Strengths and Weaknesses of Light Transmission Aggregometry in Diagnosing Hereditary Platelet Function Disorders

 , and
, and
Abstract
:1. Introduction
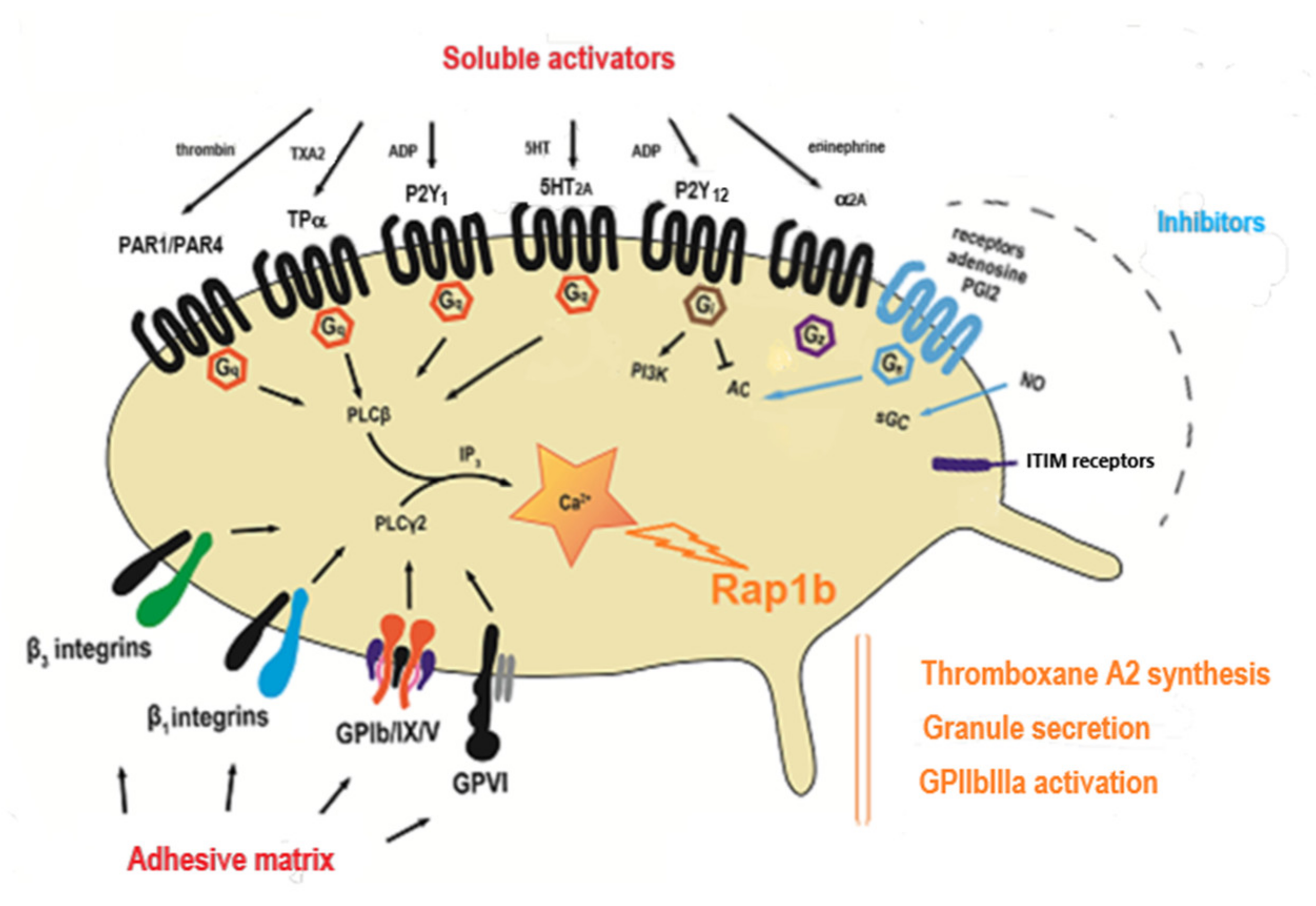
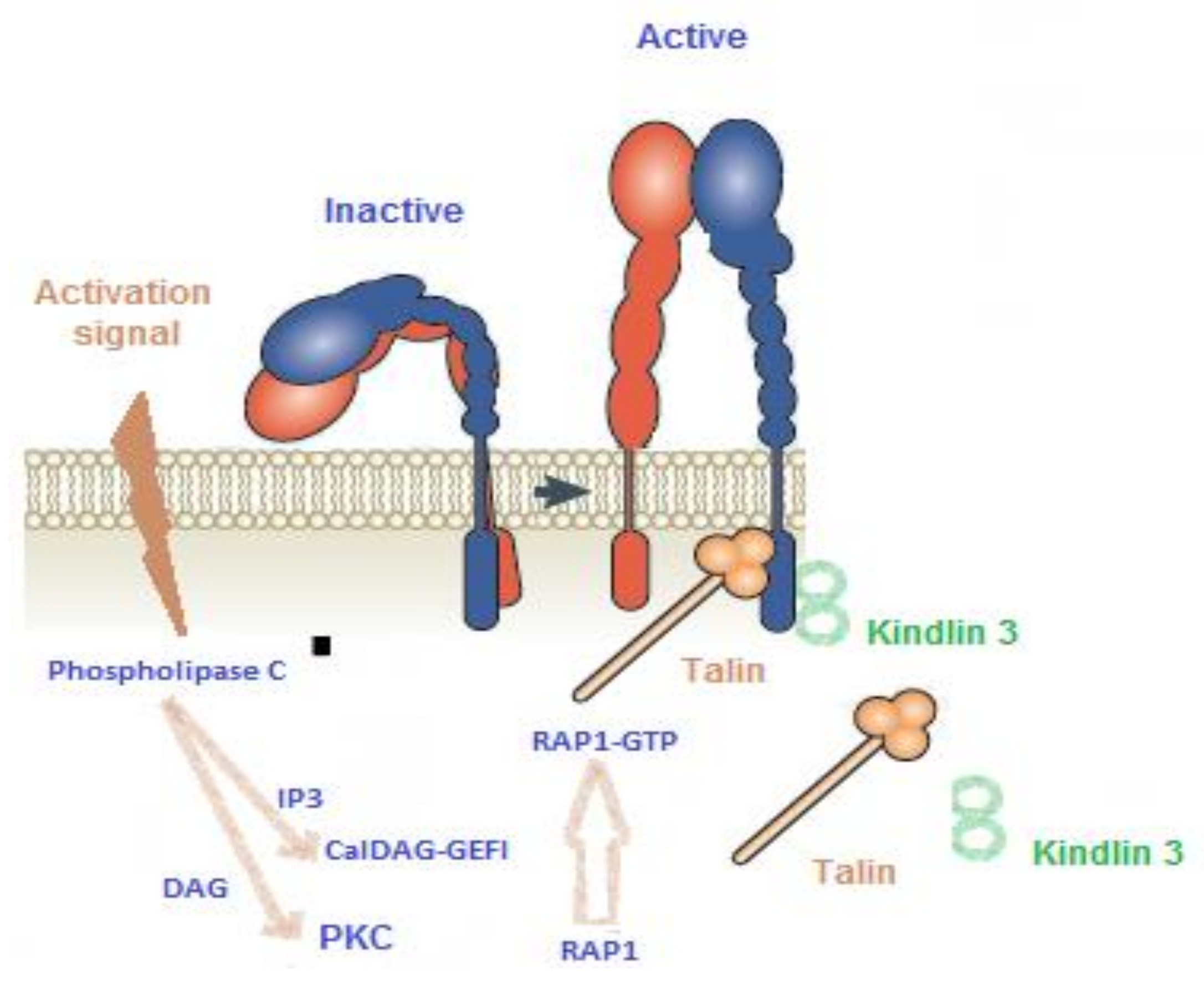
2. The Main Pathways of Platelet Activation
3. Carrying Out LTA
4. Hereditary Platelet Disorders with Known Molecular Defects
4.1. Absence of Aggregation in Response to Multiple Activators Except for Ristocetin
4.2. Reduced LTA in Response to Multiple Activators
4.2.1. CalDAG-GEFI Deficiency
4.2.2. Ephrin B2-Related Platelet Disorder
4.2.3. Abnormality of the Amplification Pathway Mediated by ADP
- P2Y12 Receptor Abnormality
- Defects in δ Granule Secretion
4.2.4. Abnormality of the Amplification Pathway Mediated by Thromboxane A2 (TXA2)
- Variation in Receptor TPα
- Defect in the Conversion of AA to TXA2
4.2.5. ARC Syndrome
4.3. Defect in Response to a Single Activator
4.3.1. Defect in Collagen-Induced Aggregation
4.3.2. Defect in Response to Ristocetin
4.3.3. The Special Case of Gαs Deficiency
5. Hereditary Platelet Disorders with No Molecular Cause Characterized Yet
5.1. Isolated Defect in Response to Epinephrine
5.2. Suspicion of Gαq Deficiency
5.3. Suspicion of Phospholipase Cβ defect
5.4. Defect Affecting the P2X1 Receptor
5.5. Defect Affecting GPIa
6. LTA Versus Other More Recently Developed Methods
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Born, G.V. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef]
- O’brien, J.R. Platelet aggregation: Part I Some effects of the adenosine phosphates, thrombin, and cocaine upon platelet adhesiveness. J. Clin. Pathol. 1962, 15, 446–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollgaard, E. Macroscopic studies of platelet aggregation. Nature of an aggregating factor in red blood cells and platelets. Thromb. Diath. Haemorrh. 1961, 6, 86–97. [Google Scholar] [PubMed]
- Hayward, C.P.M.; Moffat, K.A.; Brunet, J.; Carlino, S.A.; Plumhoff, E.; Meijer, P.; Zehnder, J.L. Update on diagnostic testing for platelet function disorders, what is practical and useful? Int. J. Lab. Hematol. 2019, 41, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifuni, S.M.; Wagner, D.D.; Bergmeier, W. CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin alphaIIbbeta3 in platelets. Blood 2008, 112, 1696–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The British Society for Haematology BCSH. Haemostasis and Thrombosis Task Force. Guidelines on platelet function testing. J. Clin. Pathol. 1988, 41, 1322–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, C.P.M.; Moffat, K.A.; Raby, A.; Israel, S.; Plumhoff, E.; Flyn, G.; Zehnder, J.L. Development of North American consensus guidelines for medical laboratories that perform and interpret platelet function testing using light transmission aggregometry. Am. J. Clin. Pathol. 2010, 134, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, M.; Cerletti, C.; Harrison, P.; Hayward, C.P.M.; Kenny, D.; Nugent, D.; Nurden, P.; Rao, A.K.; Schmaier, A.H.; Watson, P.; et al. Recommendations for the standardization of light transmission aggregometry, a consensus of the working party from the platelet physiology subcommittee of SSC/ISTH. J. Thromb. Haemost. 2013, 11, 1183–1189. [Google Scholar] [CrossRef]
- Knöfler, R.; Eberl, W.; Schulze, H.; Bakchoul, T.; Bergmann, F.; Gehrisch, S.; Geisen, C.; Gottstein, S.; Halimeh, S.; Harbrechtet, U.; et al. Diagnosis of inherited diseases of platelet function. Interdisciplinary S2K guideline of the Permanent Paediatric Committee of the Society of Thrombosis and Haemostasis Research (GTH e. V.). Hamostaseologie 2014, 34, 201–212. [Google Scholar] [CrossRef]
- Gresele, P. Subcommittee on Platelet Physiology of the International Society on Thrombosis and Hemostasis. Diagnosis of inherited platelet function disorders, guidance from the SSC of the ISTH. J. Thromb. Haemost. 2015, 13, 314–322. [Google Scholar] [CrossRef]
- Magnette, A.; Chatelain, M.; Chatelain, B.; Ten Cate, H.; Mullier, F. Pre-analytical issues in the haemostasis laboratory: guidance for the clinical Laboratories. Thromb. J. 2016, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaloro, E.J. More on preanalytical variables affecting platelet function testing using light transmittance aggregometry. Clin. Chem. Lab. Med. 2011, 49, 737–739. [Google Scholar] [CrossRef] [PubMed]
- Althaus, K.; Zieger, B.; Bakchoul, T.; Jurk, K. THROMKID-Plus Studiengruppe der Gesellschaft für Thrombose- und Hämostaseforschung (GTH) und der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH). Standardization of Light Transmission Aggregometry for Diagnosis of Platelet Disorders: An Inter-Laboratory External Quality Assessment. Thromb. Haemost. 2019, 119, 1154–1161. [Google Scholar]
- Nurden, A.T.; Pillois, X.; Wilcox, D.A. Glanzmann thrombasthenia, state of the art and future directions. Semin. Thromb. Hemost. 2013, 39, 642–655. [Google Scholar]
- Nurden, A.T. Glanzmann thrombasthenia. Orphanet. J. Rare Dis. 2006, 1, 10. [Google Scholar] [CrossRef]
- Niessner, H.; Clemetson, K.J.; Panzer, S.; Mueller-Eckhardt, C.; Santoso, S.; Bettelheim, P. Acquired thrombasthenia due to GPIIb/IIIa-specific platelet autoantibodies. Blood 1986, 68, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Bloor, A.J.; Smith, G.A.; Jaswon, M.; Parker, N.E.; Ouwehand, W.H.; Liesner, R. Acquired thrombasthenia due to GPIIbIIIa platelet autoantibodies in a 4-yr-old child. Eur. J. Haematol. 2006, 76, 89–90. [Google Scholar] [CrossRef]
- Robert, P.; Canault, M.; Farnarier, C.; Nurden, A.; Grosdidier, C.; Barlogis, V.; Bongrand, P.; Pierres, A.; Chambost, H.; Alessi, M.C. A novel leukocyte adhesion deficiency III variant, kindlin-3 deficiency results in integrin- and non integrin-related defects in different steps of leukocyte adhesion. J. Immunol. 2011, 186, 5273–5283. [Google Scholar] [CrossRef] [Green Version]
- Canault, M.; Ghalloussi, D.; Grosdidier, C.; Guinier, M.; Perret, C.E.; Chelghoum, N.; Germain, M.; Raslova, H.; Peiretti, F.; Morange, P.R.; et al. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J. Exp. Med. 2014, 211, 1349–1362. [Google Scholar] [CrossRef] [Green Version]
- Berrou, E.; Soukaseum, C.; Favier, R.; Adam, F.; Elaib, Z.; Kauskot, A.; Bordet, J.C.; Ballerini, P.; Loyau, S.; Feng, M.; et al. A mutation of the human EPHB2 gene leads to a major platelet functional defect. Blood 2018, 132, 2067–2077. [Google Scholar] [CrossRef] [Green Version]
- Gachet, C. P2 receptors, platelet function and pharmacological implications. Thromb. Haemost. 2008, 99, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Lecchi, A.; Randi, A.M.; McGregor, J.L.; Mannucci, P.M. Identification of a new congenital defect of platelet function characterized by severe impairment of platelet responses to adenosine diphosphate. Blood 1992, 80, 2787–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecchi, A.; Razzari, C.; Paoletta, S.; Dupuis, A.; Nakamura, L.; Ohlmann, P.; Gachet, C.; Jacobson, K.A.; Zieger, B.; Cattaneo, M. Identification of a new dysfunctional platelet P2Y12 receptor variant associated with bleeding diathesis. Blood 2015, 125, 1006–1013. [Google Scholar] [CrossRef]
- Patel, Y.M.; Lordkipanidze, M.; Lowe, G.C.; Nisar, S.P.; Garner, K.; Stockley, J.; Daly, M.E.; Mitchell, M.; Watson, S.P.; Austin, S.K.; et al. A novel mutation in the p2y receptor and a function-reducing polymorphism in par-1 in a patient with chronic bleeding. J. Thromb. Haemost. 2014, 12, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Shiraga, M.; Miyata, S.; Kato, H.; Kashiwagi, H.; Honda, S.; Kurata, Y.; Tomiyama, Y.; Kanakura, Y. Impaired platelet function in a patient with P2Y12 deficiency caused by a mutation in the translation initiation codon. J. Thromb. Haemost. 2005, 3, 2315–2323. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.E.; Dawood, B.B.; Lester, W.A.; Peake, I.R.; Rodeghiero, F.; Goodeve, A.C.; Makris, M.; Wilde, J.T.; Mumford, A.D.; Watson, S.P.; et al. Identification and characterization of a novel P2Y12 variant in a patient diagnosed with type 1 von Willebrand disease in the European MCMDM-1VWD study. Blood 2009, 113, 4110–4113. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, M.; Zighetti, M.L.; Lombardi, R.; Martinez, C.; Lecchi, A.; Conley, P.B.; Ware, J.; Ruggeri, Z.M. Molecular bases of defective signal transduction in the platelet P2Y12 receptor of a patient with congenital bleeding. Proc. Natl. Acad. Sci. USA 2003, 100, 1978–1983. [Google Scholar] [CrossRef] [Green Version]
- Nurden, P.; Savi, P.; Heilmann, E.; Bihour, C.; Herbert, J.M.; Maffrand, J.P.; Nurden, A. An inherited bleeding disorder linked to a defective interaction between ADP and its receptor on platelets. Its influence on glycoprotein IIb-IIIa complex function. J. Clin. Investig. 1995, 95, 1612–1622. [Google Scholar] [CrossRef] [Green Version]
- Hollopeter, G.; Jantzen, H.M.; Vincent, D.; Li, G.; England, L.; Ramakrishnan, V.; Yang, R.B.; Nurden, P.; Nurden, A.; Julius, D.; et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 2001, 409, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Nisar, S.; Daly, M.E.; Federici, A.B.; Artoni, A.; Mumford, A.D.; Watson, S.P.; Mundell, S.J. An intact PDZ motif is essential for correct P2Y12 purinoceptor traffic in human platelets. Blood 2011, 118, 5641–5651. [Google Scholar] [CrossRef]
- Cattaneo, M.; Lecchi, A.; Lombardi, R.; Gachet, C.; Zighetti, M.L. Platelets from a patient heterozygous for the defect of P2CYC receptors for ADP have a secretion defect despite normal thromboxane A2 production and normal granule stores, further evidence that some cases of platelet ‘primary secretion defect’ are heterozygous for a defect of P2CYC receptors. Thromb. Vasc. Biol. 2000, 20, E101–E106. [Google Scholar]
- Remijn, J.A.; IJsseldijk, M.J.; Strunk, A.L.; Abbes, A.P.; Engel, H.; Dikkeschei, B.; Dompeling, E.C.; de Groot, P.G.; Slingerland, R.J. Novel molecular defect in the platelet ADP receptor P2Y12 of a patient with hemorrhagic diathesis. Clin. Chem. Lab. Med. 2007, 45, 187–189. [Google Scholar] [CrossRef]
- Fontana, G.; Ware, J.; Cattaneo, M. Haploinsufficiency of the platelet P2Y12 gene in a family with congenital bleeding diathesis. Haematologica 2009, 94, 581–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundell, S.J.; Rabbolini, D.; Gabrielli, S.; Chen, Q.; Aungraheeta, R.; Hutchinson, J.L.; Kilo, T.; Mackay, J.; Ward, C.M.; Stevenson, W.; et al. Receptor homodimerization lays a critical role in a novel dominant negative P2RY12 variant identified in a family with severe bleeding. J. Thromb. Haemost. 2018, 16, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zighetti, M.L.; Carpani, G.; Sinigaglia, E.; Cattaneo, M. Usefulness of a flow cytometric analysis of intraplatelet VASP phosphorylation for the detection of patients with genetic defects of the platelet P2Y12 receptor for ADP. J. Thromb. Haemost. 2010, 8, 2332–2334. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, G.R.; Handin, R.I. Platelet Function in the Chediak-Higashi Syndrome. Blood 1976, 47, 941. [Google Scholar] [CrossRef] [Green Version]
- White, J.G.; Edson, J.R.; Desnick, S.J.; Witkop, C.J., Jr. Studies of platelets in a variant of the Hermansky-Pudlak syndrome. Am. J. Pathol. 1971, 63, 319–332. [Google Scholar]
- Glembotsky, A.C.; Bluteau, D.; Espasandin, Y.R.; Goette, N.P.; Marta, R.F.; Oyarzun, C.P.M.; Korin, L.; Lev, P.R.; Laguens, R.P.; Molinas, F.C.; et al. Mechanisms underlying platelet function defect in a pedigree with familial platelet disorder with a predisposition to acute myelogenous leukemia, potential role for candidate RUNX1 targets. J. Thromb. Haemost. 2014, 12, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Saultier, P.; Vidal, L.; Canault, M.; Bernot, D.; Falaise, C.; Pouymayou, C.; Bordet, J.C.; Saut, N.; Rostan, A.; Baccini, V.; et al. Macrothrombocytopenia and dense granule deficiency associated with FLI1 variants, ultrastructural and pathogenic features. Haematologica 2017, 102, 1006–1016. [Google Scholar] [CrossRef]
- Berrou, E.; Adam, F.; Lebret, M.; Planche, V.; Fergelot, P.; Issertial, O.; Coupry, I.; Bordet, J.C.; Nurden, P.; Bonneau, D.; et al. Gain-of-Function Mutation in Filamin A Potentiates Platelet Integrin α(IIb)β(3) Activation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1087–1097. [Google Scholar] [CrossRef] [Green Version]
- White, J.G.; Thomas, A. Platelet structural pathology in a patient with the X-linked GATA-1, R216Q mutation. Platelets 2009, 20, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Malpass, T.W.; Savage, B.; Hanson, S.R.; Slichter, S.J.; Harker, L.A. Correlation between prolonged bleeding time and depletion of platelet dense granule ADP in patients with myelodysplastic and myeloproliferative disorders. J. Lab. Clin. Med. 1984, 103, 894–904. [Google Scholar] [PubMed]
- Nieuwenhuis, H.K.; Akkerman, J.W.; Sixma, J.J. Patients with a prolonged bleeding time and normal aggregation tests may have storage pool deficiency, studies on one hundred six patients. Blood 1987, 70, 620–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawood, B.B.; Lowe, G.C.; Lordkipanidze, M.; Bem, D.; Daly, M.E.; Makris, M.; Mumford, A.; Wilde, J.T.; Watson, S.P. Evaluation of participants with suspected heritable platelet function disorders including recommendation and validation of a streamlined agonist panel. Blood 2012, 120, 5041–5049. [Google Scholar] [CrossRef] [Green Version]
- Mumford, A.D.; Dawood, B.B.; Daly, M.E.; Murden, S.L.; Williams, M.D.; Protty, M.B.; Spalton, J.C.; Wheatley, M.; Mundell, S.J.; Watson, S.P. A novel thromboxane A2 receptor D304N variant that abrogates ligand binding in a patient with a bleeding diathesis. Blood 2010, 115, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Nisar, S.P.; Lordkipanidze, M.; Jones, M.L.; Dawood, B.; Murden, S.; Cunningham, M.R.; Mumford, A.D.; Wilde, J.T.; Watson, S.P.; Mundell, S.J.; et al. A novel thromboxane A2 receptor N42S variant results in reduced surface expression and platelet dysfunction. Thromb. Haemost. 2014, 111, 923–932. [Google Scholar] [CrossRef]
- Kamae, T.; Kiyomizu, K.; Nakazawa, T.; Tadokoro, S.; Kashiwagi, H.; Honda, S.; Kanakura, Y.; Tomiyama, Y. Bleeding tendency and impaired platelet function in a patient carrying a heterozygous mutation in the thromboxane A2 receptor. J. Thromb. Haemost. 2011, 9, 1040–1048. [Google Scholar] [CrossRef]
- Hirata, T.; Kakizuka, A.; Ushikubi, F.; Fuse, I.; Okuma, M.; Narumiya, S. Arg60 to Leu mutation of the human thromboxane A2 receptor in a dominantly inherited bleeding disorder. J. Clin. Investig. 1994, 94, 1662–1667. [Google Scholar] [CrossRef] [Green Version]
- Bugert, P.; Fischer, L.; Althaus, K.; Knöfler, R.; Bakchoul, T. Platelet dysfunction caused by a novel thromboxane A (2) receptor mutation and congenital thrombocytopenia in a case of mild bleeding. Platelets 2020, 31, 276–279. [Google Scholar] [CrossRef]
- Geneviève, D.; Proulle, V.; Isidor, B.; Bellais, S.; Serre, V.; Djouadi, F.; Picard, C.; Vignon-Savoye, C.; Bader-Meunier, B.; Blanche, S.; et al. Thromboxane synthase mutations in an increased bone density disorder (Ghosal syndrome). Nat. Genet. 2008, 40, 284–286. [Google Scholar]
- Kim, S.M.; Chang, H.K.; Song, J.W.; Koh, H.; Han, S.J.; Severance Pediatric Liver Disease Research Group. Agranular platelets as a cardinal feature of ARC syndrome. J. Pediatr. Hematol. Oncol. 2010, 32, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, B.; Li, L.; Gissen, P.; Christensen, H.; McKiernan, P.J.; Ye, C.; Abdelhaleem, M.; Hayes, J.A.; Williams, M.D.; Chitayat, D.; et al. Requirement of VPS33B, a member of the Sec1/Munc18 protein family, in megakaryocyte and platelet alpha-granule biogenesis. Blood 2005, 106, 4159–4166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, B.; Lasne, D.; Rothschild, C.; Bouabdelli, M.; Ollivier, V.; Oudin, C.; Ajzenberg, N.; Grandchamp, B.; Jandrot-Persrus, M. Absence of collagen-induced platelet activation caused by compound heterozygous GPVI mutations. Blood 2009, 114, 1900–1903. [Google Scholar] [CrossRef] [PubMed]
- Matus, V.; Valenzuela, G.; Sáez, C.G.; Hidalgo, P.; Lagos, M.; Aranda, E.; Panes, O.; Pereira, J.; Pillois, X.; Nurden, A.T.; et al. An adenine insertion in exon 6 of human GP6 generates a truncated protein associated with a bleeding disorder in four Chilean families. J. Thromb. Haemost. 2013, 11, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Hermans, C.; Wittevrongel, C.; Thys, C.; Smethurst, P.A.; Van Geet, C.; Freson, K. A compound heterozygous mutation in glycoprotein VI in a patient with a bleeding disorder. J. Thromb. Haemost. 2009, 7, 1356–1363. [Google Scholar] [CrossRef]
- Arthur, J.F.; Dunkley, S.; Andrews, R.K. Platelet glycoprotein VI-related clinical defects. Br. J. Haematol. 2007, 139, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Nurden, P.; Tandon, N.; Takizawa, H.; Couzi, L.; Morel, D.; Fiore, M.; Pillois, X.; Loyau, S.; Jandrot-Perrus, M.; Nurden, A.T. An acquired inhibitor to the GPVI platelet collagen receptor in a patient with lupus nephritis. J. Thromb. Haemost. 2009, 7, 1541–1549. [Google Scholar] [CrossRef]
- Akiyama, M.; Kashiwagi, H.; Todo, K.; Moroi, M.; Berndt, M.C.; Kojima, H.; Kanakura, Y.; Tomiyama, Y. Presence of platelet-associated anti-glycoprotein (GP)VIautoantibodies and restoration of GPVI expression in patients with GPVI deficiency. J. Thromb. Haemost. 2009, 7, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Lhermusier, T.; Van Rothem, J.; Garcia, C.; Gratacap, M.P.; Payrastre, B. Targeted drug therapy, the platelet side. Platelets 2011, 22, 479–484. [Google Scholar] [CrossRef]
- Sasaki, T.; Shirai, T.; Tsukiji, N.; Otake, S.; Tamura, S.; Ichikawa, J.; Osada, M.; Satoh, K.; Ozaki, Y.; Suzuki-Inoue, K. Functional characterization of recombinant snake venom rhodocytin, rhodocytin mutant blocks CLEC-2/podoplanin-dependent platelet aggregation and lung metastasis. J. Thromb. Haemost. 2018, 16, 960–972. [Google Scholar] [CrossRef] [Green Version]
- Bellio, M.; Garcia, C.; Edouard, T.; Voisin, S.; Neel, B.G.; Cabou, C.; Valet, P.; Mori, J.; Mazharian, A.; Senis, Y.A.; et al. Catalytic dysregulation of SHP2 leading to Noonan syndromes impacts on platelet signaling and functions. Blood 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.K.; Berndt, M.C. Bernard-Soulier syndrome, an update. Semin. Thromb. Hemost. 2013, 39, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Othman, M.; Kaur, H.; Emsley, J. Platelet-type von Willebrand disease, new insights into the molecular pathophysiology of a unique platelet defect. Semin. Thromb. Hemost. 2013, 39, 663–673. [Google Scholar] [PubMed]
- Favaloro, E.J.; Patterson, D.; Denholm, A.; Mead, S.; Gilbert, A.; Collins, A.; Estell, J.; George, P.M.; Smith, M.P. Differential identification of a rare form of platelet-type (pseudo-) von Willebrand disease (VWD) from Type 2B VWD using a simplified ristocetin-induced-platelet-agglutination mixing assay and confirmed by genetic analysis. Br. J. Haematol. 2007, 139, 623–626. [Google Scholar] [CrossRef]
- Frontroth, J.P.; Favaloro, E.J. Ristocetin-Induced Platelet Aggregation (RIPA) and RIPA Mixing Studies. Methods Mol. Biol. 2017, 1646, 473–494. [Google Scholar]
- Freson, K.; Hoylaerts, M.F.; Jaeken, J.; Eyssen, M.; Arnout, J.; Vermylen, J.; Van Geet, C. Genetic variation of the extra-large stimulatory G protein alpha-subunit leads toGs hyperfunction in platelets and is a risk factor for bleeding. Thromb. Haemost. 2001, 86, 733–738. [Google Scholar] [CrossRef]
- Kambayashi, J.; Shinoki, N.; Nakamura, T.; Ariyoshi, H.; Kawasaki, T.; Sakon, M.; Monden, M. Prevalence of impaired responsiveness to epinephrine in platelets among Japanese. Thromb. Res. 1996, 81, 85–90. [Google Scholar] [CrossRef]
- Hayward, C.P.; Rivard, G.E.; Kane, W.H.; Drouin, J.; Zhengs, S.; Moore, J.C.; Kelton, J.G. An autosomal dominant, qualitative platelet disorder associated with multimerin deficiency, abnormalities in platelet factor V, thrombospondin, von Willebrand factor, and fibrinogen and an epinephrine aggregation defect. Blood 1996, 87, 4967–4978. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.K.; Koike, K.; Willis, J.; Daniel, J.L.; Beckett, C.; Hassel, B.; Day, H.J.; Smith, J.B. Holmsen H. Platelet secretion defect associated with impaired liberation of arachidonic acid and normal myosin light chain phosphorylation. Blood 1984, 64, 914–921. [Google Scholar] [CrossRef] [Green Version]
- Gabbeta, J.; Yang, X.; Kowalska, M.A.; Sun, L.; Dhanasekaran, N.; Rao, A.K. Platelet signal transduction defect with Galpha subunit dysfunction and diminished Galphaq in a patient with abnormal platelet responses. Proc. Natl. Acad. Sci. USA 1997, 94, 8750–8755. [Google Scholar] [CrossRef] [Green Version]
- Gabbeta, J.; Vaidyula, V.R.; Dhanasekaran, D.N.; Rao, A.K. Human platelet Galphaq deficiency is associated with decreased Galphaq gene expression in platelets but not neutrophils. Thromb. Haemost. 2002, 87, 129–133. [Google Scholar] [CrossRef]
- Mao, G.F.; Vaidyula, V.R.; Kunapuli, S.P.; Rao, A.K. Lineage-specific defect in gene expression in human platelet phospholipase C-beta 2 deficiency. Blood 2002, 99, 905–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oury, C.; Toth-Zsamboki, E.; Van Geet, C.; Thys, C.; Wei, L.; Nilius, B.; Vermylen, J.; Hoylaerts, M.F. A natural dominant negative P2X1 receptor due to deletion of a single amino acid residue. J. Biol. Chem. 2000, 275, 22611–22614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, M. The P2 receptors and congenital platelet function defects. Semin. Thromb. Hemost. 2005, 31, 168–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwenhuis, H.K.; Akkerman, J.W.; Houdijk, W.P.; Sixma, J.J. Human blood platelets showing no response to collagen fail to express surface glycoprotein Ia. Nature 1985, 318, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Kehrel, B.; Balleisen, L.; Kokott, R.; Mesters, R.; Stenzinger, W.; Clemetson, K.J.; van de Loo, J. Deficiency of intact thrombospondin and membrane glycoprotein Ia in platelets with defective collagen-induced aggregation and spontaneous loss of disorder. Blood 1988, 71, 1074–1078. [Google Scholar] [CrossRef] [Green Version]
- Pischel, K.D.; Hemler, M.E.; Huang, C.; Bluestein, H.G.; Woods, V.L., Jr. Use of the monoclonal antibody 12F1 to characterize the differentiation antigen VLA-2. J. Immunol. 1987, 138, 226–233. [Google Scholar]
- Handa, M.; Watanabe, K.; Kawai, Y.; Kamata, T.; Koyama, T.; Nagai, H.; Ikeda, Y. Platelet unresponsiveness to collagen, involvement of glycoprotein Ia-IIa (alpha 2 beta 1 integrin) deficiency associated with a myeloproliferative disorder. Thromb. Haemost. 1995, 73, 521–528. [Google Scholar]
- Gresele, P.; Harrison, P.; Bury, L.; Falcinelli, E.; Gachet, C.; Hayward, C.P.; Kenny, D.; Mezzano, D.; Mumford, A.D.; Nugent, D.; et al. Diagnosis of suspected inherited platelet function disorders: results of a worldwide survey. J. Thromb. Haemost. 2014, 12, 1562–1569. [Google Scholar] [CrossRef]
- Paniccia, R.; Priora, R.; Liotta, A.A.; Abbate, R. Platelet function tests: a comparative review. Vasc. Health Risk Manag. 2015, 11, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Femia, E.A.; Scavone, M.; Lecchi, A.; Cattaneo, M. Effect of platelet count on platelet aggregation measured with impedance aggregometry (Multiplate™ analyzer) and with light transmission aggregometry. J. Thromb. Haemost. 2013, 11, 2193–2196. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
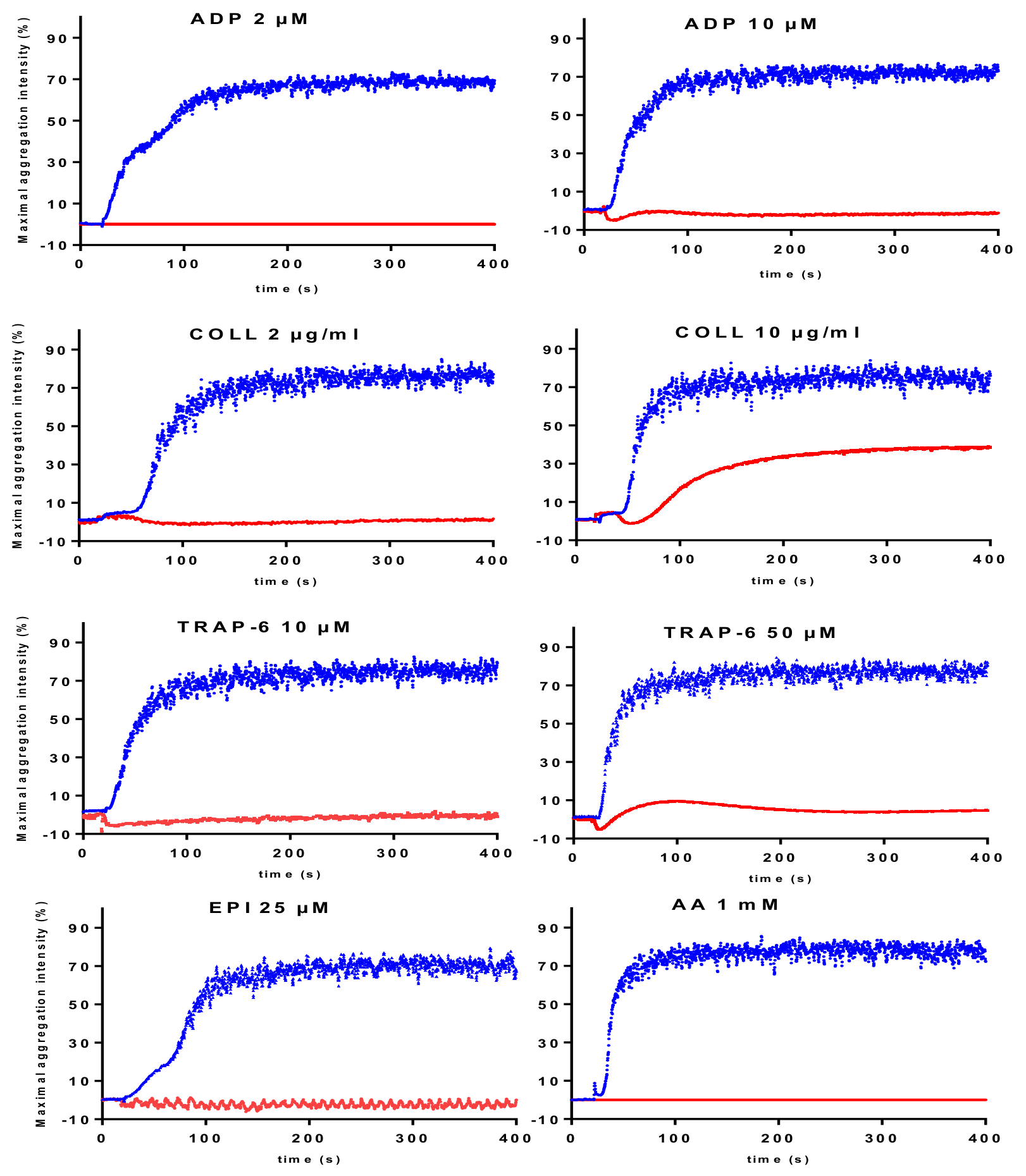
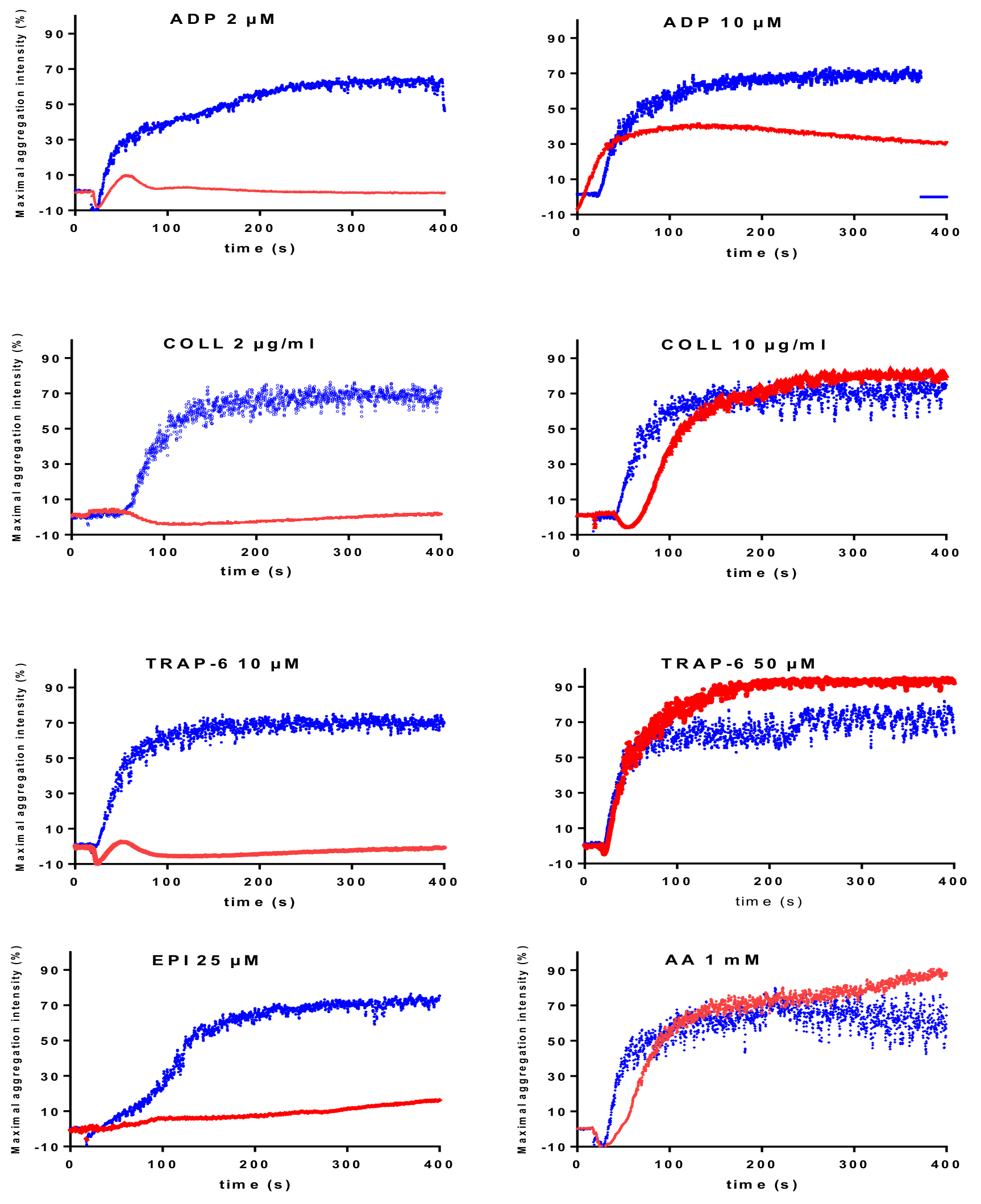
| Absence of Aggregation in Response to Multiple Activators Except Ristocetin | |
|---|---|
| LTA Traces | Diagnostic Orientation |
| Absence or marked reduction of aggregation in response to low and high agonist concentrations | If quantitative deficiency in GPIIb/IIIa: Glanzmann thrombasthenia |
| Absence or marked reduction of aggregation in response to low and high agonist concentrations | If qualitative deficiency in GPIIb/IIIa: Glanzmann thrombasthenia variant |
| If immune deficiency (infections hyperleukocytosis): Type III LAD | |
| Reduced aggregation in response to multiple activators | |
| Absence of response to low concentrations, but normal or improved response with high concentrations of activators except for epinephrine. Reduced aggregation responses to ADP (2-10 µM) and epinephrine (5–25 µM) in all reported cases is a consistent laboratory phenotype. | CalDAG-GEFI deficiency |
| Reduced response to ADP even at high concentrations (100 μM); reduced (or reversible) response to low concentrations and improved response to high concentrations with the other activators. | Severe homozygous P2Y12 deficiency |
| Reduced (or reversible) response to low concentrations of activators and improved response with high concentrations of the activators. The defective response to 10 µM ADP is often moderate. | Severe dense granule defects |
| Absence or reduced response to AA and reduced (or reversible) response to the other activators such as epinephrine and ADP. The responses improve on increasing activator concentrations. | TXA2 pathway disorder. The use of a specific TP agonist distinguishes between defect in TP receptor or TXA2 synthesis |
| Defective response to a specific activator | |
| Reduced response to collagen | GPVI disorder. The use of more specific GPVI agonist may help to confirm the diagnosis orientation. |
| Reduced response to normal or low concentration of ristocetin. | Defect in the GPIb/vWF axis. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alessi, M.-C.; Sié, P.; Payrastre, B., on behalf of the French Reference Center on Constitutional Platelet Disorders (CRPP). Strengths and Weaknesses of Light Transmission Aggregometry in Diagnosing Hereditary Platelet Function Disorders. J. Clin. Med. 2020, 9, 763. https://doi.org/10.3390/jcm9030763
Alessi M-C, Sié P, Payrastre B on behalf of the French Reference Center on Constitutional Platelet Disorders (CRPP). Strengths and Weaknesses of Light Transmission Aggregometry in Diagnosing Hereditary Platelet Function Disorders. Journal of Clinical Medicine. 2020; 9(3):763. https://doi.org/10.3390/jcm9030763
Chicago/Turabian StyleAlessi, Marie-Christine, Pierre Sié, and Bernard Payrastre on behalf of the French Reference Center on Constitutional Platelet Disorders (CRPP). 2020. "Strengths and Weaknesses of Light Transmission Aggregometry in Diagnosing Hereditary Platelet Function Disorders" Journal of Clinical Medicine 9, no. 3: 763. https://doi.org/10.3390/jcm9030763