Downregulated Platelet miR-1233-5p in Patients with Alzheimer’s Pathologic Change with Mild Cognitive Impairment is Associated with Aβ-Induced Platelet Activation via P-Selectin

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Aβ1-40 Preparation
2.3. Platelet Purification
2.4. miRNA Microarray
2.5. miRNA Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.6. Platelet Spike-in Experiment
2.7. Human MEG-01 Cells Transfection
2.8. Flow Cytometry
2.9. Adhesion Assay with Platelets or MEG-01 Cells
2.10. Statistical Analysis
3. Results
3.1. Characteristics of the Study Population
3.2. Expression Profiles of Platelet-Derived miRNAs in Aβ(−)MCI and Aβ(+)MCI Group
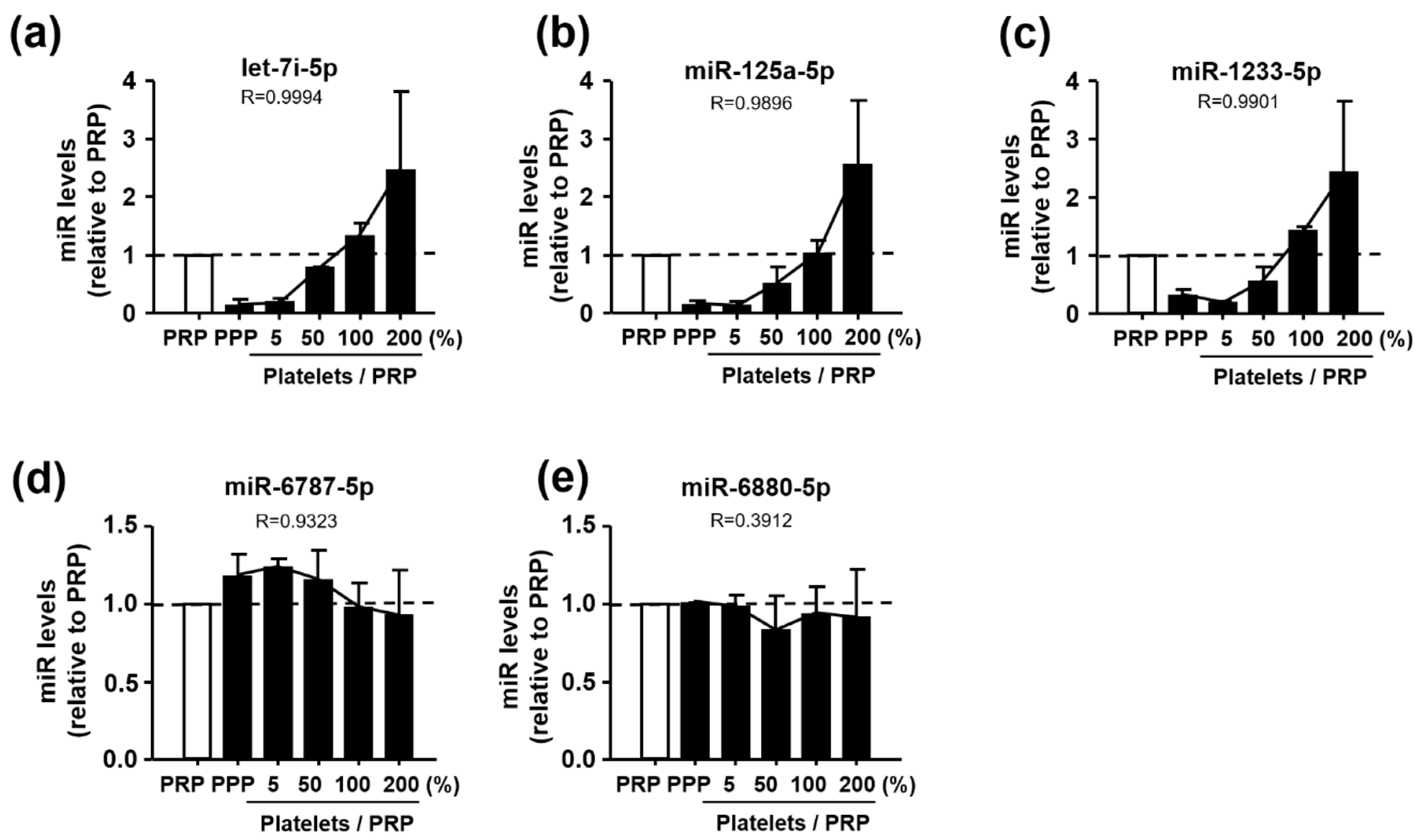
3.3. Identification of Let-7i-5p, miR-125a-5p, and miR-1233-5p as Platelet-Derived miRNAs
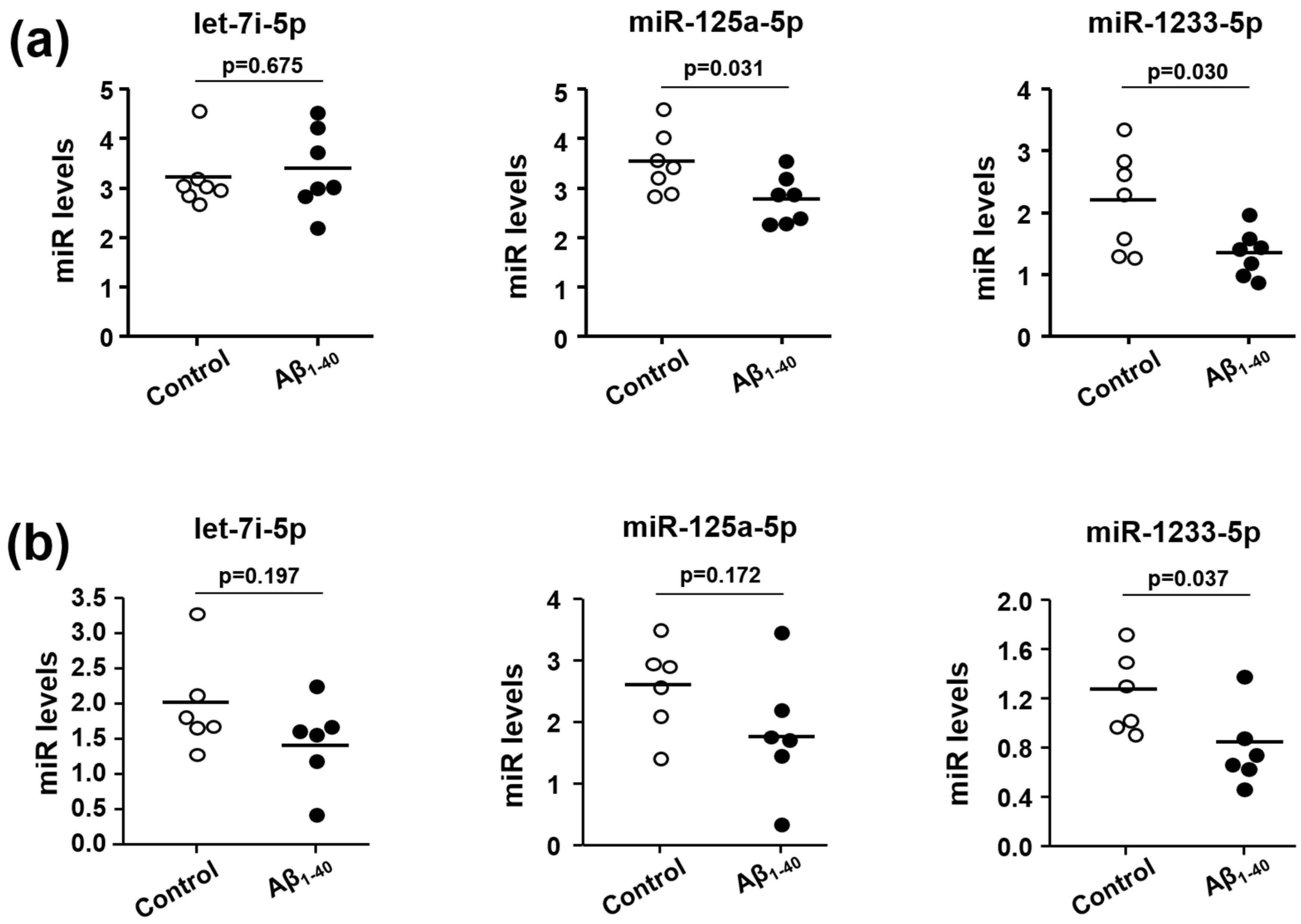
3.4. Effects of Aβ1-40 on Expression Levels of Let-7i-5p, miR-125a-5p, and miR-1233-5p in Human Platelets and MEG-01 Cells
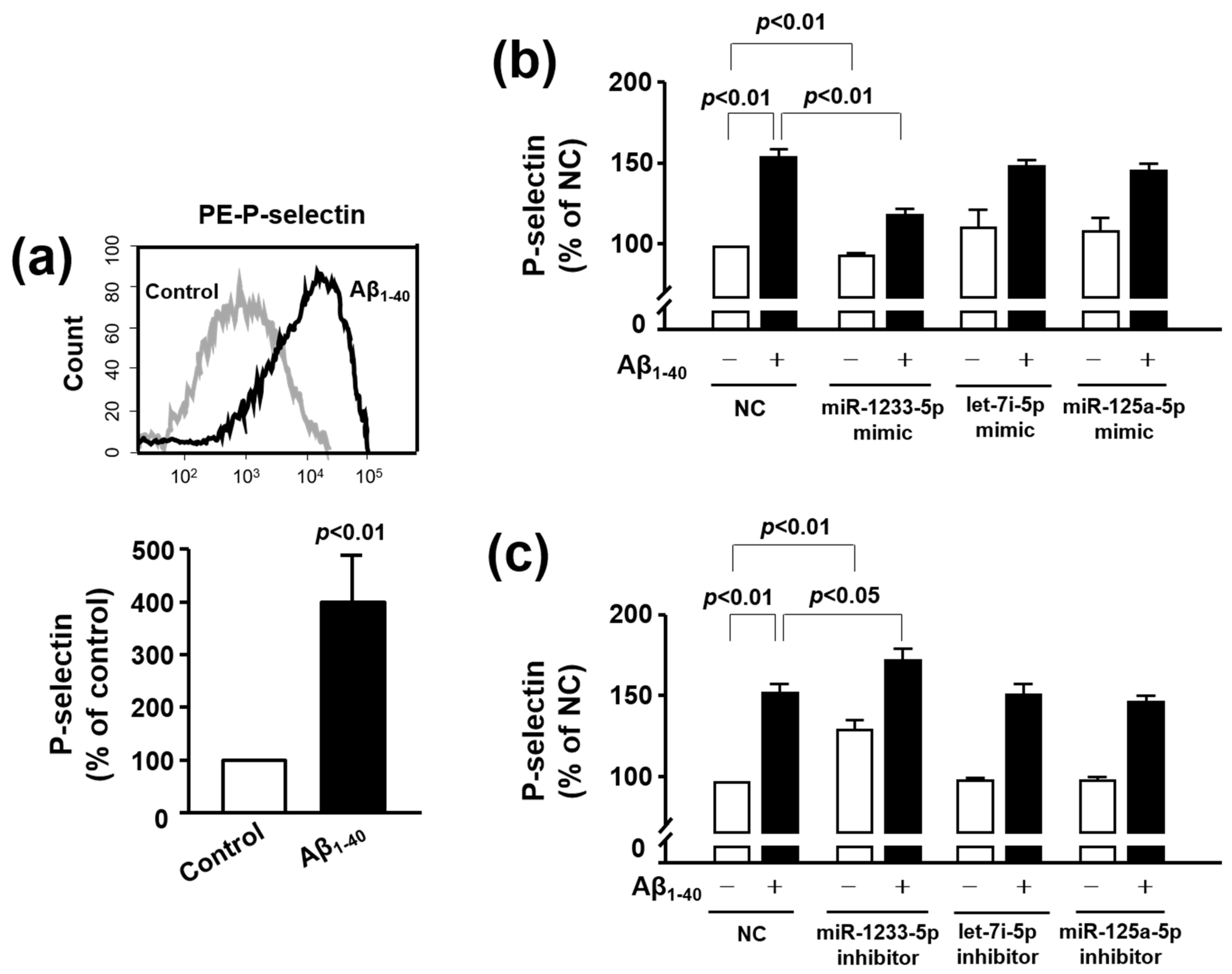
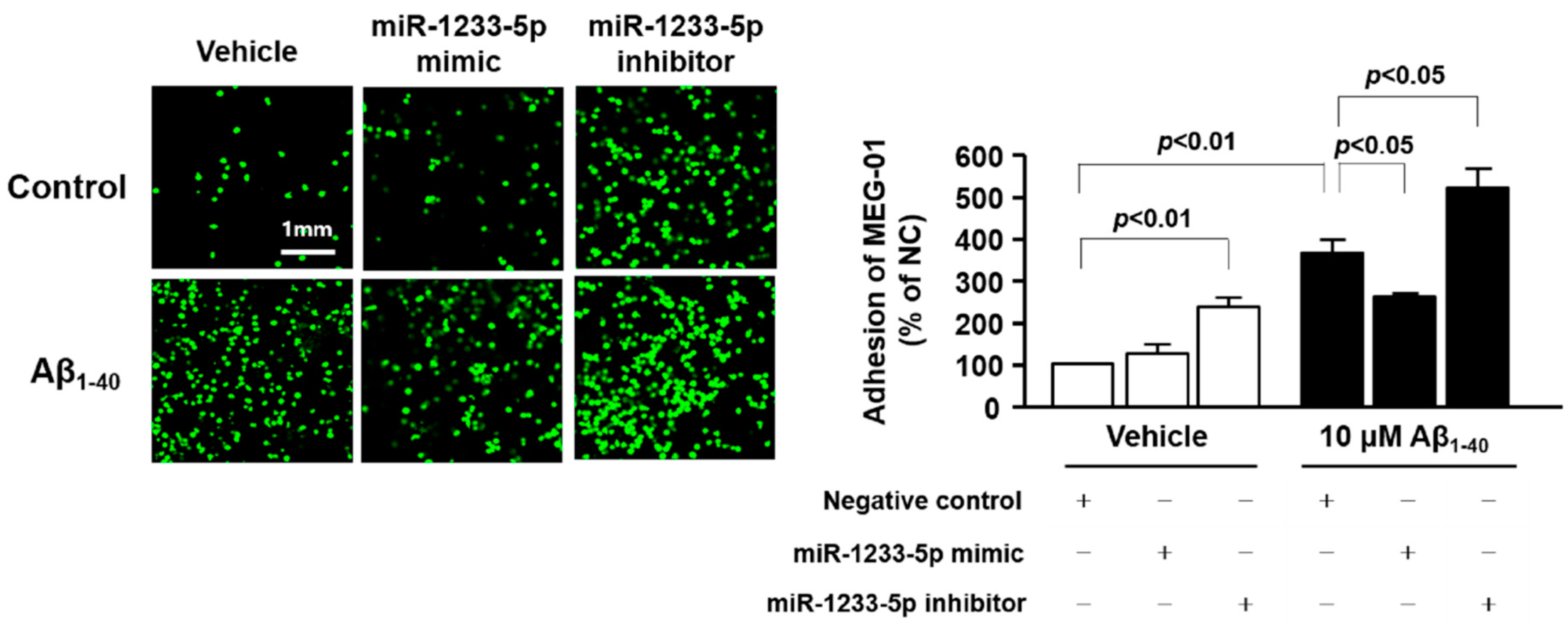
3.5. Role of miR-1233-5p in the Activation of Platelets and MEG-01 Cells
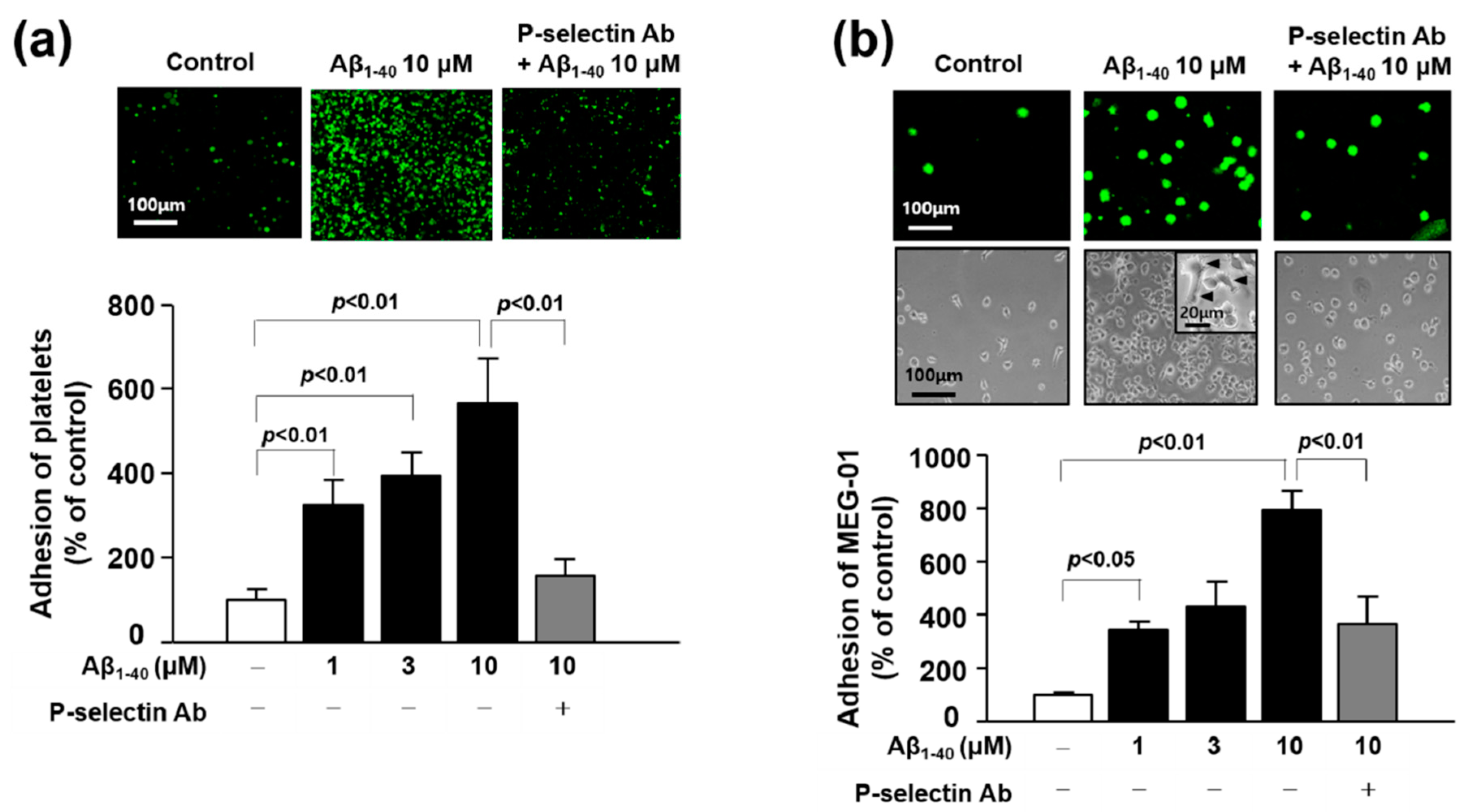
3.6. The Role of P-Selectin in Adhesion Function of Platelets and MEG-01 Cells
3.7. Role of miR-1233-5p in the Adhesion Function of Platelets and MEG-01 Cells to Fibronectin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sutphen, C.L.; Jasielec, M.S.; Shah, A.R.; Macy, E.M.; Xiong, C.; Vlassenko, A.G.; Benzinger, T.L.S.; Stoops, E.E.J.; Vanderstichele, H.M.J.; Brix, B.; et al. Longitudinal Cerebrospinal Fluid Biomarker Changes in Preclinical Alzheimer Disease During Middle Age. JAMA Neurol. 2015, 72, 1029–1042. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Epelbaum, S.; Genthon, R.; Cavedo, E.; Habert, M.O.; Lamari, F.; Gagliardi, G.; Lista, S.; Teichmann, M.; Bakardjian, H.; Hampel, H.; et al. Preclinical Alzheimer’s disease: A systematic review of the cohorts underlying the concept. Alzheimers Dement. 2017, 13, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okello, A.; Koivunen, J.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Någren, K.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.C.; et al. Conversion of amyloid positive and negative MCI to AD over 3 years. Neurology 2009, 73, 754–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Yan, C.; Wang, Z.; Wang, J.; Xia, M.; Li, K.; He, Y. Discriminative analysis of early Alzheimer’s disease using multi-modal imaging and multi-level characterization with multi-classifier (M3). NeuroImage 2012, 59, 2187–2195. [Google Scholar] [CrossRef]
- Kao, Y.H.; Chou, M.C.; Chen, C.H.; Yang, Y.H. White Matter Changes in Patients with Alzheimer’s Disease and Associated Factors. J. Clin. Med. 2019, 8, 167. [Google Scholar] [CrossRef] [Green Version]
- Kenny, A.; McArdle, H.; Calero, M. Elevated Plasma microRNA-206 Levels Predict Cognitive Decline and Progression to Dementia from Mild Cognitive Impairment. Biomolecules 2019, 9, 734. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.B.; Li, C.B.; Song, N.; Zou, Y.; Chen, S.D.; Ren, R.J.; Wang, G. Diagnostic Value of microRNA for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2016, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Ramasubramanian, B.; Kanji, S.; Chakraborty, A.R.; Haque, S.J.; Chakravarti, A. Circulating microRNAs in cancer: Hope or hype? Cancer Lett. 2016, 381, 113–121. [Google Scholar] [CrossRef]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Akingbade, O.E.S.; Gibson, C.; Kalaria, R.N.; Mukaetova-Ladinska, E.B. Platelets: Peripheral Biomarkers of Dementia? J. Alzheimers Dis. 2018, 63, 1235–1259. [Google Scholar] [CrossRef] [PubMed]
- Donner, L.; Gremer, L.; Ziehm, T.; Gertzen, C.G.W.; Gohlke, H.; Willbold, D.; Elvers, M. Relevance of N-terminal residues for amyloid-beta binding to platelet integrin alphaIIbbeta3, integrin outside-in signaling and amyloid-beta fibril formation. Cell. Signal. 2018, 50, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Gowert, N.S.; Donner, L.; Chatterjee, M.; Eisele, Y.S.; Towhid, S.T.; Munzer, P.; Walker, B.; Ogorek, I.; Borst, O.; Grandoch, M.; et al. Blood platelets in the progression of Alzheimer’s disease. PLoS ONE 2014, 9, e90523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donner, L.; Falker, K.; Gremer, L.; Klinker, S.; Pagani, G.; Ljungberg, L.U.; Lothmann, K.; Rizzi, F.; Schaller, M.; Gohlke, H.; et al. Platelets contribute to amyloid-beta aggregation in cerebral vessels through integrin alphaIIbbeta3-induced outside-in signaling and clusterin release. Sci. Signal. 2016, 9, ra52. [Google Scholar] [CrossRef]
- Nagalla, S.; Shaw, C.; Kong, X.; Kondkar, A.A.; Edelstein, L.C.; Ma, L.; Chen, J.; McKnight, G.S.; López, J.A.; Yang, L.; et al. Platelet microRNA-mRNA coexpression profiles correlate with platelet reactivity. Blood 2011, 117, 5189–5197. [Google Scholar] [CrossRef]
- Veitinger, M.; Varga, B.; Guterres, S.B.; Zellner, M. Platelets, a reliable source for peripheral Alzheimer’s disease biomarkers? Acta Neuropathol. Commun. 2014, 2, 65. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.J.; Zamolodchikov, D.; Cortes-Canteli, M.; Norris, E.H.; Glickman, J.F.; Strickland, S. Alzheimer’s disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 21812–21817. [Google Scholar] [CrossRef] [Green Version]
- Kaudewitz, D.; Skroblin, P.; Bender, L.H.; Barwari, T.; Willeit, P.; Pechlaner, R.; Sunderland, N.P.; Willeit, K.; Morton, A.C.; Armstrong, P.C.; et al. Association of MicroRNAs and YRNAs With Platelet Function. Circ. Res. 2016, 118, 420–432. [Google Scholar] [CrossRef]
- Sonkar, V.; Kulkarni, P.P.; Chaurasia, S.N.; Dash, A.; Jauhari, A.; Parmar, D.; Yadav, S.; Dash, D. Plasma Fibrinogen Is a Natural Deterrent to Amyloid Beta-Induced Platelet Activation. Mol. Med. 2016, 22, 224–232. [Google Scholar] [CrossRef]
- Catricala, S.; Torti, M.; Ricevuti, G. Alzheimer disease and platelets: How’s that relevant. Immun. Ageing 2012, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montenont, E.; Echagarruga, C.; Allen, N.; Araldi, E.; Suarez, Y.; Berger, J.S. Platelet WDR1 suppresses platelet activity and is associated with cardiovascular disease. Blood 2016, 128, 2033–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merten, M.; Thiagarajan, P. P-selectin expression on platelets determines size and stability of platelet aggregates. Circulation 2000, 102, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.; van Gils, J.M.; Mol, A.; Hordijk, P.L.; Zwaginga, J.J. Platelet binding to monocytes increases the adhesive properties of monocytes by up-regulating the expression and functionality of beta1 and beta2 integrins. J. Leukoc. Biol. 2006, 79, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Polanowska-Grabowska, R.; Wallace, K.; Field, J.J.; Chen, L.; Marshall, M.A.; Figler, R.; Gear, A.R.; Linden, J. P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2392–2399. [Google Scholar] [CrossRef]
- Ruggeri, Z.M.; Mendolicchio, G.L. Adhesion mechanisms in platelet function. Circ. Res. 2007, 100, 1673–1685. [Google Scholar] [CrossRef]
- Boyle, P.A.; Wilson, R.S.; Aggarwal, N.T.; Tang, Y.; Bennett, D.A. Mild cognitive impairment: Risk of Alzheimer disease and rate of cognitive decline. Neurology 2006, 67, 441–445. [Google Scholar] [CrossRef]
- Petersen, R.C.; Roberts, R.O.; Knopman, D.S.; Boeve, B.F.; Geda, Y.E.; Ivnik, R.J.; Smith, G.E.; Jack, C.R., Jr. Mild cognitive impairment: Ten years later. Arch. Neurol. 2009, 66, 1447–1455. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Poljak, A.; Smythe, G.A.; Sachdev, P. Plasma biomarkers for mild cognitive impairment and Alzheimer’s disease. Brain Res. Rev. 2009, 61, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Burger, K.; Teipel, S.J.; Bokde, A.L.; Zetterberg, H.; Blennow, K. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimers Dement. 2008, 4, 38–48. [Google Scholar] [CrossRef]
- Ye, B.S.; Kim, H.J.; Kim, Y.J.; Jung, N.Y.; Lee, J.S.; Lee, J.; Jang, Y.K.; Yang, J.J. Longitudinal outcomes of amyloid positive versus negative amnestic mild cognitive impairments: A three-year longitudinal study. Sci. Rep. 2018, 8, 5557. [Google Scholar] [CrossRef] [PubMed]
- Faruq, O.; Vecchione, A. microRNA: Diagnostic Perspective. Front. Med. 2015, 2, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheinerman, K.S.; Toledo, J.B.; Tsivinsky, V.G.; Irwin, D.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Chen-Plotkin, A.; Wolk, D.A.; McCluskey, L.F.; et al. Circulating brain-enriched microRNAs as novel biomarkers for detection and differentiation of neurodegenerative diseases. Alzheimers Res. Ther. 2017, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Ciarambino, T.; D’Amico, M.; Trotta, M.C.; Di Sette, A.M.; Marfella, R.; Malatino, L.; Paolisso, G.; Adinolfi, L.E. Circulating MiRNA-195-5p and -451a in Transient and Acute Ischemic Stroke Patients in an Emergency Department. J. Clin. Med. 2019, 8, 130. [Google Scholar] [CrossRef] [Green Version]
- Absalon, S.; Kochanek, D.M.; Raghavan, V.; Krichevsky, A.M. MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci. 2013, 33, 14645–14659. [Google Scholar] [CrossRef]
- Lau, P.; Bossers, K.; Janky, R.; Salta, E.; Frigerio, C.S.; Barbash, S.; Rothman, R.; Sierksma, A.S.; Thathiah, A.; Greenberg, D.; et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 1613–1634. [Google Scholar] [CrossRef] [Green Version]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Tan, L.; Yu, J.T.; Tan, M.S.; Liu, Q.Y.; Wang, H.F.; Zhang, W.; Jiang, T.; Tan, L. Genome-wide serum microRNA expression profiling identifies serum biomarkers for Alzheimer’s disease. J. Alzheimers Dis. 2014, 40, 1017–1027. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Zhang, L.; Dong, Y.; Ji, H.; Shen, L. The Potential Markers of Circulating microRNAs and long non-coding RNAs in Alzheimer’s Disease. Aging Dis. 2019, 10, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Bjorkqvist, M.; Ohlsson, M.; Minthon, L.; Hansson, O. Evaluation of a previously suggested plasma biomarker panel to identify Alzheimer’s disease. PLoS ONE 2012, 7, e29868. [Google Scholar] [CrossRef] [PubMed]
- Fejes, Z.; Poliska, S.; Czimmerer, Z.; Kaplar, M.; Penyige, A.; Gal Szabo, G.; Beke Debreceni, I.; Kunapuli, S.P.; Kappelmayer, J.; Nagy, B., Jr. Hyperglycaemia suppresses microRNA expression in platelets to increase P2RY12 and SELP levels in type 2 diabetes mellitus. Thromb. Haemost. 2017, 117, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Rahman, M.F.; Jiang, L.; Min, Y.; Tan, S.; Xie, H.; Lee, L.; Wang, M.; Malmstrom, R.E.; Lui, W.O.; et al. Thrombin-reduced miR-27b attenuates platelet angiogenic activities in vitro via enhancing platelet synthesis of anti-angiogenic thrombospondin-1. J. Thromb. Haemost. 2018, 16, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numabe, A.; Ara, N.; Hakamada-Taguchi, R.; Suzuki, N.; Hirawa, N.; Kawabata, Y.; Negoro, T.; Nagata, T.; Goto, A.; Toyo-Oka, T.; et al. Effects of the anti-platelet aggregation drug dilazep on cognitive function in Dahl salt-sensitive rats. Hypertens. Res. 2003, 26, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Canobbio, I.; Visconte, C.; Oliviero, B.; Guidetti, G.; Zara, M.; Pula, G.; Torti, M. Increased platelet adhesion and thrombus formation in a mouse model of Alzheimer’s disease. Cell. Signal. 2016, 28, 1863–1871. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Miner, J.J.; Yago, T.; Yao, L.; Lupu, F.; Xia, L.; McEver, R.P. Differential regulation of human and murine P-selectin expression and function in vivo. J. Exp. Med. 2010, 207, 2975–2987. [Google Scholar] [CrossRef] [Green Version]
- Vestweber, D.; Blanks, J.E. Mechanisms that regulate the function of the selectins and their ligands. Physiol. Rev. 1999, 79, 181–213. [Google Scholar] [CrossRef]
- Reyes-Reyes, M.E.; George, M.D.; Roberts, J.D.; Akiyama, S.K. P-selectin activates integrin-mediated colon carcinoma cell adhesion to fibronectin. Exp. Cell Res. 2006, 312, 4056–4069. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Ni, M.; Su, Y.; Wang, H.; Zhu, S.; Zhao, A.; Li, G. MicroRNAs in Serum Exosomes as Potential Biomarkers in Clear-cell Renal Cell Carcinoma. Eur. Urol. Focus 2018, 4, 412–419. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | CNI | Aβ(−)MCI | Aβ(+)MCI | p-Value | |
|---|---|---|---|---|---|
| CNI vs. Aβ(−)MCI | CNI vs. Aβ(+)MCI | ||||
| Total, n | 9 | 12 | 11 | - | - |
| Age (years) | 73.2 ± 1.43 | 74.2 ± 5.67 | 74.6 ± 2.48 | 0.681 | 0.660 |
| Female (%) | 66.7 ± 0.17 | 91.7 ± 0.29 | 72.7 ± 0.14 | 0.164 | 0.783 |
| Education (years) | 9.78 ± 1.40 | 9.50 ± 5.20 | 9.46 ± 1.27 | 0.897 | 0.870 |
| BMI (kg/m2) | 25.2 ± 1.23 | 23.7 ± 3.28 | 23.1 ± 0.49 | 0.335 | 0.109 |
| HR | 74.6 ± 4.98 | 76.8 ± 6.45 | 74.2 ± 3.13 | 0.640 | 0.949 |
| MMSE | 27.3 ± 0.29 | 24.5 ± 3.03 | 24.0 ± 0.53 | 0.014 † | < 0.001 † |
| CDR-SB | 1.22 ± 0.21 | 1.83 ± 0.69 | 2.41 ± 0.27 | 0.049 † | 0.004 † |
| GDS | 2.11 ± 0.11 | 2.75 ± 0.45 | 2.73 ± 0.14 | 0.002 † | 0.003 † |
| Platelet Count (108/mL) | 2.06 ± 0.12 | 2.29 ± 0.93 | 2.33 ± 0.28 | 0.483 | 0.450 |
| Amyloid PET (SUVr) | |||||
| Cingulate gyrus (L + R) | 0.72 ± 0.02 | 0.69 ± 0.05 | 0.92 ± 0.03 | 0.254 | < 0.001 † |
| Frontal lobe (L + R) | 0.58 ± 0.01 | 0.56 ± 0.05 | 0.79 ± 0.03 | 0.245 | < 0.001 † |
| Occipital lobe (L + R) | 0.61 ± 0.02 | 0.60 ± 0.04 | 0.68 ± 0.01 | 0.535 | 0.009 † |
| Parietal lobe (L + R) | 0.58 ± 0.01 | 0.56 ± 0.04 | 0.79 ± 0.03 | 0.255 | < 0.001 † |
| Temporal lobe (L + R) | 0.60 ± 0.01 | 0.58 ± 0.04 | 0.78 ± 0.04 | 0.226 | < 0.001 † |
| Global (L + R) | 0.59 ± 0.01 | 0.57 ± 0.04 | 0.76 ± 0.02 | 0.281 | < 0.001 † |
| ApoE ε4 carrier (%) | 0.00 ± 0.00 | 8.00 ± 0.28 | 54.4 ± 0.15 | 0.400 | 0.006 † |
| Fazekas Scale (WMH) | 1.33 ± 0.20 | 1.50 ± 0.52 | 1.27 ± 0.18 | 0.470 | 0.783 |
| VBM | |||||
| Hippocampus (L) | 0.40 ± 0.05 | 0.36 ± 0.05 | 0.37 ± 0.03 | 0.062 | 0.066 |
| Hippocampus (R) | 0.40 ± 0.03 | 0.36 ± 0.05 | 0.36 ± 0.02 | 0.063 | 0.018 † |
| miRNA | Aβ(−)MCI | Aβ(+)MCI | ||
|---|---|---|---|---|
| FC | p-value | FC | p-value | |
| hsa-let-7i-5p | −1.39 | 0.516 | −4.16 | 0.043 † |
| hsa-miR-125a-5p | 1.04 | 0.946 | −1.70 | 0.035 † |
| hsa-miR-1233-5p | −1.25 | 0.193 | −1.64 | 0.003 † |
| hsa-miR-6787-5p | −1.18 | 0.448 | −1.61 | 0.008 † |
| hsa-miR-6880 | 1.42 | 0.144 | 2.09 | 0.025 † |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.K.; Kim, M.H.; Lee, S.Y.; Son, S.J.; Hong, C.H.; Jung, Y.-S. Downregulated Platelet miR-1233-5p in Patients with Alzheimer’s Pathologic Change with Mild Cognitive Impairment is Associated with Aβ-Induced Platelet Activation via P-Selectin. J. Clin. Med. 2020, 9, 1642. https://doi.org/10.3390/jcm9061642
Lee BK, Kim MH, Lee SY, Son SJ, Hong CH, Jung Y-S. Downregulated Platelet miR-1233-5p in Patients with Alzheimer’s Pathologic Change with Mild Cognitive Impairment is Associated with Aβ-Induced Platelet Activation via P-Selectin. Journal of Clinical Medicine. 2020; 9(6):1642. https://doi.org/10.3390/jcm9061642
Chicago/Turabian StyleLee, Bo Kyung, Min Hee Kim, Sang Yoon Lee, Sang Joon Son, Chang Hyung Hong, and Yi-Sook Jung. 2020. "Downregulated Platelet miR-1233-5p in Patients with Alzheimer’s Pathologic Change with Mild Cognitive Impairment is Associated with Aβ-Induced Platelet Activation via P-Selectin" Journal of Clinical Medicine 9, no. 6: 1642. https://doi.org/10.3390/jcm9061642