Molecular Profiling in Daily Clinical Practice: Practicalities in Advanced Cholangiocarcinoma and Other Biliary Tract Cancers

 ,
,
Abstract
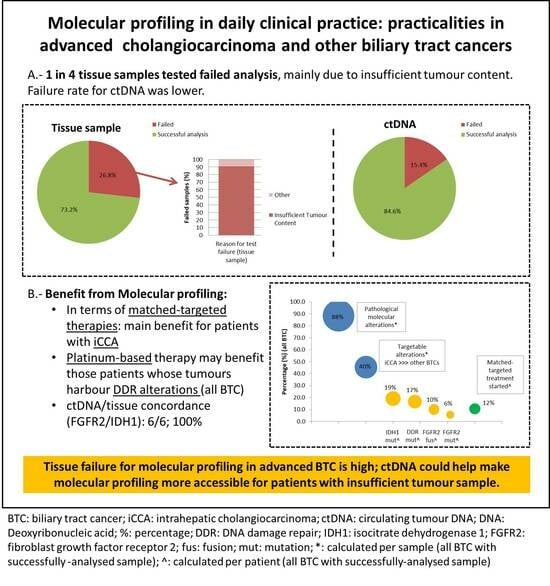
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Patient Characteristics
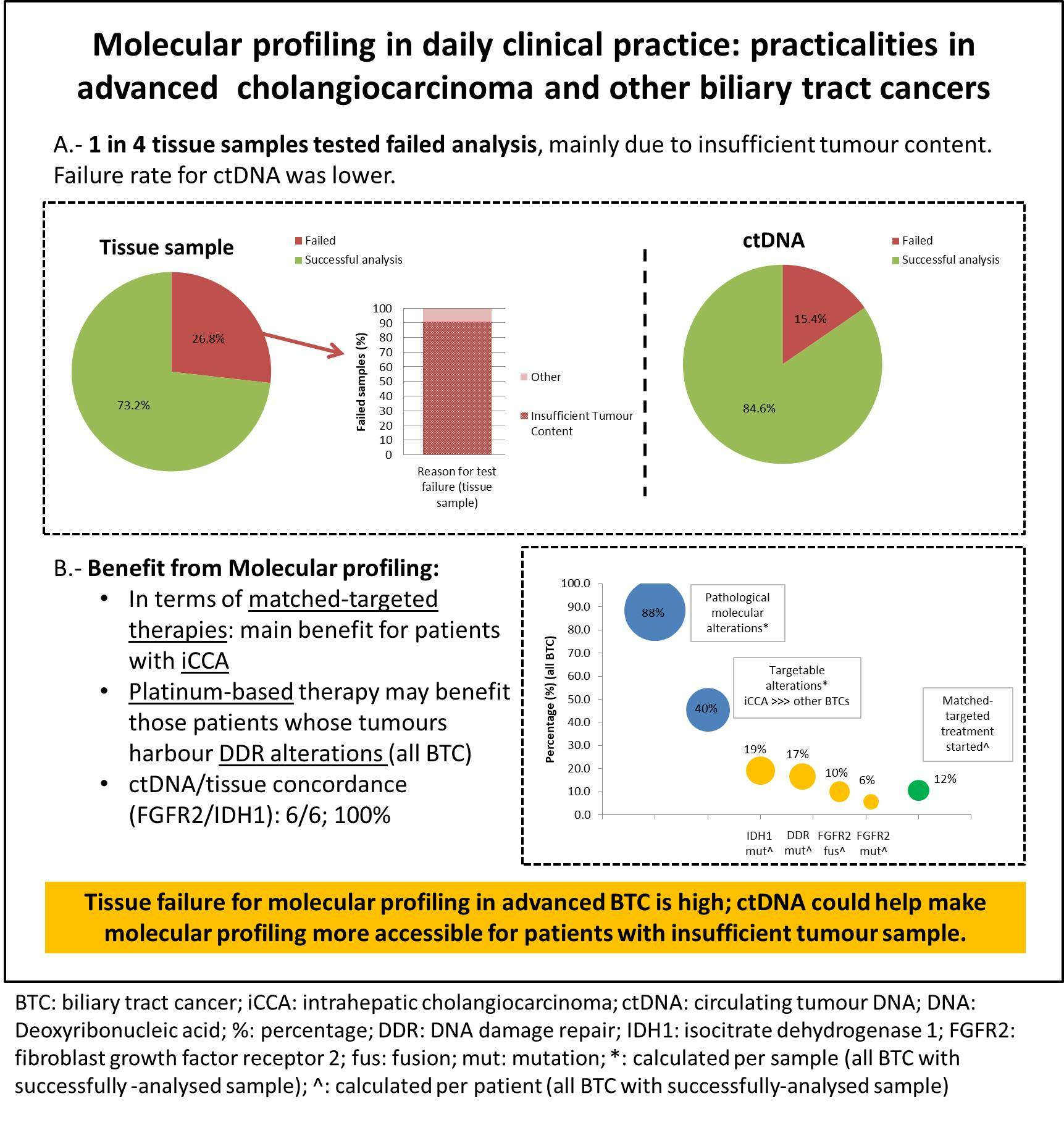
3.2. Sample Characteristics and Failure Rate
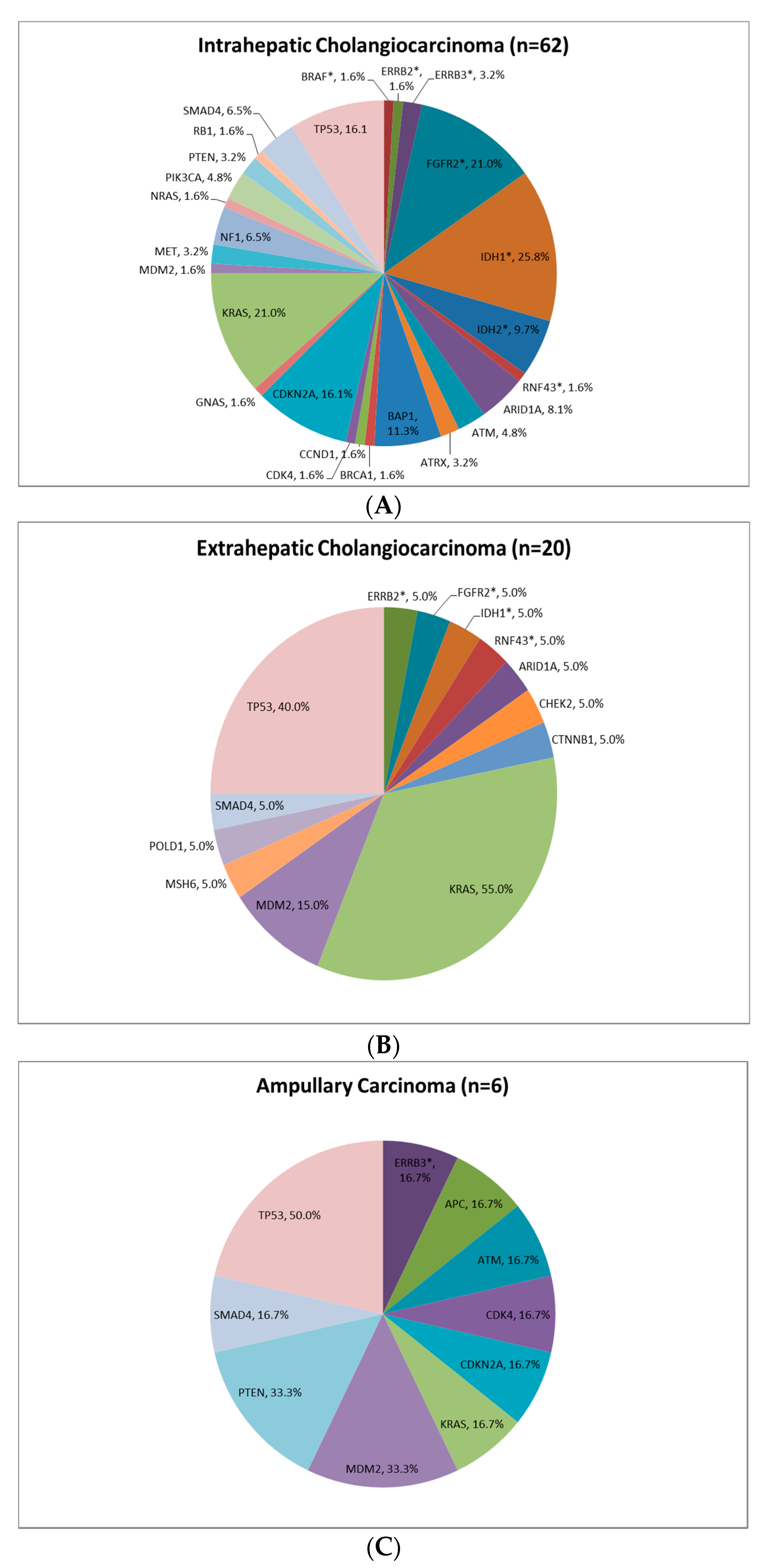
3.3. Pathological Molecular Findings
3.4. Clinical Implication of Pathological Molecular Findings
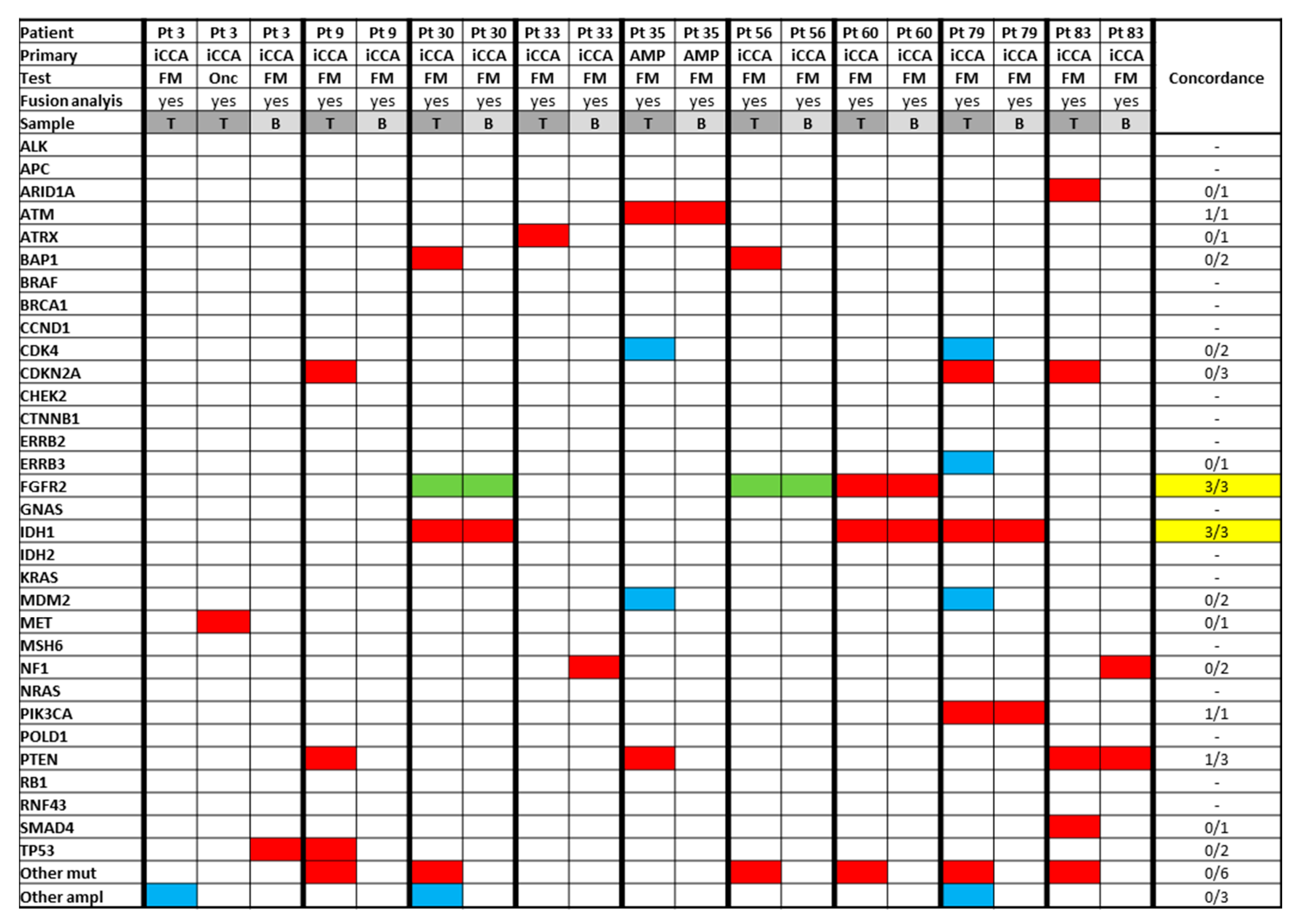
3.5. Analysis of Paired Tissue Samples
3.6. Analysis of Paired Tissue and ctDNA Samples
3.7. ctDNA Analysed Prior to Palliative Therapy Initiation
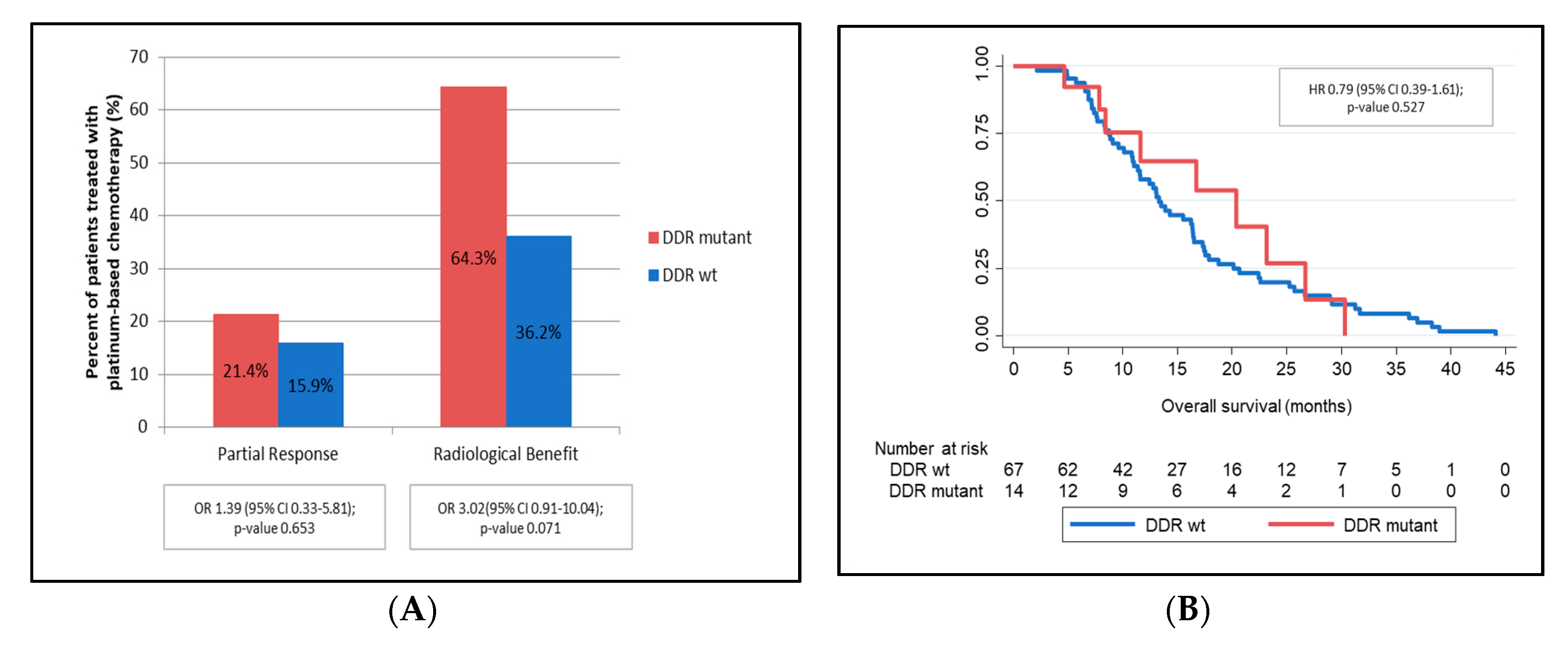
3.8. DNA Damage Repair Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patel, N.; Benipal, B. Incidence of Cholangiocarcinoma in the USA from 2001 to 2015: A US Cancer Statistics Analysis of 50 States. Cureus 2019, 11, e3962. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Gambardella, V.; Cejalvo, J.-M.; Fleitas-Kanonnikoff, T.; Cervantes, A. In the literature: June 2019. ESMO Open 2019, 4, e000547. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Kennedy, E.B.; Bachini, M.; Bekaii-Saab, T.; Crane, C.; Edeline, J.; El-Khoueiry, A.; Feng, M.; Katz, M.H.; Primrose, J.N.; et al. Adjuvant Therapy for Resected Biliary Tract Cancer: ASCO Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Kim, R. Adjuvant therapy for resected extrahepatic cholangiocarcinoma: A review of the literature and future directions. Cancer Treat. Rev. 2009, 35, 322–327. [Google Scholar] [CrossRef] [PubMed]
- DeOliveira, M.L.; Cunningham, S.C.; Cameron, J.L.; Kamangar, F.; Winter, J.M.; Lillemoe, K.D.; Choti, M.A.; Yeo, C.J.; Schulick, R.D. Cholangiocarcinoma: Thirty-one-year experience with 564 patients at a single institution. Ann. Surg. 2007, 245, 755–762. [Google Scholar] [CrossRef]
- Valle, J.W.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for Biliary Tract Cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Okusaka, T.; Nakachi, K.; Fukutomi, A.; Mizuno, N.; Ohkawa, S.; Funakoshi, A.; Nagino, M.; Kondo, S.; Nagaoka, S.; Funai, J.; et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: A comparative multicentre study in Japan. Br. J. Cancer 2010, 103, 469–474. [Google Scholar] [CrossRef]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. ABC-06 | A randomised phase III, multi-centre, open-label study of Active Symptom Control (ASC) alone or ASC with oxaliplatin / 5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced/metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem) chemotherapy. J. Clin. Oncol. 2019, 37, 4003. [Google Scholar]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Molecular targeted therapies: Ready for “prime time” in biliary tract cancer. J. Hepatol. 2020, 73, 170–185. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New Horizons for Precision Medicine in Biliary Tract Cancers. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Fizazi, K.; Maillard, A.; Penel, N.; Baciarello, G.; Allouache, D.; Daugaard, G.; Van De Wouw, A.; Soler, G.; Vauleon, E.; Chaigneau, L.; et al. A phase III trial of empiric chemotherapy with cisplatin and gemcitabine or systemic treatment tailored by molecular gene expression analysis in patients with carcinomas of an unknown primary (CUP) site (GEFCAPI 04). Ann. Oncol. 2019, 30, v851. [Google Scholar] [CrossRef]
- Javle, M.; Lowery, M.A.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients with FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Kelley, R.; Roychowdhury, S.; Weiss, K.; Abou-Alfa, G.; Macarulla, T.; Sadeghi, S.; Waldschmidt, D.; Zhu, A.; Goyal, L.; et al. Updated results from a phase II study of infigratinib (BGJ398), a selective pan-FGFR kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma containing FGFR2 fusions. Ann. Oncol. 2018, 29, mdy424-030. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Arkenau, H.; Tran, B.; Bahleda, R.; Kelley, R.; Hierro, C.; Ahn, D.; Zhu, A.; Javle, M.; Winkler, R.; et al. Efficacy of TAS-120, an irreversible fibroblast growth factor receptor (FGFR) inhibitor, in cholangiocarcinoma patients with FGFR pathway alterations who were previously treated with chemotherapy and other FGFR inhibitors. Ann. Oncol. 2018, 29, v100. [Google Scholar] [CrossRef]
- Tran, B.; Meric-Bernstam, F.; Arkenau, H.-T.; Bahleda, R.; Kelley, R.; Hierro, C.; Ahn, D.; Zhu, A.; Javle, M.; Winkler, R.; et al. Efficacy of TAS-120, an irreversible fibroblast growth factor receptor inhibitor (FGFRi), in patients with cholangiocarcinoma and FGFR pathway alterations previously treated with chemotherapy and other FGFRi’s. Ann. Oncol. 2018, 29, ix49–ix50. [Google Scholar] [CrossRef]
- Mazzaferro, V.; El-Rayes, B.F.; Cotsoglou, C.; Harris, W.P.; Damjanov, N.; Masi, G.; Rimassa, L.; Personeni, N.; Braiteh, F.S.; Zagonel, V.; et al. ARQ 087, an oral pan-fibroblast growth factor receptor (FGFR) inhibitor, in patients (pts) with advanced intrahepatic cholangiocarcinoma (iCCA) with FGFR2 genetic aberrations. J. Clin. Oncol. 2017, 35, 4017. [Google Scholar] [CrossRef]
- Mazzaferro, V.; El-Rayes, B.F.; Busset, M.D.D.; Cotsoglou, C.; Harris, W.P.; Damjanov, N.; Masi, G.; Rimassa, L.; Personeni, N.; Braiteh, F.; et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br. J. Cancer 2018, 120, 165–171. [Google Scholar] [CrossRef]
- Cleary, J.M.; Voss, M.H.; Meric-Bernstam, F.; Hierro, C.; Heist, R.S.; Ishii, N.; Kirpicheva, Y.; Nicolas-Metral, V.; Pokorska-Bocci, A.; Vaslin, A.; et al. Safety and efficacy of the selective FGFR inhibitor debio 1347 in phase I study patients with FGFR genomically activated advanced biliary tract cancer (BTC). J. Clin. Oncol. 2018, 36, 447. [Google Scholar] [CrossRef]
- Ng, M.C.H.; Goyal, L.; Bang, Y.-J.; Oh, -Y.; Chao, T.-Y.; Cleary, J.M.; Voss, M.H.; Meric-Bernstam, F.; Iyer, G.; Heist, R.S.; et al. AB065. P-36. Debio 1347 in patients with cholangiocarcinoma harboring an FGFR gene alteration: Preliminary results. HepatoBiliary Surg. Nutr. 2019, 8, AB065. [Google Scholar] [CrossRef]
- Bahleda, R.; Italiano, A.; Hierro, C.; Mita, A.C.; Cervantes, A.; Chan, N.; Awad, M.M.; Calvo, E.; Moreno, V.; Govindan, R.; et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced or Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 4888–4897. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Strickler, J.H.; Govindan, R.; Chai, S.; Chan, N.; Quiroga-Garcia, V.; Bahleda, R.; Hierro, C.; Zhong, B.; Gonzalez, M.; et al. Safety and activity of the pan-fibroblast growth factor receptor (FGFR) inhibitor erdafitinib in phase 1 study patients (Pts) with molecularly selected advanced cholangiocarcinoma (CCA). J. Clin. Oncol. 2017, 35, 4074. [Google Scholar] [CrossRef]
- Tannapfel, A.; Sommerer, F.; Benicke, M.; Katalinic, A.; Uhlmann, D.; Witzigmann, H.; Hauss, J.; Wittekind, C. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut 2003, 52, 706–712. [Google Scholar] [CrossRef]
- Koo, B.K.; Van Es, J.H.; Born, M.V.D.; Clevers, H. Porcupine inhibitor suppresses paracrine Wnt-driven growth of Rnf43; Znrf3-mutant neoplasia. Proc. Natl. Acad. Sci. USA 2015, 112, 7548–7550. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Galdy, S.; Barriuso, J.; Moghadam, S.; Beckett, E.; Rogan, J.; Backen, A.; Billington, C.; McNamara, M.G.; Hubner, R.A.; et al. The HER3 pathway as a potential target for inhibition in patients with biliary tract cancers. PLoS ONE 2018, 13, e0206007. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Siena, S.; Ou, S.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and Antitumor Activity of the Multi-Targeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib (RXDX-101): Combined Results from Two Phase 1 Trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New Routes to Targeted Therapy of Intrahepatic Cholangiocarcinomas Revealed by Next-Generation Sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef]
- Mukherjee, S. The Search for Cancer Treatment Beyond Mutant-Hunting. New York Times Magazine. 13 June 2018.
- Brand, M.; Measures, A.M.; Wilson, B.G.; Cortopassi, W.A.; Alexander, R.; Höss, M.; Hewings, D.S.; Rooney, T.P.C.; Paton, R.; Conway, S.J. Small Molecule Inhibitors of Bromodomain–Acetyl-lysine Interactions. ACS Chem. Biol. 2014, 10, 22–39. [Google Scholar] [CrossRef]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Biliary Tract Cancer: State of the Art and potential role of DNA Damage Repair. Cancer Treat. Rev. 2018, 70, 168–177. [Google Scholar] [CrossRef]
- Chae, H.; Kim, D.; Yoo, C.; Kim, K.-P.; Jeong, J.H.; Chang, H.-M.; Lee, S.S.; Park, D.H.; Song, T.J.; Hwang, S.; et al. Therapeutic relevance of targeted sequencing in management of patients with advanced biliary tract cancer: DNA damage repair gene mutations as a predictive biomarker. Eur. J. Cancer 2019, 120, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Lamarca, A.; Frizziero, M.; McNamara, M.G.; Valle, J.W. Clinical and translational research challenges in biliary tract cancers. Curr. Med. Chem. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvise, M.; Lamarca, A.; Forner, A. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 2019, 39, 98–107. [Google Scholar] [CrossRef]
- Levit, L.A.; Peppercorn, J.M.; Tam, A.L.; Marron, J.M.; Mathews, D.J.; Levit, K.; Roach, N.; Ratain, M.J. Ethical Framework for Including Research Biopsies in Oncology Clinical Trials: American Society of Clinical Oncology Research Statement. J. Clin. Oncol. 2019, 37, 2368–2377. [Google Scholar] [CrossRef]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive molecular profiling of intra- and extrahepatic cholangiocarcinomas: Potential targets for intervention. Clin Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Ramos, I.; Trigo, J.; Palka, M.; Gómez-Rueda, A.; Jantus-Lewintre, E.; Camps, C.; Isla, D.; Iranzo, P.; Ponce-Aix, S.; et al. Clinical utility of plasma-based digital next-generation sequencing in patients with advance-stage lung adenocarcinomas with insufficient tumor samples for tissue genotyping. Ann. Oncol. 2019, 30, 290–296. [Google Scholar] [CrossRef]
- Ettrich, T.J.; Schwerdel, D.; Dolnik, A.; Beuter, F.; Blätte, T.J.; Schmidt, S.A.; Stanescu-Siegmund, N.; Steinacker, J.; Marienfeld, R.; Kleger, A.; et al. Genotyping of circulating tumor DNA in cholangiocarcinoma reveals diagnostic and prognostic information. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Aguado-Fraile, E.; Abou-Alfa, G.K.; Zhu, A.X.; Macarulla, T.; Fan, B.; Nejad, P.; Choe, S.; Jiang, L.; Gliser, C.; Pandya, S.S.; et al. IDH1 mutation detection in plasma circulating tumor DNA (ctDNA) and association with clinical response in patients with advanced intrahepatic cholangiocarcinoma (IHC) from the phase III ClarIDHy study. J. Clin. Oncol. 2020, 38, 4576. [Google Scholar] [CrossRef]
- Paweletz, C.P.; Lau, C.J.; Oxnard, G.R. Does Testing Error Underlie Liquid Biopsy Discordance? JCO Precis. Oncol. 2019, 3, 1–3. [Google Scholar] [CrossRef]
- Zill, O.A.; Greene, C.; Sebisanovic, D.; Siew, L.M.; Leng, J.; Vu, M.; Hendifar, A.E.; Wang, Z.; Atreya, C.E.; Kelley, R.K.; et al. Cell-Free DNA Next-Generation Sequencing in Pancreatobiliary Carcinomas. Cancer Discov. 2015, 5, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [PubMed]
- Galdy, S.; Lamarca, A.; McNamara, M.G.; Hubner, R.A.; Cella, C.A.; Fazio, N.; Valle, J.W. HER2/HER3 pathway in biliary tract malignancies; systematic review and meta-analysis: A potential therapeutic target? Cancer Metastasis Rev. 2017, 36, 141–157. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; ElZawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.-C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- Javle, M.M.; Murugesan, K.; Shroff, R.T.; Borad, M.J.; Abdel-Wahab, R.; Schrock, A.B.; Chung, J.; Goyal, L.; Frampton, G.M.; Kelley, R.K.; et al. Profiling of 3634 cholangiocarcinomas (CCA) to identify genomic alterations (GA), tumor mutational burden (TMB), and genomic loss of heterozygosity (gLOH). J. Clin. Oncol. 2019, 37, 4087. [Google Scholar] [CrossRef]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive parallel sequencing uncovers actionable FGFR2–PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef]
- Churi, C.R.; Shroff, R.; Wang, Y.; Rashid, A.; Kang, H.C.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation Profiling in Cholangiocarcinoma: Prognostic and Therapeutic Implications. PLoS ONE 2014, 9, e115383. [Google Scholar] [CrossRef]
- Borad, M.J.; Champion, M.D.; Egan, J.B.; Liang, W.S.; Fonseca, R.; Bryce, A.H.; McCullough, A.E.; Barrett, M.T.; Hunt, K.; Patel, M.D.; et al. Integrated Genomic Characterization Reveals Novel, Therapeutically Relevant Drug Targets in FGFR and EGFR Pathways in Sporadic Intrahepatic Cholangiocarcinoma. PLoS Genet. 2014, 10, e1004135. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Borad, M.J.; Kelley, R.K.; Wang, Y.; Abdel-Wahab, R.; Meric-Bernstam, F.; Baggerly, K.A.; Kaseb, A.; Al-Shamsi, H.O.; Ahn, D.H.; et al. Cholangiocarcinoma With FGFR Genetic Aberrations: A Unique Clinical Phenotype. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Jain, A.; Kwong, L.N.; Javle, M. Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice. Curr. Treat. Options Oncol. 2016, 17, 58–0432. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.P.; Fritcher, E.G.B.; Pestova, K.; Schulz, J.; Sitailo, L.A.; Vasmatzis, G.; Murphy, S.J.; McWilliams, R.R.; Hart, S.N.; Halling, K.C.; et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum. Pathol. 2014, 45, 1630–1638. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | All Patients (n = 104) | ctDNA Prior to Treatment Cohort (n = 12) | |||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Gender | Female | 52 | 50.0 | 10 | 83.3 |
| Male | 52 | 50.0 | 2 | 16.7 | |
| Age (years) | Median (range) | 62.5 (18.6–83.5) | 67.4 (52.8–80.6) | ||
| Ethnic group | British | 87 | 91.6 | 9 | 75.0 |
| Other | 8 | 8.4 | 3 | 25.0 | |
| Primary tumour | iCCA | 71 | 68.2 | 8 | 66.7 |
| eCCA | 24 | 23.1 | 1 | 8.3 | |
| GBC | 3 | 2.9 | 1 | 8.3 | |
| Amp | 6 | 5.8 | 2 | 16.7 | |
| Stage | Advanced (metastatic) | 104 | 100 | 12 | 100 |
| Received palliative therapy | Yes | 98 | 94.2 | 12 | 100 |
| No | 6 | 5.8 | 0 | 0.0 | |
| Line of therapy (if palliative therapy) | First-line | 94 | 96.0 | 5 | 41.7 |
| Second-line | 2 | 2.0 | 5 | 41.7 | |
| Third-line | 2 | 2.0 | 2 | 16.6 | |
| Which palliative therapy (if palliative therapy) | Cisplatin-gemcitabine | 90 | 92.0 | 8 | 66.7 |
| FOLFIRINOX | 2 | 2.0 | 0 | 0.0 | |
| FOLFOX | 2 | 2.0 | 3 | 25.0 | |
| Cisplatin + NUC1031 | 2 | 2.0 | 0 | 0.0 | |
| Gemcitabine | 1 | 1.0 | 1 | 8.3 | |
| SIRT | 1 | 1.0 | 0 | 0.0 | |
| Follow-up (months) | Median (range) | 12.5 (1.0–50.9) | 8.4 (5.1–11.3) | ||
| Progression | Yes | 85 | 86.7 | 19 | 83.3 |
| PFS (months) | Median (95% CI) | 8.2 (6.9–9.0) | 4.6 (2.4–8.7) | ||
| Died | Yes | 74 | 71.1 | 7 | 58.3 |
| OS from sample (months) | Median (95% CI) | 14.8 (12.3–20.3) | 7.7 (5.9-not reached) | ||
| OS from palliative therapy initiation (months) | Median (95% CI) | 16.4 (13.3–19.7) | 7.4 (4.6-not reached) | ||
| All Samples (n = 112) | Tissue Sample (n = 90) | ctDNA (n = 22) | p-Value (Blood vs. ctDNA) | ||
|---|---|---|---|---|---|
| Pathological molecular findings | Yes | 99; 88.4% | 80; 88.9% | 19; 86.4% | 0.740 |
| Targetable findings | Yes | 45; 40.2% ^; 45.5% * | 39; 43.3% ^; 48.6% *; | 6; 27.3% ^; 31.6% *; | 0.177 |
| Presence of variants of unknown significance | Yes | 76; 67.9% | 60; 66.7% | 16; 72.7% | 0.585 |
| Max MAF reported | Median (range) | n/a | n/a | 0.89 (0.21–76.4) | n/a |
| TMB (Mut/Mb) | Median (range) | n/a | 2 (0–6) | n/a | n/a |
| All Samples (n = 112) | Tissue Sample (n = 90) | ctDNA (n = 22) | ctDNA Prior to Palliative Therapy (n = 12) | Individual Patients (n = 89) | Targetable | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | n | % | n | % | |||
| ALK | Fusion | 1 | 0.9 | 0 | 0.0 | 1 | 4.5 | 1 | 8.3 | 1 | 1.1 | * |
| APC | Mutation | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | |
| ARID1A | Mutation | 6 | 5.4 | 6 | 6.7 | 0 | 0.0 | 0 | 0.0 | 6 | 6.7 | |
| ATM | Mutation | 5 | 4.5 | 3 | 3.3 | 2 | 9.1 | 1 | 8.3 | 4 | 4.5 | |
| ATRX | Mutation | 2 | 1.8 | 2 | 2.2 | 0 | 0.0 | 0 | 0.0 | 2 | 2.2 | |
| BAP1 | Mutation | 8 | 7.1 | 8 | 8.9 | 0 | 0.0 | 0 | 0.0 | 8 | 9.0 | |
| BRAF | Mutation | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | * |
| BRCA1 | Mutation | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | |
| CCDN1 | Amplification | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | |
| CDK4 | Amplification | 2 | 1.8 | 2 | 2.2 | 0 | 0.0 | 0 | 0.0 | 2 | 2.2 | |
| CDKN2A | Mutation | 11 | 9.8 | 11 | 12.2 | 0 | 0.0 | 0 | 0.0 | 11 | 12.4 | |
| CHECK2 | Mutation | 1 | 0.9 | 0 | 0.0 | 1 | 4.5 | 0 | 0.0 | 1 | 1.1 | |
| CTNNB1 | Mutation | 1 | 0.9 | 0 | 0.0 | 1 | 4.5 | 0 | 0.0 | 1 | 1.1 | |
| ERRB2 | Amplification | 2 | 1.8 | 2 | 2.2 | 0 | 0.0 | 0 | 0.0 | 2 | 2.2 | * |
| ERRB3 | Amplification | 3 | 2.7 | 3 | 3.3 | 0 | 0.0 | 0 | 0.0 | 3 | 3.4 | * |
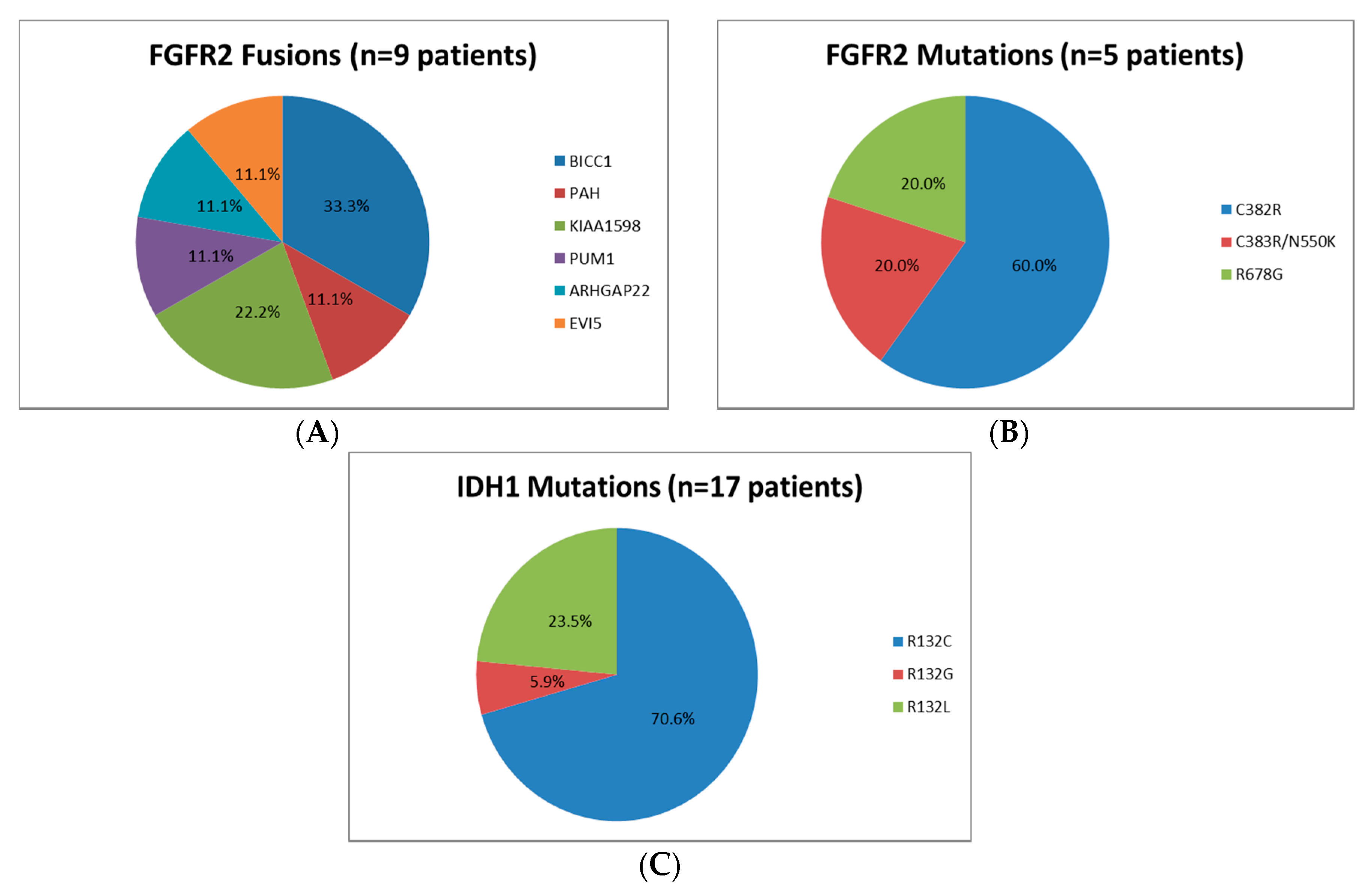
| FGFR2 | Mutation | 7 | 6.3 | 6 | 6.7 | 1 | 4.5 | 1 | 8.3 | 5 | 5.6 | * |
| FGFR2 | Fusion | 12 | 10.7 | 8 | 8.9 | 4 | 18.2 | 1 | 8.3 | 9 | 10.1 | * |
| GNAS | Mutation | 1 | 0.9 | 0 | 0.0 | 1 | 4.5 | 1 | 8.3 | 1 | 1.1 | |
| IDH1 | Mutation | 21 | 18.8 | 17 | 18.9 | 4 | 18.2 | 2 | 16.7 | 17 | 19.1 | * |
| IDH2 | Mutation | 6 | 5.4 | 6 | 6.7 | 0 | 0.0 | 0 | 0.0 | 5 | 5.6 | * |
| KRAS | Mutation | 28 | 25.0 | 27 | 30.0 | 1 | 4.5 | 0 | 0.0 | 24 | 27.0 | |
| MDM2 | Amplification | 6 | 5.4 | 6 | 6.7 | 0 | 0.0 | 0 | 0.0 | 6 | 6.7 | |
| MET | Mutation | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | |
| MET | Amplification | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | |
| MSH6 | Mutation | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | |
| NF1 | Mutation | 4 | 3.6 | 1 | 1.1 | 3 | 13.6 | 2 | 16.7 | 4 | 4.5 | |
| NRAS | Mutation | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | |
| PI3KCA | Mutation | 4 | 3.6 | 3 | 3.3 | 1 | 4.5 | 0 | 0.0 | 3 | 3.4 | |
| POLD1 | Mutation | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | |
| PTEN | Mutation | 5 | 4.5 | 4 | 4.4 | 1 | 4.5 | 0 | 0.0 | 4 | 4.5 | |
| RB1 | Mutation | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | |
| RNF43 | Mutation | 1 | 0.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 1 | 1.1 | * |
| SMAD4 | Mutation | 6 | 5.4 | 6 | 6.7 | 0 | 0.0 | 0 | 0.0 | 6 | 6.7 | |
| TP53 | Mutation | 22 | 19.6 | 14 | 15.6 | 8 | 36.4 | 7 | 58.3 | 22 | 24.7 | |
| Other | Mutation | 36 | 32.1 | 33 | 36.7 | 3 | 13.6 | 1 | 8.3 | 36 | 40.4 | |
| Other | Amplification | 16 | 14.3 | 16 | 17.8 | 0 | 0.0 | 0 | 0.0 | 16 | 18.0 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamarca, A.; Kapacee, Z.; Breeze, M.; Bell, C.; Belcher, D.; Staiger, H.; Taylor, C.; McNamara, M.G.; Hubner, R.A.; Valle, J.W. Molecular Profiling in Daily Clinical Practice: Practicalities in Advanced Cholangiocarcinoma and Other Biliary Tract Cancers. J. Clin. Med. 2020, 9, 2854. https://doi.org/10.3390/jcm9092854
Lamarca A, Kapacee Z, Breeze M, Bell C, Belcher D, Staiger H, Taylor C, McNamara MG, Hubner RA, Valle JW. Molecular Profiling in Daily Clinical Practice: Practicalities in Advanced Cholangiocarcinoma and Other Biliary Tract Cancers. Journal of Clinical Medicine. 2020; 9(9):2854. https://doi.org/10.3390/jcm9092854
Chicago/Turabian StyleLamarca, Angela, Zainul Kapacee, Michael Breeze, Christopher Bell, Dean Belcher, Helen Staiger, Claire Taylor, Mairéad G. McNamara, Richard A. Hubner, and Juan W. Valle. 2020. "Molecular Profiling in Daily Clinical Practice: Practicalities in Advanced Cholangiocarcinoma and Other Biliary Tract Cancers" Journal of Clinical Medicine 9, no. 9: 2854. https://doi.org/10.3390/jcm9092854