New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges


1
Department of Clinical and Experimental Medicine, University of Messina, 98125 Messina, Italy
2
NEuroMuscular Omnicentre (NEMO) Sud Clinical Centre, University Hospital “G. Martino”, 98125 Messina, Italy
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2020, 9(7), 2222; https://doi.org/10.3390/jcm9072222
Submission received: 3 June 2020
/
Revised: 1 July 2020
/
Accepted: 10 July 2020
/
Published: 13 July 2020
(This article belongs to the Special Issue Innovative Therapies in Neuromuscular Diseases)
Abstract
:Spinal muscular atrophy (SMA) is one of the most common autosomal recessive diseases with progressive weakness of skeletal and respiratory muscles, leading to significant disability. The disorder is caused by mutations in the survival motor neuron 1 (SMN1) gene and a consequent decrease in the SMN protein leading to lower motor neuron degeneration. Recently, Food and Drug Administration (FDA) and European Medical Agency (EMA) approved the antisense oligonucleotide nusinersen, the first SMA disease-modifying treatment and gene replacement therapy by onasemnogene abeparvovec. Encouraging results from phase II and III clinical trials have raised hope that other therapeutic options will enter soon in clinical practice. However, the availability of effective approaches has raised up ethical, medical and financial issues that are routinely faced by the SMA community. This review covers the available data and the new challenges of SMA therapeutic strategies.
1. Introduction
Spinal muscular atrophy (SMA) is a heterogeneous hereditary neuromuscular disease, presenting with progressive weakness of skeletal and respiratory muscles, leading to muscle atrophy and significant disability. The disease is caused by a homozygous deletion or a heterozygous deletion combined with point mutation on the other allele on the survival motor neuron 1 (SMN1) gene on chromosome 5q and consequential lack of the SMN proteins, causing degeneration of lower motor neurons [1,2]. SMA encompasses a wide range of clinical severity and has been classified into subtypes according to age at onset and the maximum motor milestones achieved, ranging from the most severe SMA I to the mildest SMA IV.
SMA I patients never acquire the sitting position and present clinical onset before six months as “floppy infant” with reduced spontaneous movements and paradoxical breathing pattern. The life expectancy related to respiratory muscle failure is under two years without drug treatment [3]. SMA II shows a milder course with onset between six and 18 months. Patients are able to sit, but not walk independently and develop respiratory involvement that usually requires the use of non-invasive ventilation before adulthood and orthopedic complications such as severe scoliosis and joint contractures. SMA III and IV are the mildest forms with later onset, achievement of independent walking and variable clinical course, in SMA type IV usually without life-threatening events. SMA I and II patients often present failure to thrive and dysphagia and require to increase nutritional uptake with the use of hyperproteic and hypercaloric oral supplements or, in the more severe cases, gastrostomy placement. These patients require a multidisciplinary rehabilitative approach covering respiratory, orthopedic, psychological, physio- and speech-therapist and nutritional care [4,5].
SMA is one of the most common autosomal recessive diseases and causes of mortality in childhood with a carrier frequency of one in 40–67 adults and an incidence of one in 11,000 live births [6]. However, the epidemiologic burden of SMA differs among the subtypes. Several studies approached this issue showing a lower incidence for SMA III compared to the other forms. Ogino et al., in their review, proposed in SMA type I an incidence rate of 5.83 per 100,000 live births, in SMA type II 2.66 per 100,000 livebirths and in SMA type III 1.20 per 100,000 live births. Therefore, SMA type I, II and III constituted, respectively, 60%, 27% and 12% of all SMA cases [7]. A more recent review showed for SMA type I, II and III incidence rates of around 5.5, 1.9 and 1.7 per 100,000, respectively [8].
The clinical variability in SMA is mainly attributable to variable copy numbers of SMN2, which is a SMN1 paralogous gene, producing a protein lacking exon 7 (SMNΔ7) due to alternative splicing, but also small amounts of functional SMN proteins. The number of SMN2 copies is inversely related to clinical phenotypes [9]. However, several genetic modifiers, such as plastin-3 and neurocalcin delta, have been hypothesized to explain the clinical variability between patients with the same number of SMN2 copies and within discordant families [10].
Recently, the antisense oligonucleotide (ASO) nusinersen and the gene replacement therapy by onasemnogene abeparvovec have become available for SMA patients. Moreover, encouraging results from phase II and III clinical trials have raised hope that other therapeutic options will enter soon in clinical practice [11,12,13,14,15]. However, the availability of effective approaches has raised up ethical, medical and financial issues that are routinely faced by the SMA community.
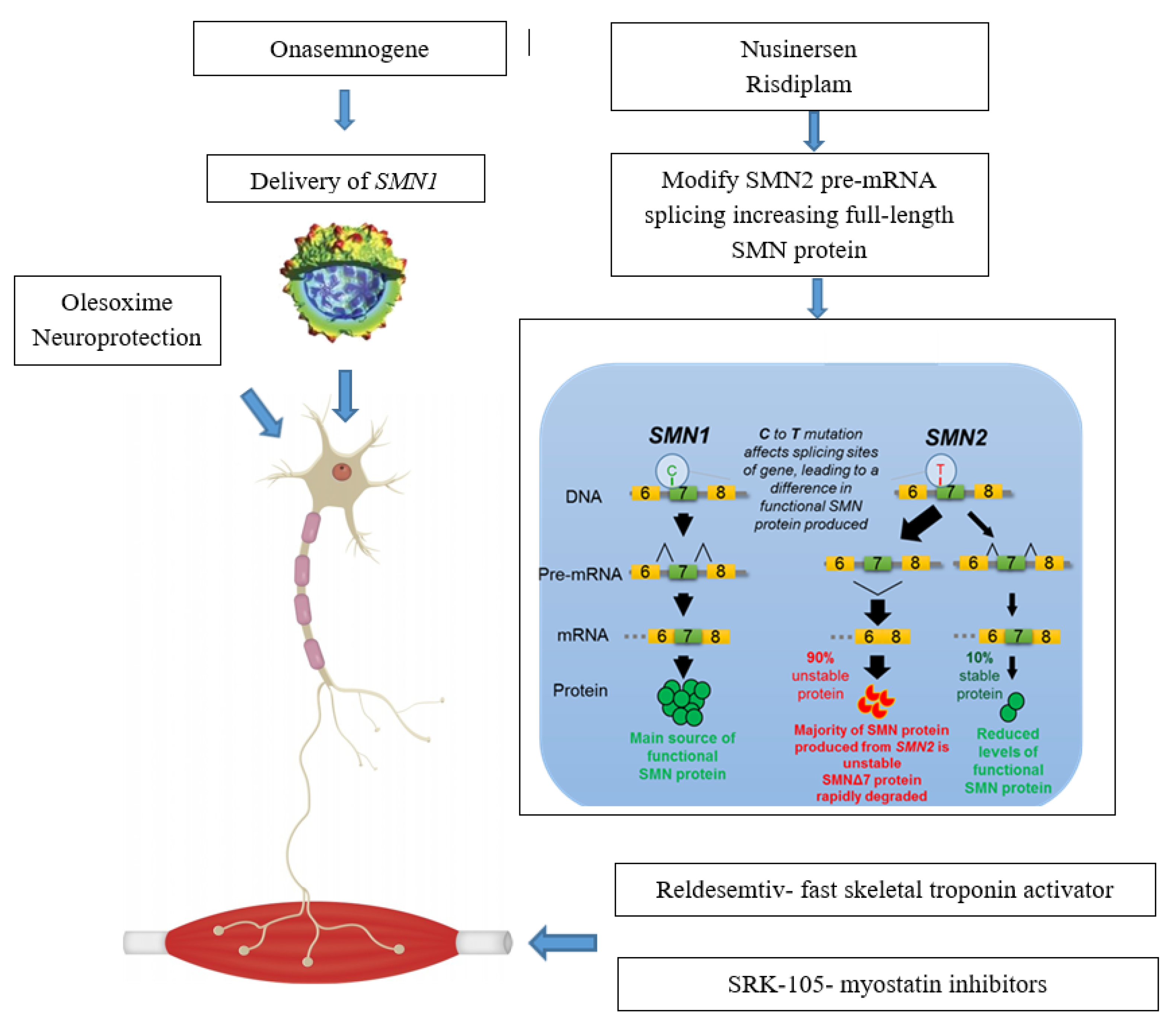
Actual approaches can be subdivided in Survival Motor Neuron (SMN)—Dependent Gene Therapies, which act as splicing modificator of SMN2 (nusinersen, small molecules) or replacing SMN1 gene (onasemnogene abeparvovec) and in treatments targeting SMN—Independent Factors (muscle enhancing therapies and neuroprotection) (Figure 1).
This review covers the available data of SMA therapeutic strategies in pre-clinical development, currently tested in clinical trials and available in clinical practice.
2. SMN—Dependent Gene Therapies
2.1. Splicing Modification of SMN2
2.1.1. Nusinersen
The first approved drug for SMA was nusinersen, which is an ASO that promotes the inclusion of exon 7 in mRNA transcripts of SMN2. Nusinersen binds to an intronic splice-silencing-site in intron 7 of SMN2 and inhibits the action of other splice-factors, promoting exon 7 incorporation into the mRNA. This mechanism allows the translation of a higher level of fully functional SMN protein with a significant amelioration of survival and pathology in different SMA experimental models [16,17,18]. The journey of nusinersen towards approval and commercialization has been supported by several trials demonstrating efficacy without any major drug-related adverse event. ASOs do not cross the blood-brain barrier, therefore in all clinical trials nusinersen was administered intrathecally with a frequency of four times over two months in the initial loading period and every four months in the maintenance period.
After promising results for nusinersen in phase I and II trials in children with SMA type II and III [19,20], two phase III, randomized, double-blind, sham-procedure controlled studies were initiated consequently. ENDEAR (ClinicalTrials.gov identifier: NCT02193074, years 2014–2016) assessed safety and clinical efficacy of nusinersen in 121 infants with infantile-onset SMA and younger than seven months. In the interim analysis, infants treated with nusinersen had higher improvement in the motor milestone categories of the Hammersmith Infant Neurological Examination (HINE) than controls (41% vs. 0%, p < 0.001). Moreover, the nusinersen group demonstrated a prolonged time to death (hard ratio for death 0.37; p = 0.004) or need for permanent ventilation compared to controls and six out of 73 treated patients achieved independent sitting over a one year treatment period. Furthermore, infants with shorter disease duration at screening had better response to treatment [21].
CHERISH (ClinicalTrials.gov identifier: NCT02292537, years 2014–2017) involved 126 children with later-onset SMA. The median age at baseline was four years (two to nine years) in the treated group and three years (two to seven years) in the controls. Interim analysis after 15 months of treatment showed in the nusinersen group a mean increase of 4.0 points vs. a mean decrease of 1.9 points in controls (p < 0.001) in the Hammersmith Functional Motor Scale-Expanded (HFMSE) score. Regarding upper limb function, there was a least-squares mean increase from baseline to month 15 in the Revised Upper Limb Module (RULM) score in the nusinersen group of 4.2 points and a mean decrease in the control group of −0.5 points (p < 0.001). In the final analysis, 57% of nusinersen patients vs. 26% in the sham group had a rise of three points in HFMSE scores after 15 months of treatment [22]. Both trials were terminated prematurely on the basis of these positive results and patients continued treatment in an open-label extension study (SHINE, ClinicalTrials.gov identifier: NCT02594124, started in 2017 and ongoing) with the aim of assessing long-term effects of nusinersen in terms of safety and tolerability and clinical changes (HFMSE, RULM and WHO motor milestones). Interim analysis showed a continued benefit of nusinersen on HFMSE and RULM score over day 1170 of treatment and confirmed a more evident effect in younger patients at first dose (2.06 to <3.69 years) [23].
The ad interim results of the NURTURE open-label study (ClinicalTrials.gov identifier: NCT02386553, started in 2017 and ongoing) emphasized the importance of proactive treatment with nusinersen as soon as reached the genetic diagnosis in presymptomatic infants and strengthened the rationale for newborn screening (NBS). This study involved 25 presymptomatic SMA infants below six weeks of age and, after a median of 2.9 years of follow up, the effects of nusinersen were very encouraging. At a mean age of 34.8 (25.7–45.4) months of age, 100% of patients were alive, 92% achieved walking with assistance and 88% independently [24].
Nusinersen was approved by Food and Drug Administration (FDA) in December 2016 and by European Medical Agency (EMA) in June 2017. While waiting for regulatory approval, nusinersen was made available for compassionate use for SMA type I infants by the pharmaceutical company. The advent of an expanded access programs (EAP) using an intrathecal administration in fragile infants such as type 1 SMA has raised a number of issues, related to its planning and management, that have been dealt differently among countries [25]. However, “real-world data” confirmed therapeutic benefits with motor function improvements in SMA I [26,27,28,29]. We recently demonstrated in 85 SMA I patients (aged from two months to 15 years) after a year of treatment an amelioration on both the functional scales, Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) and HINE-2 for the whole group (p < 0.001) and the subgroups with two and three SMN2 copies (p < 0.001). The difference was found not only in patients younger than 210 days at baseline (p < 0.001), but also on the CHOP-INTEND in those younger than five years and on the HINE-2 younger than two years [30].
2.1.2. Small Molecules
Risdiplam, previously known as RO703406 and RG7916, is a small molecule that modulates SMN2 gene splicing, binding two sites in SMN2 pre-mRNA: 5′ splice site (5′ ss) of intron 7 and exonic splicing enhancer 2 (ESE2) in exon 7. The unique specificity of binding two sites increases levels of full-length SMN mRNA and protein, while reducing the impact on splicing of other pre-mRNA and avoiding the possibility of off-target effects [31]. Preclinical studies showed that risdiplam can reach the central nervous system and peripheral organs in vivo and can lead to a significant increase of SMN protein in blood, brain and muscles, with an increase survival in different SMA mouse models [32,33]. One of the advantage of this drug is the oral route of administration. While the intrathecal administration route of nusinersen mainly limits its effect to motoneurons of the central nervous system, the systemic distribution demonstrated in preclinical studies with risdiplam by oral administration allows to hypothesize a possible effect in other tissues. This is of relevance, as numerous studies in human and murine models indicate that SMA may actually be considered as a multi-system disorder with an involvement of neuromuscular junction, gastrointestinal-tract, cardio-vascular system, lung and liver [34,35].
A phase I study in healthy volunteers (ClinicalTrials.gov identifier: NCT02633709, years 2016 and 2017) identified the optimal dosage and demonstrated that risdiplam increases SMN2/SMN∆7 mRNA ratio in a dose-dependent manner [36].
Four ongoing phase II trials are assessing safety and efficacy of risdiplam in different SMA types. The results of these studies have been presented in several conferences but are not yet published. FIREFISH (ClinicalTrials.gov identifier: NCT02913482) is an open-label trial testing the effectiveness in SMA I patients between one and seven months of age with two copies of SMN2. Part 1 of FIREFISH has tested for a year the drug at two different dosages, while Part 2 assesses the effectiveness at the selected higher dose. Part 1 involved 21 patients: Four received the lower dose and 17 the higher dose that is currently being used in Part 2 of the trial. After receiving risdiplam for 12 months, 90.5% of infants were alive with no permanent ventilation, 33% in the whole cohort and 41% of infants treated with the higher dose were able to sit without support for at least 5 s and 86% of all infants showed a ≥4-point improvement in CHOP-INTEND score from baseline. Moreover, 94.7% of infants are able to feed orally or in combination with a feeding tube ad no infant has lost the ability to swallow [37]. At the cut-off point after 16 months of treatment, the median change from baseline in CHOP-INTEND score was 19 over 64 of maximum score and 5% of infants were able to stand supporting their weight and 10% were able to bounce [38]. Preliminary final results of the Part 2 have been recently presented. Event-free survival time was greatly improved in infants treated with risdiplam compared to natural history. Treated patients showed a significant improvement in HINE-2 and CHOP-INTEND after 12 months. Swallowing and feeding ability was maintained by the majority of infants. Nearly half of the treated patients did not require hospitalization up to 12 months [39].
SUNFISH (ClinicalTrials.gov identifier: NCT02908685, started in 2016 and open-label extension ongoing) is assessing safety, tolerability and effectiveness in SMA type II and III cases aged two to 25 years who are not ambulatory. Part 1 of SUNFISH (n = 51) evaluated safety, pharmacodynamics (PD), pharmacokinetic (PK) and optimal dosage of risdiplam, while its efficacy is tested vs. placebo with a 2:1 randomization in Part 2 (n = 180). The results of the SUNFISH Part II were presented very recently and the primary end-point was met with a least squares mean changes at the 32-item motor function measure from baseline significantly greater in patients receiving risdiplam vs. placebo (Δ = 1.55; p = 0.0156). Moreover, RULM total change from baseline was significantly greater in patients receiving risdiplam vs. placebo (Δ = 1.59; = 0.0028), whereas the difference was not significant at the HFMSE. At the SMA Independence Scale (SMAIS) caregivers and patients (≥12 years) in the treated group reported improvement in independence when completing activities of daily living [40].
JEWELFISH (ClinicalTrials.gov identifier: NCT03032172, started in 2018 and ongoing) is an open-label, exploratory study to assess the safety, tolerability, PD and PK in patients with a broad spectrum of age (six months–60 years), who have previously participated in a study with a therapy targeting SMN2 pre-mRNA splicing.
RAINBOW FISH (ClinicalTrials.gov identifier: NCT03779334, started in 2019 and ongoing) is an open-label, single-arm, multicenter clinical study to investigate the efficacy, safety, PK and PD in pre-symptomatic infants enrolled between birth and six weeks of age.
Based on the above-mentioned positive results, risdiplam is under review for approval by FDA and a recent press release announced that FDA has extended the Prescription Drug User Fee Act date for its review of the New Drug Application of risdiplam till August 24 2020 [41]. In parallel, in many countries an EAP has been started for SMA type I patients (and in US also for SMA type II; ClinicalTrials.gov Identifier: NCT04256265), who were not eligible for treatment with currently approved treatments for SMA or cannot continue treatment as documented by the treating physician.
2.2. SMN1 Gene Replacement
Onasemnogene abeparvovec (previously known as AVXS-101) is a SMN1 gene replacement therapy, which uses a non-replicating adeno-associated virus capsid (scAAV9) to efficiently deliver wild-type SMN1 gene to motor neuron cells. This construct, an AAV9 vector carrying SMN1 complementary recombinant DNA, can cross the brain–blood barrier, produces a sustained expression of SMN protein and prolongs survival of treated SMA-mice [42,43,44]. The main advantages of this approach are that a one-time injection is needed and it would lead to systemic expression of the SMN protein. Safety and tolerability has to be strictly monitored as acute hepatotoxicity and sensory neuron toxicity were reported in primates and piglets following high-dose intravenous administration of AAV vectors expressing human SMN [45]. Another issue could be the reported presence of pre-existing anti-AAV9 antibody in the SMA population [46].
AVXS-101-CL-101 (ClinicalTrials.gov identifier: NCT02122952, years 2014–2017) is an open-label study of 15 SMA type I cases with bi-allelic SMN1 mutations (deletion or point mutations) and two copies of SMN2. Onasemnogene abeparvovec was administered as a single intravenous injection at two different doses: Twelve patients receiving 2.0 × 1014 vector genomes (vg) per kg and three receiving 6.7 × 1013 vg per kg. Serum aminotransferase levels increased in four cases, but returned to normal levels after treatment with corticosteroids. After the elevation in the first patient, the protocol was amended with addition of oral prednisolone for four weeks after drug administration. In the high-dose group, the scores of the CHOP-INTEND showed a rapid increase of 9.8 points after one month and 15.4 points after three months, compared to the decrease observed in SMA type I natural history [47]. Recently, a comparison with SMA type I untreated patients of the NeuroNEXT (NN101) study (ClinicalTrials.gov identifier: NCT01736553, started in 2017) [48] showed that after 24 months of follow-up the survival rate was 100% in AVXS-101-treated infants and 38% in the NN101 study cohort. Baseline mean CHOP-INTEND score was 28.2, improving to 56.5 in the treated group, compared to 20.3 with a decrease to 5.3 in controls. Moreover, 11 (92%) of the AVXS-101–treated infants were able to sit unassisted for ≥5 s, 10 (83%) for ≥10 s, nine (75%) for ≥30 s and two (17%) could stand and walk independently. CHOP-INTEND scores suggested that patients in the NN101 cohort did not achieve any motor milestones [49]. Further analysis demonstrated that the best predictors of functional amelioration were age below three months and high CHOP-INTEND scores at baseline [50].
The results of the STR1VE study (ClinicalTrials.gov Identifier: NCT03306277, 2018–2020) have been recently presented [51]. STR1VE-US is a part of the global phase 3 STR1VE clinical program. This includes open-label, phase 3, single-arm, single-dose, multi-center trials (STR1VE-US in the United States, STR1VE-AP in Asia Pacific and STR1VE-EU in Europe) designed to assess safety and efficacy in symptomatic patients with SMA type I < six months of age with one or two copies of the SMN2 gene. In STR1VE-US, 20 of 22 patients (91%) met the co-primary efficacy endpoint of event-free survival at 14 months and 13 (59%) met the co-primary efficacy endpoint of functional sitting for ≥30 s at 18 months of age. Thirteen patients (59%) could sit independently for ≥30 s (p < 0.0001 vs. natural history) at the 18 months of age. Fifteen patients (68.2%) remain free of non-invasive ventilatory support during the study. Eighteen (81.8%) were free of ventilatory support at 18 months of age. CHOP-INTEND scores ameliorated by a mean of 6.9 points at one month, 11.7 points at three months and 14.6 points at six months after treatment. Twenty-one patients (95%) reached a CHOP-INTEND score ≥40 and 14 (64%) a score ≥50.
SPR1NT (ClinicalTrials.gov Identifier: NCT03505099, started in 2018) is an ongoing Phase 3, open-label, single-arm, multi-center trial designed to assess safety and efficacy of a one-time intravenous infusion in presymptomatic patients with SMA below six weeks of age and two or three copies of SMN2. Fourteen patients with two copies and 15 patients with three copies of SMN2 were treated.
The completed clinical trial, START (ClinicalTrials.gov Identifier: NCT02122952, 2018–2020), enrolled 15 patients with infantile-onset SMA, 12 in a high-dose and three in a low-dose cohort. By 24 months following infusion, none of the patients the high-dose cohort required permanent ventilation. Patients in the low-dose cohort did not reach the ability to sit without support; in the high-dose cohort, nine of 12 patients (75%) reached the ability to sit without support for ≥ 30 s and two patients (17%) to stand and walk independently. These results showed a dose-response relationship.
The longer follow-up data were provided from the START Long-Term Follow-Up (LTFU) (ClinicalTrials.gov Identifier: NCT03421977). This is an ongoing, observational, long-term follow-up study of patients who completed START and electively enrolled in the study. The mean age of patients was 4.8 years (range 4.3–5.6 years) and the mean time since gene therapy treatment was 4.5 years (range 4.1–5.2 years). Of the 10 patients from cohort 2 (high dose of START) who enrolled in LTFU, all are free of permanent ventilation. No changes in the previously achieved milestones have been reported during the follow up period. Two patients have reached the ability of standing with assistance (neither of whom have received treatment with nusinersen) during the follow up period.
Based on these affirmative results, in May 2019 FDA approved onasemnogene abeparvovec for the treatment of SMA patients with less than two years of age with bi-allelic mutations in the SMN1 gene, including those who are pre-symptomatic at diagnosis [52]. Furthermore, in May 2020 the European Commission (EC) granted conditional approval for onasemnogene abeparvovec for SMA type I patients with a bi-allelic mutation in the SMN1 gene or for SMA patients with a bi-allelic mutation in the SMN1 gene and up to three copies of the SMN2 gene. In both cases, the approval covers SMA patients with a weight up to 21 kg according to the dosing guidance [53].
3. Treatments Targeting Survival Motor Neuron (SMN)—Independent Factors
3.1. Muscle Enhancing Therapies
Reldesemtiv (previously known as Tirasemtiv and CK-2127107) is a selective small-molecule troponin activator in fast skeletal muscles. The rationale for its use in SMA stands on several lines of evidence. This molecule increases the affinity of troponin C to calcium, sensitizes the sarcomere to calcium effects and reinforces contraction [54,55]. Moreover, Reldesemtiv has also been demonstrated to promote muscle response to nervous stimulus in humans [56]. Following a phase I study confirming its safety, a phase II, double-blind, randomized, placebo-controlled trial (ClinicalTrials.gov identifier: NCT02644668, years 2015–2018) on 70 patients with SMA type II to IV examined its effect on functional and respiratory performances. The compound were administered orally at two different doses (150 mg × 2/day and 450 mg × 2/day). The results showed, in the higher dosage group, a trend towards an increase from baseline in the six-minute walk test (6MWT) and of the maximal expiratory pressure (MEP). Adverse events were similar between treated and placebo groups [57].
SRK-015 is a monoclonal antibody, which selectively inhibits myostatin, promoting muscle cells growth and differentiation and improving muscle force in SMA mice [58,59]. A phase I trial (ClinicalTrials.gov identifier: NCT02644777, years 2017–2018) confirmed its safety and tolerability. A phase II study (TOPAZ, ClinicalTrials.gov identifier: NCT03921528, started in 2019 and ongoing), involved 58 SMA type II and SMA III patients, aged two to 21 years. Patients have received treatment by intravenous infusion every four weeks for one year. The six-month interim results will be available by the end of 2020.
3.2. Future Prospectives in SMN Independent Therapeutic Targets
Several SMN independent factors have been identified over the last years as involved in SMA pathogenesis on the basis of in vitro and in vivo studies and therefore they could represent future therapeutic targets.
Autophagy is the process by which cytoplasmic contents are delivered by autophagosomes to the lysosomes for degradation. In vitro and in vivo studies reported an increase of autophagosomes in the cytoplasm of SMA motoneurons, suggesting that autophagy dysregulation might alter intracellular trafficking, leading to cytotoxicity [60,61,62]. Moreover, intramuscular injections of the neurotrophic factor tetanus toxin heavy chain (TTC) can reduce the expression of autophagy markers (Becn1, Atg5, Lc3, and p62) in SMNΔ7 mice muscles without effects on weight and survival time [63]. Furthermore, inhibition of autophagy by intracerebroventricular administration of 3-methyladenine (3-MA) has been shown to ameliorate autophagic features, increase lifespan and improve motor performances in SMA pups [62].
Autophagy and apoptosis are linked in SMA as shown by the fact that 3-MA administration reduces also apoptotic cell death in the lumbar spinal cord [62]. Apoptosis has been demonstrated to be involved in SMA pathogenesis by several evidence. SMN protein decrement promotes apoptosis in vitro [64]. The c-Jun NH2-terminal kinase (JNK) cascades, known to have a pro-apoptotic role, is activated in SMNΔ7 mice and in SMA patients [65]. Moreover, the double JNK3-SMNΔ7 knockout (KO) mouse model shows a milder SMA phenotype [65]. Furthermore, JNK pharmacological inhibition ameliorates morphological features, improves motor performances and lifespan of SMA mice [66].
Another possible therapeutic target is agrin, a synaptic organizer relevant for the efficiency of neuromuscular transmission. A reduction of 50% in agrin expression levels has been found in muscle of SMNΔ7 mice. The administration of C-terminal fragment of mouse agrin to SMA pups can restore the crosstalk between muscles and motoneurons with positive effects on the maturation of the neuromuscular junction and on muscle tropism [67]. The repletion of agrin parallels to an amelioration on the overall disease phenotype and to a prolonged survival in severely affected SMA model mice [68].
Interestingly, the nuclear factor-kappa B pathway, deeply studied in another neuromuscular disorder in childhood, such as Duchenne muscular dystrophy, seems to be implicated also in SMA pathogenesis. This pathway modulates cell survival in mice spinal cord motor neurons induced by neurotrophic factors and its inhibition causes SMN reduction in SMA motoneurons [69,70,71].
Therefore, in conclusion, the results of these studies are promising, but additional evidence is needed before clinical translation of new compounds acting on these cellular/molecular pathways.
4. Discussion
The management of SMA is deeply changing and several issues have to be considered as clinicians use these innovative, effective and expensive new treatments (Table 1).
Regardless of the approach, the presented studies demonstrated better efficacy in SMA children pre-symptomatic or with the shortest disease duration [72]. This is a consequence of the rapid denervation process occurring in the first six months of life and the aim of pre-symptomatic treatment is the precocious rescue of motoneurons. However, we recently demonstrated that the mean age at diagnosis is 4.70 months (SD ± 2.82) in SMA type I, 15.6 months (SD ± 5.88) in SMA type II, and 4.34 years (SD ± 4.01) in SMA type III [73]. Therefore, these findings support the need of NBS to achieve a better efficacy of the available therapeutic options. NBS is nowadays performed in Italy, Taiwan, some States in US, Belgium and Germany as pilot studies [74,75,76,77,78]. The decision of how to handle newborns who test positive in the screening is crucial. An algorithm, based upon SMN2 copy number, has been proposed by the SMA NBS Multidisciplinary working group, sponsored by CureSMA. The experts reached a consensus to start immediately treatment in infants with one, two and three SMN2 copies, regardless of the presence of symptoms and to strictly follow patients with four copies till symptoms’ onset and then start treatment. However, it is still under debate how to handle families and infants with four or more copies and expected to have mild and late-onset disease [79,80,81]. Moreover, this aspect is further complicated by the fact that the correlation between SMN2 copies and phenotype can be influenced by genetic modifiers, as demonstrated in siblings with the same SMA genotype [82]. A qualified genetic counselling and psychological support are mandatory to help parents to face the stressful situation to receive a severe diagnosis in an apparently healthy baby and to take relevant treatment decisions.
The variability in treatment response among patients could be better understood with the availability of reliable biomarkers. This knowledge would help to identify prognostic factors, to avoid long-term exposure to expensive drugs with still unknown long-term drug-related adverse events. A variety of biomarkers is currently under investigation including epigenetic, genetic, proteomic, electrophysiological and imaging tools [83]. Neurofilaments (NFs) muscle-specific miRNAs (myomiRs) and CSF proteomic profile, although biomarkers are not yet validated, have recently drawn attention as promising tools in SMA. NFs are markers of axonal degeneration. In infants with SMA type I the level of CSF NFs was significantly higher than in controls, with a response to nusinersen treatment that correlated with clinical improvement [84]. Similar results were confirmed in plasma in the ENDEAR study in symptomatic SMA type I patients [85]. In older patients with less severe forms of SMA, the role of NFs has not yet been confirmed probably as consequence of a slower disease progression [86,87]. SMN protein regulates RNA metabolism and biogenesis of microRNA (miRNA), which are gene expression modulators, and their dysregulation is implicated in a variety of neuromuscular diseases. Recently, a reduced expression level of circulating myomiRs miR-133a, -133b, miR-206 and −1 has been demonstrated in SMA type II and III patients under nusinersen treatment. Moreover, miR-133a decrement could be a predictor of motor function response to therapy [88]. A recent study evaluated, by mass spectrometry, non-targeted CSF proteomic profiles in SMA type II and III patients. The analysis highlighted two groups with differences in age and expression of proteins related to neurodegeneration and neuroregeneration. Moreover, intraindividual CSF differences were present between non-responders and responders to nusinersen treatment, with a correlation with motor functional improvement in the latter group [89].Considering the high costs of the approved new treatments and the limited data on long-term efficacy and safety, the scientific community feels the burning need to systematically collect “real-world data” to provide evidence for clinical decision-making and reimbursement. Only one study reported the results of a number needed to treat (NNT) analysis comparing the efficacy of nusinersen and onasemnogene abeparvovec on several outcomes using data from AVXS-101-CL-101 and ENDEAR studies in symptomatic SMA type I infants. Authors demonstrated an efficacy advantage of onasemnogene abeparvosec in terms of motor milestones achieved, motor function (CHOP-INTEND score) improvement and independence from permanent assisted ventilation. Moreover, the probability of preventing death was 20% higher in the onasemnogene abeparvosec treated group [90]. However, the main drawback of this study is the use of an unanchored indirect analysis of NNT between two studies. Head-to-head clinical trials should be performed to estimate comparative efficacy of the available approaches, avoiding possible biases such as differences in study design and patient characteristics. Moreover, a recent study highlights wide variations in cost and benefit estimates of nusinersen and indicates that onasemnogene abeparvosec is unlikely to represent “value for money” according to current standards of reimbursement for the UK NHS [91]. On the contrary, a US study demonstrated an incremental cost-effectiveness ratio (ICER), expressed as cost/quality-adjusted life year ($/QALY), of $46,947 for chronic treatment with nusinersen and of $31,379 base case at a price of $5M for onasemnogene abeparvosec, indicating that the latter was cost-effective with prices of ≤$5M [92]. Several international disease-specific registries have now been established (e.g., the International SMA Consortium Spinal Muscular Atrophy Patient Registry (iSMAC), the TREAT-NMD registry and the SMArtCARE project), collecting “real-world data” on treated and untreated SMA patients with standardized outcome measures, including patient-oriented tools on a longitudinal setting [93,94,95,96,97,98].
All studies so far have been conducted in patients followed in tertiary referral centers with the recommended standards of care and this likely contributed to the positive results of the trials. Nowadays, families have different therapeutic options, but it has to be highlighted by clinicians that the efficacy of these new compounds is significantly related to the adherence to a careful multidisciplinary management of care. Moreover, increment in muscle strength, acquisition of new milestones and increased survival are bringing attention to emerging phenotypes [15]. Treated SMA type I patients can be able to sit also unsupported and; therefore, with the effect of gravity, they exhibit a higher rate of scoliosis with often severe kyphoscoliosis in the first years of life. A careful X-ray and clinical monitoring, the use of braces and eventually the surgical option with “growing rods” are crucial. Moreover, the increase in muscle strength promotes the worsening of contractures, therefore intensive stretching and the use of standing frame or knee-ankle-foot-orthoses when possible have to be envisaged. The reported cognitive involvement might be more frequent in longer-surviving SMA type I patients and this aspect has to be elucidated in long term follow-up [99].
5. Conclusions
We are facing an exciting era with three available therapeutic options in a disease considered incurable for more than a century. NBS leading to early treatment is vital to provide optimal care. Each therapeutic strategy has its weaknesses and strengths and clinicians need to know them to optimize clinical care. Once these approaches will be available for all SMA types, the final choice will be based on patient’s clinical features and compliance and on the feasibility of drug administration. Since post-symptomatically treated patients are not cured, patients and parents ask for combined approaches. Additional therapies to complement present and forthcoming SMN-targeted treatments are needed in order to optimize their effects. Combinations of different drugs that increase SMN level or with muscle enhancing therapies must be tested in clinical trials.
The validation of prognostic factors and biomarkers to precisely detect evidence of response would decrement the time of exposure to expensive medications with unknown long-term drug-related adverse events, identifying in non-responders accurate treatment discontinuation criteria. The complementary effort by patients, care-givers, SMA advocacy groups and policymakers is required to continue promoting innovation for the benefit of SMA patients.
Author Contributions
S.M. provided the conception, draft and writing of this review. M.S. provided the conception and revision of this review. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
S.M. is member of advisory boards and has consulted for Avexis, Roche, Biogen Idec. She was also principal investigator for trials funded by Ionis Pharmaceuticals. M.S. declares no conflict of interest.
References
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercuri, E.; Bertini, E.; Iannaccone, S.T. Childhood spinal muscular atrophy: Controversies and challenges. Lancet Neurol. 2012, 11, 443–452. [Google Scholar] [CrossRef]
- Farrar, M.A.; Vucic, S.; Johnston, H.M.; du Sart, D.; Kiernan, M.C. Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J. Pediatr. 2013, 162, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Mercuri, E.; Meyer, O.H.; Simonds, A.K.; Schroth, M.K.; Graham, R.J.; Kirschner, J.; Iannaccone, S.T.; Crawford, T.O.; Woods, S.; et al. Diagnosis and management ofspinal muscular atrophy: Part 2: Pulmonary and acutecare; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 2018, 28, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Sugarman, E.A.; Nagan, N.; Zhu, H. Pan-ethnic carrier screening and Prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72,400 specimens. Eur. J. Hum. Genet. 2012, 20, 27–32. [Google Scholar] [CrossRef]
- Ogino, S.; Wilson, R.B.; Gold, B. New insights on the evolution of the SMN1 and SMN2 region: Simulation and meta-analysis for allele and haplotype frequency calculations. Eur. J. Hum. Genet. 2004, 12, 1015–1023. [Google Scholar] [CrossRef]
- Ingrid, E.C.; Verhaart, I.E.C.; Robertson, A.; Wilson, I.J.; Aartsma-Rus, A.; Cameron, S.; Jones, C.C.; Cook, S.F.; Lochmüller, H. Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy–A literature review. Orphanet J. Rare Dis. 2017, 2, 124. [Google Scholar] [CrossRef] [Green Version]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Chen, T.H. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? Int. J. Mol. Sci. 2020, 21, 3297. [Google Scholar] [CrossRef] [PubMed]
- Messina, S. New Directions for SMA Therapy. J. Clin. Med. 2018, 7, 251. [Google Scholar] [CrossRef] [Green Version]
- Wadman, R.I.; van der Pol, W.L.; Bosboom, W.M.; Asselman, F.L.; van den Berg, L.H.; Iannaccone, S.T.; Vrancken, A.F. Drug treatment for spinal muscular atrophy type I. Cochrane Database Syst. Rev. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Wadman, R.I.; van der Pol, W.L.; Bosboom, W.M.; Asselman, F.L.; van den Berg, L.H.; Iannaccone, S.T.; Vrancken, A.F. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst. Rev. 2020, 1. [Google Scholar] [CrossRef]
- Ramdas, S.; Servais, L. New treatments in spinal muscular atrophy: An overview of currently available data. Expert Opin. Pharmacother. 2020, 21, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Schorling, D.C.; Pechmann, A.; Kirschner, J. Advances in Treatment of Spinal Muscular Atrophy—New Phenotypes, New Challenges, New Implications for Care. J. Neuromuscul. Dis. 2020, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol. Cell Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Rigo, F.; Hua, Y.; Krainer, A.R.; Bennett, C.F. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol. 2012, 199, 21–25. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet Lond. Engl. 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, J.; Darras, B.; Farrar, M.; Mercuri, E.; Chiriboga, C.; Kuntz, N.; Shieh, P.; Tulinius, M.; Montes, J.; Reyna, S.; et al. Interim report on the safety and efficacy of longer-term treatment with nusinersen in later-onset spinal muscular atrophy (SMA): Results from the SHINE study. Presented at the World-Muscle-Society Conference 2019. Neuromuscul. Disord. 2019, 29, S184. [Google Scholar] [CrossRef] [Green Version]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen Initiated in Infants During the Presymptomatic Stage of Spinal Muscular Atrophy: Interim Efficacy and Safety Results From the Phase 2 NURTURE Study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef] [Green Version]
- Messina, S.; Pane, M.; Sansone, V.; Bruno, C.; Catteruccia, M.; Vita, G.; Palermo, C.; Albamonte, E.; Pedemonte, M.; Bertini, E.; et al. Expanded access program with Nusinersen in SMA type I in Italy: Strengths and pitfalls of a successful experience. Neuromuscul. Disord. 2017, 27, 1084–1086. [Google Scholar] [CrossRef] [PubMed]
- Farrar, M.A.; Teoh, H.L.; Carey, K.A.; Cairns, A.; Forbes, R.; Herbert, K.; Holland, S.; Kristi, K.J.; Menezes, M.P.; Morrison, M.; et al. Nusinersen for SMA: Expanded access programme. J. Neurol. Neurosurg. Psychiatry 2018, 89, 937–942. [Google Scholar] [CrossRef]
- Pechmann, A.; Langer, T.; Schorling, D.; Stein, S.; Vogt, S.; Schara, U.; Kölbel, H.; Schwartz, O.; Hahn, A.; Giese, K.; et al. Evaluation of children with SMA type 1 under treatment with nusinersen within the expanded access program in Germany. J. Neuromuscul. Dis. 2018, 5, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Aragon-Gawinska, K.; Seferian, A.M.; Daron, A.; Gargaun, E.; Vuillerot, C.; Cances, C.; Ropars, J.; Chouchane, M.; Cuppen, Y.; Hughes, I.; et al. Nusinersen in patients older than 7 months with spinal muscular atrophy type 1: A cohort study. Neurology 2018, 91, 1312–1318. [Google Scholar] [CrossRef]
- Pane, M.; Palermo, C.; Messina, S.; Sansone, V.A.; Bruno, C.; Catteruccia, M.; Sframeli, M.; Albamonte, E.; Pedemonte, M.; D’Amico, A.; et al. Nusinersen in type 1 SMA infants, children and young adults: Preliminary results on motor function. Neuromuscul. Disord. 2018, 28, 582–585. [Google Scholar] [CrossRef]
- Pane, M.; Coratti, G.; Sansone, V.A.; Messina, S.; Bruno, C.; Catteruccia, M.; Sframeli, M.; Albamonte, E.; Pedemonte, M.; D’Amico, A.; et al. Nusinersen in type 1 spinal muscular atrophy: Twelve-month real-world data. Ann. Neurol. 2019, 86, 443–451. [Google Scholar] [CrossRef]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to SMN2 pre-mRNA-protein Complex Elicits Specificity for Small Molecule Splicing Modifiers. Nat. Commun. 2017, 8, 1476. [Google Scholar] [CrossRef]
- Poirier, A.; Weetall, M.; Heinig, K.; Bucheli, F.; Schoenlein, K.; Alsenz, J.; Bassett, S.; Ullah, M.; Senn, C.; Ratni, H.; et al. Risdiplam Distributes and Increases SMN Protein in Both the Central Nervous System and Peripheral Organs. Pharmacol. Res. Perspect. 2018, 29, e00447. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 9, 6501–6517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, G.; Gillingwater, T.H. Spinal Muscular Atrophy: Going beyond the Motor Neuron. Trends Mol. Med. 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Yeo, C.J.J.; Darras, B.T. Overturning the Paradigm of Spinal Muscular Atrophy as just a Motor Neuron Disease. Pediatr. Neurol. 2020. [Google Scholar] [CrossRef]
- Sturm, S.; Günther, A.; Jaber, B.; Jordan, P.; Al Kotbi, N.; Parkar, N.; Cleary, Y.; Frances, N.; Bergauer, T.; Heinig, K.; et al. A Phase 1 Healthy Male Volunteer Single Escalating Dose Study of the Pharmacokinetics and Pharmacodynamics of Risdiplam (RG7916, RO7034067), a SMN2 Splicing Modifier. Br. J. Clin. Pharmacol. 2019, 85, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranello, G.; Servais, L.; Day, J.W.; Deconinck, N.; Mercuri, E.; Klein, A.; Darras, B.; Masson, R.; Kletzl, H.; Cleary, Y.; et al. FIREFISH Part 1: 1-Year Results on Motor Function in Babies with Type 1 SMA. Presented at the American Academy of Neurology Conference 2019. Neurology 2019, 92, 3. [Google Scholar]
- Baranello, G.; Servais, L.; Day, J.W.; Deconinck, N.; Mercuri, E.; Klein, A.; Darras, B.; Masson, R.; Kletzl, H.; Cleary, Y.; et al. FIREFISH Part 1: 16-Month safety and exploratory outcomes of risdiplam (RG7916) treatment in infants with Type 1 spinal muscular atrophy (SMA). Presented at the World-Muscle-Society Conference 2019. Neuromuscul. Disord. 2019, 29, S184. [Google Scholar] [CrossRef]
- Servais, L.; Baranello, G.; Masson, R.; Mazurkiewicz-Bełdzińska, M.; Rose, K.; Vlodavets, D.; Xiong, H.; Zanoteli, E.; El-Khairi, M.; Fuerst-Recktenwald, S.; et al. FIREFISH Part 2: Efficacy and Safety of Risdiplam (RG7916) in Infants with Type 1 Spinal Muscular Atrophy (SMA). Presented at the American Academy of Neurology Conference 2020. Neurology 2020, 94, 1302. [Google Scholar]
- Mercuri, E.; Barisic, N.; Boespflug-Tanguy, O.; Deconinck, N.; Kostera-Pruszczyk, A.; Masson, R.; Mazzone, E.; Nascimento, R.; Osorio, A.; Saito, K.; et al. SUNFISH Part 2: Efficacy and safety of risdiplam (RG7916) in patients with Type 2 or non-ambulant Type 3 spinal muscular atrophy (SMA) Presented at the American Academy of Neurology Conference 2020. Neurology 2020, 94, 1260. [Google Scholar]
- Roche Press Release 07/04/2020: Roche Provides Regulatory Update on Risdiplam for the Treatment of Spinal Muscular Atrophy (SMA). Available online: https://www.roche.com/media/releases/med-cor-2020-04-07.htm (accessed on 30 May 2020).
- Le, T.T.; McGovern, V.L.; Alwine, I.E.; Wang, X.; Massoni-Laporte, A.; Rich, M.M.; Burghes, A.H.M. Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 2011, 20, 3578–3591. [Google Scholar] [CrossRef] [Green Version]
- Valori, C.F.; Ning, K.; Wyles, M.; Mead, R.J.; Grierson, A.J.; Shaw, P.J.; Azzouz, M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010, 2, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.M.; et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an AAV vector expressing human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sproule, D.M.; Al-Zaidy, S.A.; Shell, R. AVXS-101 phase 1 gene therapy clinical trial in SMA type 1: Experience with pre-existing anti-AAV9 antibody in the SMA1 population (S13.001). Neurology 2017, 88, 1. [Google Scholar]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Kolb, S.J.; Coffey, C.S.; Yankey, J.W.; Krosschell, K.; Arnold, W.D.; Rutkove, S.B.; Swoboda, K.J.; Reyna, S.P.; Sakonju, A.; Darras, B.T. Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol. 2017, 82, 883–891. [Google Scholar] [CrossRef]
- Al-Zaidy, S.A.; Kolb, S.J.; Lowes, L.; Alfano, L.N.; Shell, R.; Church, K.R.; Nagendran, S.; Sproule, D.M.; Feltner, D.E.; Wells, C.; et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: Comparative Study with a Prospective Natural History Cohort. J. Neuromuscul. Dis. 2019, 6, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of age andmotor function in a phase 1/2A study of infants with SMA Type 1 receiving single-dose gene replacement therapy. Pediatr. Neurol. 2019, 19, 30280–30282. [Google Scholar] [CrossRef] [Green Version]
- Day, J.D.; Chiriboga, C.A.; Crawford, T.O.; Darras, B.T.; Finkel, R.S.; Connolly, A.M.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.N.; Shieh, P.B.; et al. Onasemnogene Abeparvovec-xioi Gene-Replacement Therapy for Spinal Muscular Atrophy Type 1 (SMA1): Phase 3 US Study (STR1VE) Update (1828) Presented at the American Academy of Neurology Conference 2020. Neurology 2020, 94, 1828. [Google Scholar]
- Press Release Novartis 2019. Available online: https://www.novartis.com/news/media-releases/avexis-receives-fda-approval-zolgensma-first-and-only-gene-therapy-pediatric-patients-spinal-muscular-atrophy-sma (accessed on 30 May 2020).
- Press Release Novartis 2020. Available online: https://www.globenewswire.com/news-release/2020/05/19/2035354/0/en/AveXis-receives-EC-approval-and-activates-Day-One-access-program-for-Zolgensma-the-only-gene-therapy-for-spinal-muscular-atrophy-SMA.html (accessed on 30 May 2020).
- Hwee, D.T.; Kennedy, A.; Ryans, J.; Russell, A.J.; Jia, Z.; Hinken, A.C.; Morgans, D.J.; Malik, F.I.; Jasper, J.R. Fast skeletal muscle troponin activator tirasemtiv increases muscle function and performance in the B6SJL-SOD1G93A ALS mouse model. PLoS ONE 2014, 9, e96921. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.J.; Hwee, D.T.; Kim, L.H.; Durham, N.; Yang, H.T.; Hinken, A.C.; Kennedy, A.R.; Terjung, R.L.; Jasper, J.R.; Malik, F.I.; et al. Fast skeletal muscle troponin activator CK-2066260 increases fatigue resistance by reducing the energetic cost of muscle contraction. J. Physiol. 2019, 597, 4615–4625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, A.J.; Miller, T.M.; Vijayakumar, V.; Stoltz, R.; James, J.K.; Meng, L.; Wolff, A.A.; Malik, F. CK-2127107 Amplifies Skeletal Muscle Response to Nerve Activation in Humans. Muscle Nerve 2018, 57, 729–734. [Google Scholar] [CrossRef]
- Rudnicki, S.A.; Andrews, J.A.; Malik, F.I. CY 5021 A phase 2, double-blind, randomized, placebo-controlled, multiple-dose study of reldesemtiv 2 ascending-dose cohorts of patients with Spinal Muscular Atrophy (SMA). In Proceedings of the Cure SMA 2018, Dallas, TX, USA, 16 June 2018. [Google Scholar]
- Feng, Z.; Ling, K.K.; Zhao, X.; Zhou, C.; Karp, G.; Welch, E.M.; Naryshkin, N.; Ratni, H.; Chen, K.S.; Metzger, F.; et al. Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum. Mol. Genet. 2016, 25, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef] [Green Version]
- Garcera, A.; Bahi, N.; Periyakaruppiah, A.; Arumugam, S.; Soler, R.M. Survival motor neuron protein reduction deregulates autophagy in spinal cord motoneurons in vitro. Cell. Death Dis. 2013, 4, e686. [Google Scholar] [CrossRef] [Green Version]
- Periyakaruppiah, A.; de la Fuente, S.; Arumugam, S.; Bahí, N.; Garcera, A.; Soler, R.M. Autophagy modulators regulate survival motor neuron protein stability in motoneurons. Exp. Neurol. 2016, 283, 287–297. [Google Scholar] [CrossRef]
- Piras, A.; Boido, M. Autophagy inhibition: A new therapeutic target in spinal muscular atrophy. Neural Regen. Res. 2018, 13, 813–814. [Google Scholar] [CrossRef]
- Oliván, S.; Calvo, A.C.; Rando, A.; Herrando-Grabulosa, M.; Manzano, R.; Zaragoza, P.; Tizzano, E.F.; Aquilera, J.; Osta, R. Neuroprotective effect of non-viral gene therapy treatment based on tetanus toxin C-fragment in a severe mouse model of spinal muscular atrophy. Front. Mol. Neurosci. 2016, 9, 76. [Google Scholar] [CrossRef]
- Parker, G.C.; Li, X.; Anguelov, R.A.; Toth, G.; Cristescu, A.; Acsadi, G. Survival motor neuron protein regulates apoptosis in an in vitro model of spinal muscular atrophy. Neurotox. Res. 2008, 13, 39–48. [Google Scholar] [CrossRef]
- Genabai, N.K.; Ahmad, S.; Zhang, Z.; Jiang, X.; Gabaldon, C.A.; Gangwani, L. Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Hum. Mol. Genet. 2015, 15, 6986–7004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schellino, R.; Boido, M.; Borsello, T.; Vercelli, A. Pharmacological c-Jun NH2-Terminal Kinase (JNK) Pathway Inhibition Reduces Severity of Spinal Muscular Atrophy Disease in Mice. Front. Mol. Neurosci. 2018, 11, 308. [Google Scholar] [CrossRef] [Green Version]
- Boido, M.; De Amicis, E.; Valsecchi, V.; Trevisan, M.; Ala, U.; Ruegg, M.A.; Hettwer, S.; Vercelli, A. Increasing Agrin Function Antagonizes Muscle Atrophy and Motor Impairment in Spinal Muscular Atrophy. Front. Cell Neurosci. 2018, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Caine, C.; Awano, T.; Herbst, R.; Monani, U.R. Motor neuronal repletion of the NMJ organizer, Agrin, modulates the severity of the spinal muscular atrophy disease phenotype in model mice. Hum. Mol. Genet. 2017, 26, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Vita, G.L.; Aguennouz, M.; Sframeli, M.; Romeo, S.; Rodolico, C.; Vita, G. Activation of NF-κB pathway in Duchenne muscular dystrophy: Relation to age. Acta Myol. 2011, 30, 16–23. [Google Scholar] [PubMed]
- Mincheva, S.; Garcera, A.; Gou-Fabregas, M.; Encinas, M.; Dolcet, X.; Soler, R.M. The canonical nuclear factor-kappa B pathway regulates cell survival in a developmental model of spinal cord Motoneurons. J. Neurosci. 2011, 31, 6493–6503. [Google Scholar] [CrossRef]
- Arumugam, S.; Mincheva-Tasheva, S.; Periyakaruppiah, A.; de la Fuente, S.; Soler, R.M.; Garcera, A. Regulation of Survival Motor Neuron Protein by the Nuclear Factor-Kappa B Pathway in Mouse Spinal Cord Motoneurons. Mol. Neurobiol. 2018, 55, 5019–5030. [Google Scholar] [CrossRef]
- Dangouloff, T.; Servais, L. Clinical evidence supporting early treatment in spinal muscular atrophy: Current perspectives. Therap. Clin. Risk Manag. 2019, 15, 1153–1161. [Google Scholar] [CrossRef] [Green Version]
- Pera, M.C.; Coratti, G.; Berti, B.; D’Amico, A.; Sframeli, M.; Albamonte, E.; de Sanctis, R.; Messina, S.; Catteruccia, M.; Brigati, G.; et al. Diagnostic journey in Spinal Muscular Atrophy: Is it still an odyssey? PLoS ONE 2020, 15, e0230677. [Google Scholar] [CrossRef] [PubMed]
- Kraszewski, J.N.; Kay, D.M.; Stevens, C.F.; Koval, C.; Haser, B.; Ortiz, V.; Albertorio, A.; Cohen, L.L.; Jain, R.; Andrew, S.P.; et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet. Med. 2018, 20, 608–613. [Google Scholar] [CrossRef] [Green Version]
- Vill, K.; Kolbel, H.; Schwartz, O.; Blaschek, A.; Olgemoller, B.; Harms, E.; Burggraf, S.; Roschinger, W.; Durner, J.; Glaser, D.; et al. One year of newborn screening for SMA – results of a German Pilot Project. J. Neuromuscul. Dis. 2019, 6, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boemer, F.; Caberg, J.H.; Dideberg, V.; Dardenne, D.; Bours, V.; Hiligsmann, M.; Dangouloff, T.; Servais, L. Newborn screening for SMA in Southern Belgium. Neuromuscul. Dis. 2019, 29, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Chiang, S.C.; Weng, W.C.; Lee, N.C.; Lin, C.J.; Hsieh, W.S.; Lee, W.T.; Jong, Y.J.; Ko, T.M.; Hwu, W.L. Presymptomatic diagnosis of Spinal Muscular Atrophy through newborn screening. J. Pediatr. 2017, 190, 124–129.e1. [Google Scholar] [CrossRef] [PubMed]
- Czibere, L.; Burggraf, S.; Fleige, T.; Gluck, B.; Keitel, L.M.; Landt, O.; Durner, J.; Roschinger, W.; Hohenfellner, K.; Wirth, B.; et al. High-throughput genetic newborn screening for spinal muscular atrophy by rapid nucleic acid extraction from dried blood spots and 384-well qPCR. Eur. J. Hum. Gen. 2020, 28, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Glascock, J.; Sampson, J.; Haidet-Phillips, A.; Connolly, A.; Darras, B.; Day, J.; Finkel, R.; Howell, R.R.; Klinger, K.; Kuntz, N.; et al. Treatment algorithm for infants diagnosed with Spinal Muscular Atrophy through Newborn Screening. J. Neuromuscul. Dis. 2018, 5, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller-Felber, W.; Vill, K.; Schwartz, O.; Gläser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Röschinger, W.; Becker, M.; Durner, J.; et al. Infants Diagnosed with Spinal Muscular Atrophy and 4 SMN2 Copies through Newborn Screening—Opportunity or Burden? J. Neuromuscul. Dis. 2020, 7, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Institute for Quality and Efficiency in Health Care (IQWiG). Newborn Screening for 5q-Linked Spinal Muscular Atrophy: IQWiG Reports. Commission No. S18-02; Institute for Quality and Efficiency in Health Care (IQWiG): Cologne, Germany, 2020. [Google Scholar]
- Wirth, B.; Garbes, L.; Riessland, M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr. Opin. Genet. Dev. 2013, 23, 330–338. [Google Scholar] [CrossRef]
- Kariyawasam, D.S.T.; D’Silva, A.; Lin, C.; Ryan, M.M.; Farrar, M.A. Biomarkers and the development of a personalized medicine approach in Spinal Muscular Atrophy. Front. Neurol. 2019, 10, 898. [Google Scholar] [CrossRef]
- Olsson, B.; Alberg, L.; Cullen, N.C.; Michael, E.; Wahlgren, L.; Kroksmark, A.K.; Rostasy, K.; Blennow, K.; Zetterberg, H.; Tulinius, M. NFL is a marker of treatment response in children with SMA treated with nusinersen. J. Neurol. 2019, 266, 2129–2136. [Google Scholar] [CrossRef] [Green Version]
- Darras, B.T.; Crawford, T.O.; Finkel, R.S.; Mercuri, E.; De Vivo, D.C.; Oskoui, M.; Tizzano, E.F.; Ryan, M.M.; Muntoni, F.; Zhao, G.; et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 932–944. [Google Scholar] [CrossRef]
- Totzeck, A.; Stolte, B.; Kizina, K.; Bolz, S.; Schlag, M.; Thimm, A.; Kleinschnitz, C.; Hagenacker, T. Neurofilament Heavy Chain and Tau Protein are not elevated in cerebrospinal fluid of adult patients with Spinal Muscular Atrophy during loading with Nusinersen. Int. J. Mol. Sci. 2019, 20, 5397. [Google Scholar] [CrossRef] [Green Version]
- Faravelli, I.; Meneri, M.; Saccomanno, D.; Velardo, D.; Abati, E.; Gagliardi, D.; Parente, V.; Petrozzi, L.; Ronchi, D.; Stocchetti, D.; et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: An explorative study on Spinal Muscular Atrophy type 3 patients. J. Cell. Mol. Med. 2020, 24, 3034–3039. [Google Scholar] [CrossRef] [Green Version]
- Bonanno, S.; Marcuzzo, S.; Malacarne, C.; Giagnorio, E.; Masson, R.; Zanin, R.; Arnoldi, M.T.; Andreetta, F.; Simoncini, O.; Venerando, A.; et al. Circulating MyomiRs as Potential Biomarkers to Monitor Response to Nusinersen in Pediatric SMA Patients. Biomedicines 2020, 26, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, T.; Latzer, P.; Schmid, D.; Warnken, U.; Saffari, A.; Ziegler, A.; Kollmer, J.; Möhlenbruch, M.; Ulfert, C.; Herweh, C.; et al. Cerebrospinal fluid proteomic profiling in nusinersen-treated patients with spinal muscular atrophy. J. Neurochem. 2020, 153, 650–661. [Google Scholar] [CrossRef]
- Dabbous, O.; Maru, B.; Jansen, J.P.; Lorenzi, M.; Cloutier, M.; Guérin, A.; Pivneva, I.; Wu, E.Q.; Arjunji, R.; Feltner, D.; et al. Survival, motor function, and motor milestones: Comparison of AVXS-101 relative to Nusinersen for the treatment of infants with Spinal Muscular Atrophy Type 1. Adv. Ther. 2019, 36, 1164–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connock, M.; Andronis, L.; Auguste, P.; Dussart, C.; Armoiry, X. Will the US$5 million onasemnogene abeparvosec treatment for spinal muscular atrophy represent ‘value for money’ for the NHS? A rapid inquiry into suggestions that it may be cost-effective. Expert. Opin. Biol. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Malone, D.C.; Dean, R.; Arjunji, R.; Arjunji, R.; Jensen, I.; Cyr, P.; Miller, B.; Maru, B.; Sproule, D.M.; Feltner, D.E.; et al. Cost-effectiveness analysis of using onasemnogene abeparvocec (AVXS-101) in spinal muscular atrophy type 1 patients. J. Mark. Access Health Policy 2019, 7, 1601484. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. International SMA Patient Registry—Full Text View. Available online: https://clinicaltrials.gov/ct2/show/NCT00466349 (accessed on 30 May 2020).
- TREAT-NMD: National SMA Registries. Available online: http://www.treat-nmd.eu/sma/patient-registries/sma/ (accessed on 30 May 2020).
- Pechmann, A.; Konig, K.; Bernert, G.; Schachtrup, K.; Schara, U.; Schorling, D.; Schwersenz, I.; Stein, S.; Tassoni, A.; Vogt, S.; et al. SMArtCARE—A platform to collect real-life outcome data of patients with spinal muscular atrophy. Orphanet J. Rare Dis. 2019, 14, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercuri, E.; Finkel, R.; Scoto, M.; Hall, S.; Eaton, S.; Rashid, A.; Balashkina, J.; Coratti, G.; Pera, M.C.; Samsuddin, S.; et al. Development of an academic disease registry for spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Frongia, A.L.; Antonaci, L.; Pera, M.C.; Coratti, G.; Pane, M.; Pasternak, A.; Civitello, M.; Montes, J.; Mayhew, A.; et al. A critical review of patient and parent caregiver oriented tools to assess health-related quality of life, activity of daily living and caregiver burden in spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, 940–950. [Google Scholar] [CrossRef]
- Mercuri, E.; Messina, S.; Montes, J.; Muntoni, F.; Sansone, V.A.; All Participants and the SMA PROM Working Group. Patient and parent oriented tools to assess health-related quality of life, activity of daily living and caregiver burden in SMA. Rome, 13 July 2019. Neuromuscul. Disord. 2020. [Google Scholar] [CrossRef] [PubMed]
- Polido, G.J.; de Miranda, M.M.V.; Carvas, N.; Mendonça, R.H.; Caromano, F.A.; Reed, U.C.; Zanoteli, E.; Voos, M.C. Cognitive performance of children with spinal muscular atrophy: A systematic review. Dement. Neuropsychol. 2019, 13, 436–443. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Main available therapeutic approaches and their mechanisms of action. SMN1 = survival motor neuron 1; SMN2 = survival motor neuron 2; SMN = survival motor neuron.
Figure 1.
Main available therapeutic approaches and their mechanisms of action. SMN1 = survival motor neuron 1; SMN2 = survival motor neuron 2; SMN = survival motor neuron.


{kind=link}
Table 1.
Main clinical developments in spinal muscular atrophy (SMA).
| Approach /Compound | Sponsor | Mechanism | Trials’ Phase (SMA Type) | Administration | FDA Approval |
|---|---|---|---|---|---|
| Splicing modifiers of SMN2 gene | |||||
| Nusinersen | Ionis-Biogen | ASO | I, II and III (I, II, III) | Intrathecal | X |
| Risdiplam | Roche | Small molecule | I, II and III (I, II, III) | Oral | pending |
| Albuterol | Beta-adrenergic agonist | Off-label | Oral | ||
| Replacing SMN1 gene | |||||
| Onasemnogene abeparvosec | Novartis-Avexis | AAV-9-vector construct | I, II and III (I, II) | Intravenous | X |
| Onasemnogene abeparvosec | Novartis-Avexis | AAV-9-vector construct | I | Intrathecal | |
| Muscle enhancing | |||||
| Reldesemtiv | Cytokinetics | Troponin activator | I and II (II, III, IV) | Oral | |
| SRK-105 | Scholar Rock | Myostatin inhibitor | I and II (II, III) | Intravenous | |
| Neuroprotection | |||||
| Olesoxime | Hoffmann-La Roche | Anti-apoptotic agent | I and II (II, III) (development ended in 2018) | Oral | |
ASO = antisense-oligonucleotide; AAV = adeno-associated virus; FDA= Food and Drug Administration.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222. https://doi.org/10.3390/jcm9072222
AMA Style
Messina S, Sframeli M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. Journal of Clinical Medicine. 2020; 9(7):2222. https://doi.org/10.3390/jcm9072222
Chicago/Turabian StyleMessina, Sonia, and Maria Sframeli. 2020. "New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges" Journal of Clinical Medicine 9, no. 7: 2222. https://doi.org/10.3390/jcm9072222
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.