S.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; Sambhara, S.; Lal, S.K. Human Heat shock protein 40 (Hsp40/DnaJB1) promotes influenza A virus replication by assisting nuclear import of viral ribonucleoproteins. Sci. Rep. 2016, 6, 19063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafirassou, M.L.; Meertens, L.; Umaña-Diaz, C.; Labeau, A.; Dejarnac, O.; Bonnet-Madin, L.; Kümmerer, B.M.; Delaugerre, C.; Roingeard, P.; Vidalain, P.O.; et al. A global interactome map of the Dengue virus NS1 identifies virus restriction and dependency host factors. Cell Rep. 2017, 21, 3900–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchkovich, N.J.; Maguire, T.G.; Yu, Y.; Paton, A.W.; Paton, J.C.; Alwine, J.C. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78, which is required for virion assembly. J. Virol. 2008, 82, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruri-Avidal, L.; López, S.; Arias, C.F. Endoplasmic reticulum chaperones are involved in the morphogenesis of rotavirus infectious particles. J. Virol. 2008, 82, 5368–5380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khachatoorian, R.; Riahi, R.; Ganapathy, E.; Shao, H.; Wheatley, N.M.; Sundberg, C.; Jung, C.L.; Ruchala, P.; Dasgupta, A.; Arumugaswami, V.; et al. Allosteric heat shock protein 70 inhibitors block hepatitis C virus assembly. Int. J. Antimicrob. Agents 2016, 47, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Knowlton, J.J.; de Castro, I.F.; Ashbrook, A.W.; Gestaut, D.R.; Zamora, P.F.; Bauer, J.A.; Forrest, J.C.; Frydman, J.; Risco, C.; Dermody, T.S. The TRiC chaperonin controls reovirus replication through outer-capsid folding. Nat. Microbiol. 2018, 3, 481–493. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. ER chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 2004, 28, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Royle, J.; Ramírez-Santana, C.; Akpunarlieva, S.; Donald, C.L.; Gestuveo, R.J.; Anaya, J.M.; Merits, A.; Burchmore, R.; Kohl, A.; Varjak, M. Glucose-Regulated Protein 78 interacts with Zika virus envelope protein and contributes to a productive infection. Viruses 2020, 12, 524. [Google Scholar] [CrossRef]
- Schröder, M. Endoplasmic reticulum stress responses. Cell Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Lumley, E.C.; Osborn, A.R.; Scott, J.E.; Scholl, A.G.; Mercado, V.; McMahan, Y.T.; Coffman, Z.G.; Brewster, J.L. Moderate endoplasmic reticulum stress activates a PERK and p38-dependent apoptosis. Cell Stress Chaperones 2017, 22, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Su, H.L.; Liao, C.L.; Lin, Y.L. Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 2002, 76, 4162–4171. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Feng, Z.; He, B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Schmechel, S.C.; Raghavan, A.; Abelson, M.; Reilly, C.; Katze, M.G.; Kaufman, R.J.; Bohjanen, P.R.; Schiff, L.A. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 2006, 80, 2019–2033. [Google Scholar] [CrossRef] [Green Version]
- Bukrinsky, M.; Zhao, Y. Heat-shock proteins reverse the G2 arrest caused by HIV-1 viral protein R. DNA Cell Biol 2004, 23, 223–225. [Google Scholar] [CrossRef]
- Iordanskiy, S.; Zhao, Y.; Dubrovsky, L.; Iordanskaya, T.; Chen, M.; Liang, D.; Bukrinsky, M. Heat shock protein 70 protects cells from cell cycle arrest and apoptosis induced by human immunodeficiency virus type 1 viral protein R. J. Virol. 2004, 78, 9697–9704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, D.; Benko, Z.; Agbottah, E.; Bukrinsky, M.; Zhao, R.Y. Anti-vpr activities of heat shock protein 27. Mol. Med. 2007, 13, 229–239. [Google Scholar] [CrossRef]
- Lee, S.H.; Song, R.; Lee, M.N.; Kim, C.S.; Lee, H.; Kong, Y.Y.; Kim, H.; Jang, S.K. A molecular chaperone glucose-regulated protein 94 blocks apoptosis induced by virus infection. Hepatology 2008, 47, 854–866. [Google Scholar] [CrossRef]
- Wen, K.W.; Damania, B. Hsp90 and Hsp40/Erdj3 are required for the expression and anti-apoptotic function of KSHV K1. Oncogene 2010, 29, 3532–3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neckers, L.; Tatu, U. Molecular chaperones in pathogen virulence: Emerging new targets for therapy. Cell Host Microbe 2008, 4, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Pack, C.D.; Gierynska, M.; Rouse, B.T. An intranasal heat shock protein-based vaccination strategy confers protection against mucosal challenge with herpes simplex virus. Hum. Vaccin 2008, 4, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Chase, G.; Deng, T.; Fodor, E.; Leung, B.W.; Mayer, D.; Schwemmle, M.; Brownlee, G. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 2008, 377, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.P.; Shan, G.Z.; Peng, Z.G.; Zhu, J.H.; Meng, S.; Zhang, T.; Gao, L.Y.; Tao, P.Z.; Gao, R.M.; Li, Y.H.; et al. Synthesis and biological evaluation of heat-shock protein 90 inhibitors: Geldanamycin derivatives with broad antiviral activities. Antivir. Chem. Chemother. 2010, 20, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Oglesbee, M. Virus-heat shock protein interaction and a novel axis for innate antiviral immunity. Cells 2012, 1, 646–666. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Wunderink, R.G. MERS, SARS and other coronaviruses as causes of pneumonia. Respirology 2018, 23, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A structural view of SARS-CoV-2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Hendershot, L.M. The mammalian endoplasmic reticulum as a sensor for cellular stress. Cell Stress Chaperones 2002, 7, 222–229. [Google Scholar] [CrossRef]
- Chu, H.; Chan, C.M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.H.; Sze, K.H.; Yang, D.; Shuai, H.; et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 2014, 25, 528–537. [Google Scholar] [CrossRef]
- Lai, C.C.; Jou, M.J.; Huang, S.Y.; Li, S.W.; Wan, L.; Tsai, F.J.; Lin, C.W. Proteomic analysis of up-regulated proteins in human promonocyte cells expressing severe acute respiratory syndrome coronavirus 3C-like protease. Proteomics 2007, 7, 1446–1460. [Google Scholar] [CrossRef] [Green Version]
- Fukushi, M.; Yoshinaka, Y.; Matsuoka, Y.; Hatakeyama, S.; Ishizaka, Y.; Kirikae, T.; Sasazuki, T.; Miyoshi-Akiyama, T. Monitoring of S protein maturation in the endoplasmic reticulum by calnexin is important for the infectivity of severe acute respiratory syndrome coronavirus. J. Virol. 2012, 86, 11745–11753. [Google Scholar] [CrossRef] [Green Version]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, F.; Ye, S.; Guo, X.; Muhanmmad Memon, A.; Wu, M.; He, Q. Comparative proteome analysis of porcine jejunum tissues in response to a virulent strain of porcine epidemic diarrhea virus and its attenuated strain. Viruses 2016, 8, 323. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yang, X.; Xu, P.; Wu, X.; Zhou, L.; Wang, H. Heat shock protein 70 in lung and kidney of specific-pathogen-free chickens is a receptor-associated protein that interacts with the binding domain of the spike protein of infectious bronchitis virus. Arch. Virol. 2017, 162, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, A. Effect of the unfolded protein response on ER protein export: A potential new mechanism to relieve ER stress. Cell Stress Chaperones 2018, 23, 797–806. [Google Scholar] [CrossRef]
- Aoe, T. Pathological aspects of COVID-19 as a conformational disease and the use of pharmacological chaperones as a potential therapeutic strategy. Front. Pharmacol. 2020, 11, 1095. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.P.; Siu, K.L.; Chin, K.T.; Yuen, K.Y.; Zheng, B.; Jin, D.Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isler, J.A.; Maguire, T.G.; Alwine, J.C. Production of infectious human cytomegalovirus virions is inhibited by drugs that disrupt calcium homeostasis in the endoplasmic reticulum. J. Virol. 2005, 79, 15388–15397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhal, B.D.; Nguyen, H.; Dang, M.; Gopallawa, I.; Jiang, J.; Dang, V.; Ono, S.; Morimoto, K. Abrogation of ER stress-induced apoptosis of alveolar epithelial cells by angiotensin 1-7. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L33–L41. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Gao, Y.; Zhao, W.; Yu, G.; Jin, F. ACE-2/ANG1-7 ameliorates ER stress-induced apoptosis in seawater aspiration-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L1015–L1027. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [Green Version]
- Honda, T.; Horie, M.; Daito, T.; Ikuta, K.; Tomonaga, K. Molecular chaperone BiP interacts with Borna disease virus glycoprotein at the cell surface. J. Virol. 2009, 83, 12622–12625. [Google Scholar] [CrossRef] [Green Version]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Báez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Lin, C.W.; Lin, K.H.; Hsieh, T.H.; Shiu, S.Y.; Li, J.Y. Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol. Med. Microbiol. 2006, 46, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Nagy, P.D.; Wang, R.Y.; Pogany, J.; Hafren, A.; Makinen, K. Emerging picture of host chaperone and cyclophilin roles in RNA virus replication. Virology 2011, 411, 374–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, D.; Bagchi, P.; Chatterjee, A.; Nayak, M.K.; Mukherjee, A.; Chattopadhyay, S.; Nagashima, S.; Kobayashi, N.; Komoto, S.; Taniguchi, K.; et al. The molecular chaperone heat shock protein-90 positively regulates rotavirus infection. Virology 2009, 391, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geller, R.; Taguwa, S.; Frydman, J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta 2012, 1823, 698–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakovac, H. COVID-19 and hypertension—Is the HSP60 culprit for the severe course and worse outcome? Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H793–H796. [Google Scholar] [CrossRef]
- Mayer, M.P. Recruitment of Hsp70 chaperones: A crucial part of viral survival strategies. Rev. Physiol. Biochem. Pharmacol. 2005, 153, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kim, J.H.; Lee, C.H.; Ahn, Y.J.; Song, J.H.; Baek, S.H.; Kwon, D.H. Antiviral activity of quercetin 7-rhamnoside against porcine epidemic diarrhea virus. Antivir. Res. 2009, 81, 77–81. [Google Scholar] [CrossRef]
- Fan, C.Y.; Lee, S.; Cyr, D.M. Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones 2008, 8, 309–316. [Google Scholar] [CrossRef]
- Shah, S.; Danda, D.; Kavadichanda, C.; Das, S.; Adarsh, M.B.; Negi, V.S. Autoimmune and rheumatic musculoskeletal diseases as a consequence of SARS-CoV-2 infection and its treatment. Rheumatol. Int. 2020, 40, 1539–1554. [Google Scholar] [CrossRef]
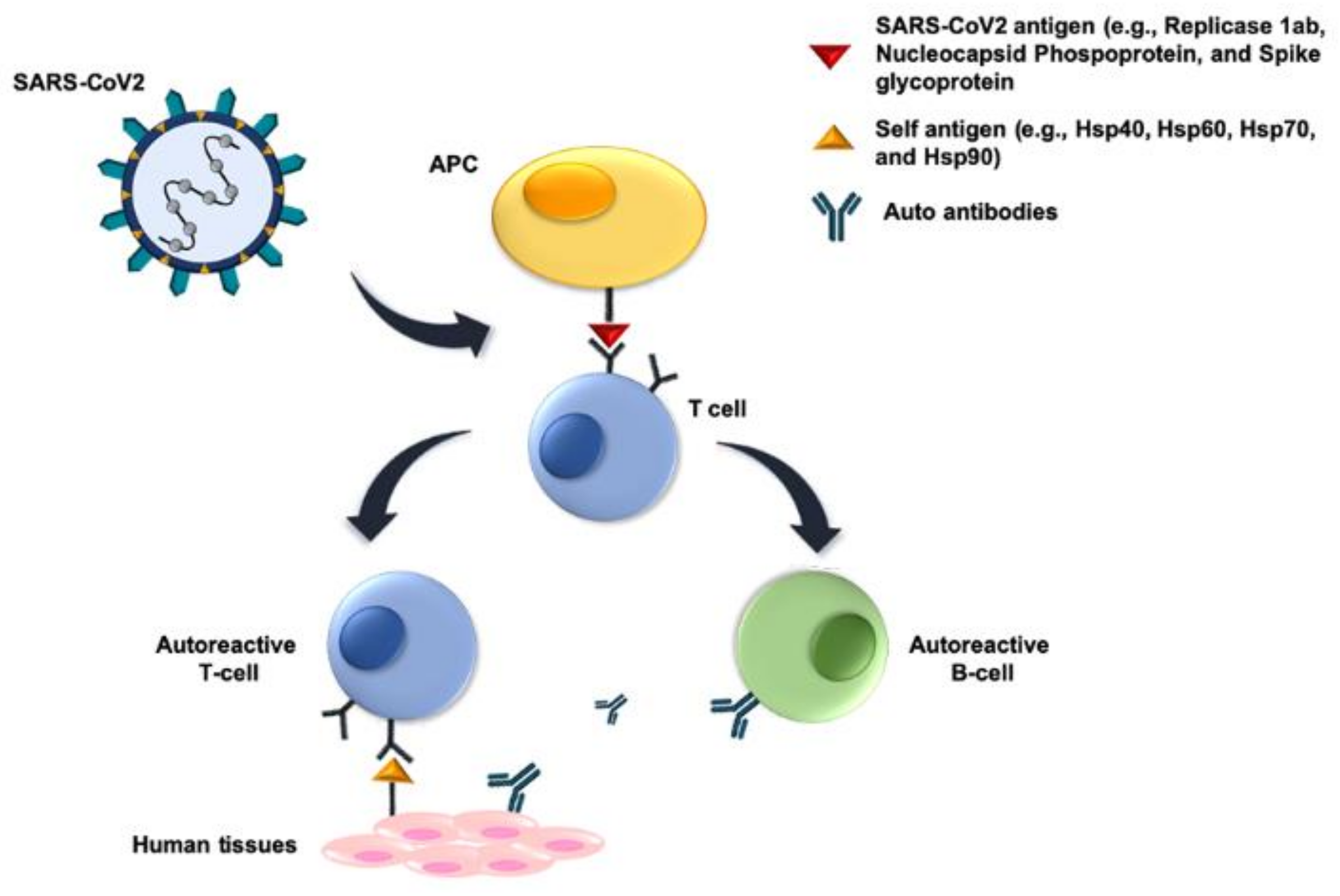
- Cappello, F.; Marino Gammazza, A.; Dieli, F.; Conway de Macario, E.; Macario, A.J.L. Does SARS-CoV-2 trigger stress-induced autoimmunity by molecular mimicry? A hypothesis. J. Clin. Med. 2020, 9, 2038. [Google Scholar] [CrossRef] [PubMed]
- Marino Gammazza, A.; Légaré, S.; Lo Bosco, G.; Fucarino, A.; Angileri, F.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F. Human molecular chaperones share with SARS-CoV-2 antigenic epitopes potentially capable of eliciting autoimmunity against endothelial cells: Possible role of molecular mimicry in COVID-19. Cell Stress Chaperones 2020, 25, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Lucchese, G.; Flöel, A. SARS-CoV-2 and Guillain-Barré syndrome: Molecular mimicry with human heat shock proteins as potential pathogenic mechanism. Cell Stress Chaperones 2020, 25, 731–735. [Google Scholar] [CrossRef]
- Cappello, F.; Marino Gammazza, A.; Palumbo Piccionello, A.; Campanella, C.; Pace, A.; Conway de Macario, E.; Macario, A.J.L. Hsp60 chaperonopathies and chaperonotherapy: Targets and agents. Expert Opin. Ther. Targets 2014, 18, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Kusaczuk, M. Tauroursodeoxycholate-Bile acid with chaperoning activity: Molecular and cellular effects and therapeutic perspectives. Cells 2019, 8, 1471. [Google Scholar] [CrossRef] [Green Version]
- Rellmann, Y.; Gronau, I.; Hansen, U.; Dreier, R. 4-Phenylbutyric acid reduces endoplasmic reticulum stress in chondrocytes that is caused by loss of the protein disulfide isomerase ERp57. Oxid. Med. Cell Longev. 2019, 2019, 6404035. [Google Scholar] [CrossRef]
- Rojas, M.; Restrepo-Jiménez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [CrossRef]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Mol. Chap. | Virus | Role | Reference |
|---|---|---|---|
| Hsp40 | HIV-1 | Participates in Nef-mediated enhancement of viral gene expression and replication | [42,46] |
| IAV | Assists nuclear import of viral ribonucleoprotein | [51] | |
| Hsp60 | HBV | Participates in polymerase activation and replication initiation. Promotes virus-mediated apoptosis of infected cells | [33,41] |
| HCV | Promotes infected cells apoptosis | [34] | |
| Hsc/Hsp70 | Rotaviruses | Is part of host-cell membrane receptor and promotes virus entry and infectivity | [35] |
| DENV | Is part of host-cell membrane receptor and promotes virus entry and infectivity | [36] | |
| JEV | Is part of host-cell membrane receptor and contributes to virus entry. Enhances virus replication | [37,48] | |
| ZKV | Modulates virus entry, replication, and egress | [39] | |
| Reoviruses | Participates in outer capsid disassembly | [40] | |
| HCV | Regulates viral genome translation, virions assembly and release | [55] | |
| HIV-1 | Inhibits viral gene expression. Delay Vpr induced cell cycle arrest and cell apoptosis | [46,65,66] | |
| Hsp90 | DENV | Is part of host-cell membrane receptor and promotes virus entry and infectivity | [36] |
| HCV | Positively regulates virus replication | [43] | |
| Influenza virus | Promotes nuclear import of the genome and the RNA-dependent RNA polymerase, ensuring virus replication | [44,72] | |
| EBV | Mediates the assembly and the nuclear import of the DNA polymerase | [47] | |
| KSHV | Promotes anti-apoptotic signaling | [69] | |
| BiP/GRP78 | CMV | Regulates virions assembly | [53] |
| Rotavirus | Ensures proper assembly and maturation of viral particles | [54] | |
| GRP94 | HCV | Blocks infected cells apoptosis | [68] |
| Calnexin | Rotavirus | Ensures proper assembly and maturation of viral particles | [54] |
| Calreticulin | Rotavirus | Ensures proper assembly and maturation of viral particles | [54] |
| CCT | Influenza virus | Promotes virus replication and transcription | [45] |
| Rabies virus | Promotes virus replication and transcription | [49,50] | |
| DENV | Promotes virus replication and transcription | [52] | |
| Reovirus | Participates in viral proteins folding | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paladino, L.; Vitale, A.M.; Caruso Bavisotto, C.; Conway de Macario, E.; Cappello, F.; Macario, A.J.L.; Marino Gammazza, A. The Role of Molecular Chaperones in Virus Infection and Implications for Understanding and Treating COVID-19. J. Clin. Med. 2020, 9, 3518. https://doi.org/10.3390/jcm9113518
Paladino L, Vitale AM, Caruso Bavisotto C, Conway de Macario E, Cappello F, Macario AJL, Marino Gammazza A. The Role of Molecular Chaperones in Virus Infection and Implications for Understanding and Treating COVID-19. Journal of Clinical Medicine. 2020; 9(11):3518. https://doi.org/10.3390/jcm9113518
Chicago/Turabian StylePaladino, Letizia, Alessandra Maria Vitale, Celeste Caruso Bavisotto, Everly Conway de Macario, Francesco Cappello, Alberto J.L. Macario, and Antonella Marino Gammazza. 2020. "The Role of Molecular Chaperones in Virus Infection and Implications for Understanding and Treating COVID-19" Journal of Clinical Medicine 9, no. 11: 3518. https://doi.org/10.3390/jcm9113518