The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondria: Key Players in Innate Immunity
2.1. Mitochondrial Dynamics During Viral Infection
2.2. Mitochondrial Dynamics in NLRP3 Inflammasome Activation
2.3. Mitochondrial Dynamics during Bacterial Infection
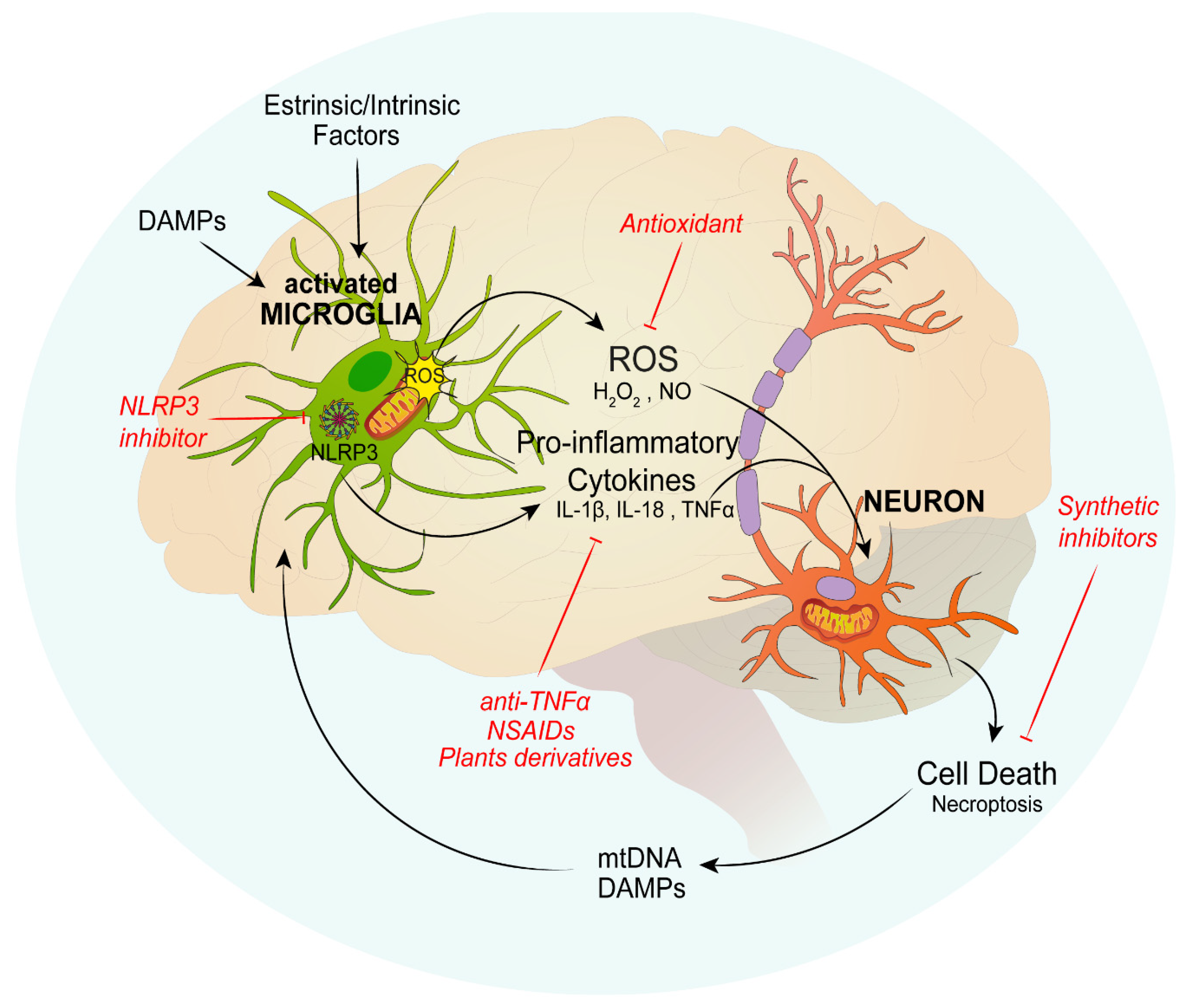
3. Role of Mitochondria and Neuroinflammation in Neurodegenerative Diseases
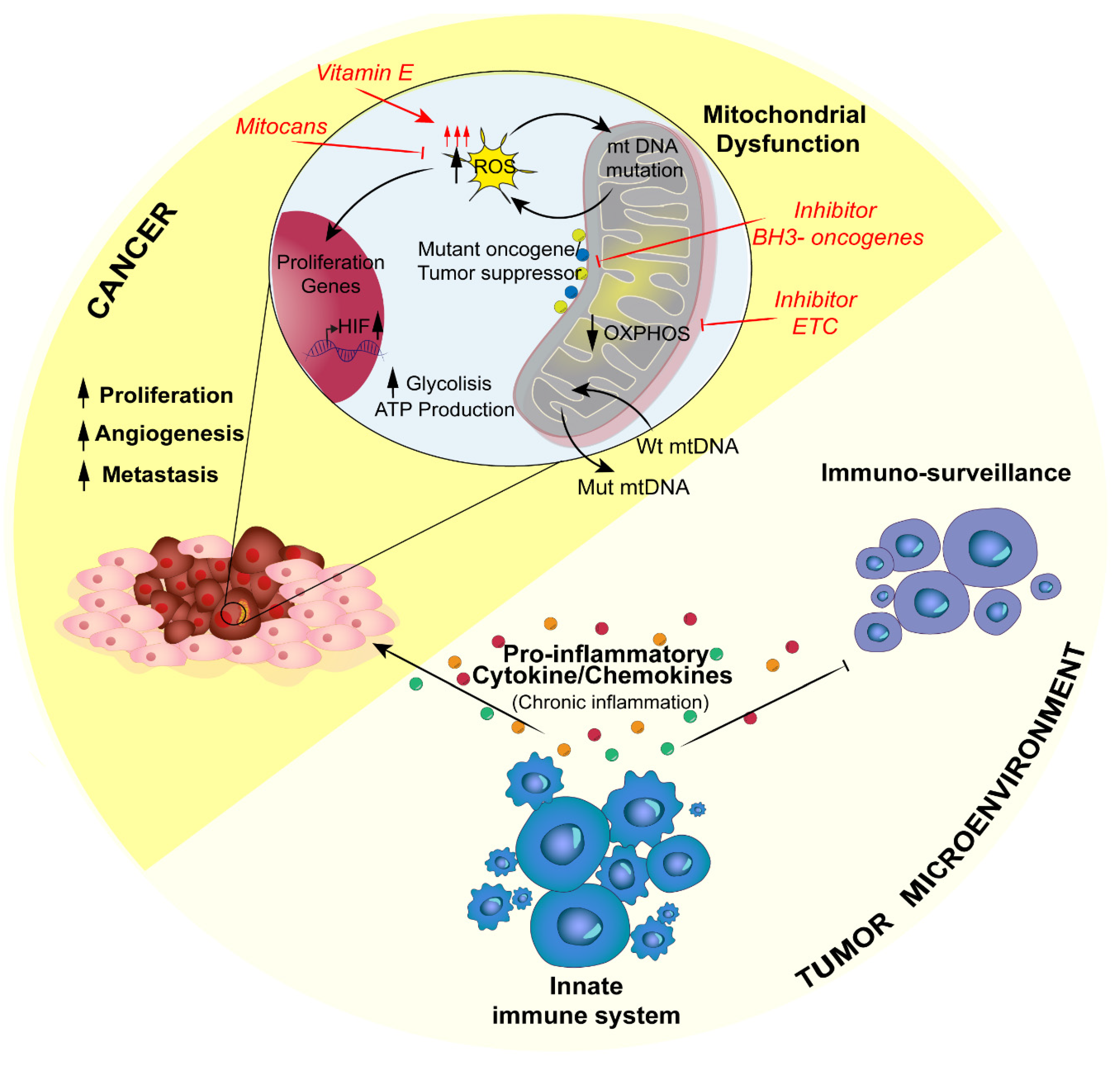
4. Inflammation-Related Mitochondrial Dysfunction in Cancer: A Negative Loop
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease; |
| ALR | AIM2-like receptor; |
| ALS | amyotrophic lateral sclerosis; |
| ANT | adenine nucleotide translocase; |
| ATP | adenosine triphosphate; |
| Ca2+ | calcium ion; |
| CARDs | caspase recruitment domains; |
| CASR | Ca2+ sensing receptor; |
| CLRs | C-type lectin receptors; |
| COX | cyclooxygenase; |
| CMV | cytomegalovirus; |
| DAMPs | damage-associated molecular patterns; |
| DENV | dengue virus; |
| ETC | electron transport chain; |
| ER | endoplasmic reticulum; |
| FADH2 | flavin adenine dinucleotide; |
| HIF | hypoxia-inducible factors; |
| HK | hexokinase; |
| IFN | type I interferon |
| IL-1β | interleukin-1beta; |
| IRF-3 | interferon regulatory factor 3; |
| JNK | c-Jun amino-terminal kinase; |
| KGDH | alpha-ketoglutarate dehydrogenase; |
| MAMs | mitochondria associated ER membranes; |
| MAVS | mitochondrial antiviral signaling; |
| MCU | mitochondrial calcium uniporter; |
| MDA-5 | melanoma differentiation-associated gene 5; |
| MDVs | mitochondrial-derived vesicles; |
| MFN1 | mitofusin-1; |
| MFN2 | mitofusin-2; |
| MMP | mitochondrial membrane potential; |
| mROS | mitochondrial reactive oxygen species; |
| mtDNA | mitochondrial DNA; |
| MRC | mitochondrial respiration complex; |
| MS | multiple sclerosis; |
| NADH | nicotinamide adenine dinucleotide; |
| NDDs | neurodegenerative disorders; |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NSAID | nonsteroidal anti-inflammatory drugs; |
| NO | nitric oxide; |
| NLRs | nucleotide-binding oligomerization domain-like receptors; |
| NLRP3 | NOD-, LRR- and pyrin domain containing 3; |
| OMM | outer mitochondrial membrane; |
| OPA1 | optic atrophy 1; |
| OXPHOS | mitochondrial oxidative phosphorylation; |
| PAMPs | pathogen-associated molecular patterns; |
| PD | Parkinson’s disease; |
| PDH | pyruvate dehydrogenase; |
| PKM2 | pyruvate kinase M2; |
| PRRs | pattern recognition receptor; |
| RIG-1 | retinoic acid-inducible gene I; |
| RIPK1 | receptor-interacting kinases 1; |
| RLR | RIG-I-like receptor; |
| ROS | reactive oxygen species; |
| SOSS | silencing of survival signals; |
| STING | stimulator of interferon genes; |
| TAMs | tumor-associated macrophages; |
| TCA | tricarboxylic acid cycle; |
| TLRs | Toll-like receptors; |
| TRAF6 | tumor necrosis factor receptor associated factor 6; |
| VDAC | voltage-dependent anionic channel; |
| VopE | Type 3 secretion system effector. |
References
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-ros crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 2012, 329635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, A.; Tiwari-Pandey, R.; Pandey, N.R. Mitochondria: The indispensable players in innate immunity and guardians of the inflammatory response. J. Cell Commun. Signal 2019, 13, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhir, R.; Halder, A.; Sunkaria, A. Mitochondria as a centrally positioned hub in the innate immune response. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81, 281–293. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.; Lindsley, T.A.; Sheridan, A.; Bhoiwala, D.L.; Hushmendy, S.F.; Yager, E.J.; Ruggiero, E.A.; Crawford, D.R. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J. Alzheimers Dis. 2012, 30, 617–627. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.T.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.W.; Olefsky, J.M. JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J. Biol. Chem. 2005, 280, 35361–35371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.L.; Coyne, C.B. Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol. 2013, 425, 5009–5019. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, K.; Oshiumi, H.; Takeda, M.; Ishihara, N.; Yanagi, Y.; Seya, T.; Kawabata, S.; Koshiba, T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal 2009, 2, ra47. [Google Scholar] [CrossRef]
- Koshiba, T.; Yasukawa, K.; Yanagi, Y.; Kawabata, S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci. Signal 2011, 4, ra7. [Google Scholar] [CrossRef]
- Choi, H.J.; Park, A.; Kang, S.; Lee, E.; Lee, T.A.; Ra, E.A.; Lee, J.; Lee, S.; Park, B. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat. Commun. 2018, 9, 125. [Google Scholar] [CrossRef]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Soares, F.; Tattoli, I.; Girardin, S.E. Mitochondria in innate immunity. EMBO Rep. 2011, 12, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Lei, C.Q.; Zhang, Y.; Li, M.; Jiang, L.Q.; Zhong, B.; Kim, Y.H.; Shu, H.B. ECSIT bridges RIG-I-like receptors to VISA in signaling events of innate antiviral responses. J. Innate Immun. 2015, 7, 153–164. [Google Scholar] [CrossRef]
- Yu, C.Y.; Liang, J.J.; Li, J.K.; Lee, Y.L.; Chang, B.L.; Su, C.I.; Huang, W.J.; Lai, M.M.; Lin, Y.L. Dengue Virus Impairs Mitochondrial Fusion by Cleaving Mitofusins. PLoS Pathog. 2015, 11, e1005350. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Liao, Y.; Zhou, L.; Peng, S.; Chen, H.; Yuan, Z. Amplified RLR signaling activation through an interferon-stimulated gene-endoplasmic reticulum stress-mitochondrial calcium uniporter protein loop. Sci. Rep. 2016, 6, 20158. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat. Immunol. 2008, 9, 866–872. [Google Scholar] [CrossRef]
- Van Bruggen, R.; Koker, M.Y.; Jansen, M.; van Houdt, M.; Roos, D.; Kuijpers, T.W.; van den Berg, T.K. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood 2010, 115, 5398–5400. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [PubMed]
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteom. 2013, 79, 219–230. [Google Scholar] [CrossRef]
- Zhang, A.; Williamson, C.D.; Wong, D.S.; Bullough, M.D.; Brown, K.J.; Hathout, Y.; Colberg-Poley, A.M. Quantitative proteomic analyses of human cytomegalovirus-induced restructuring of endoplasmic reticulum-mitochondrial contacts at late times of infection. Mol. Cell Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef]
- Missiroli, S.; Danese, A.; Iannitti, T.; Patergnani, S.; Perrone, M.; Previati, M.; Giorgi, C.; Pinton, P. Endoplasmic reticulum-mitochondria Ca(2+) crosstalk in the control of the tumor cell fate. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 858–864. [Google Scholar] [CrossRef]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal 2015, 22, 995–1019. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014, 35, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Won, J.H.; Hwang, I.; Hong, S.; Lee, H.K.; Yu, J.W. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 2015, 5, 15489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Bae, S.H.; Ryu, J.C.; Kwon, Y.; Oh, J.H.; Kwon, J.; Moon, J.S.; Kim, K.; Miyawaki, A.; Lee, M.G.; et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Danilchanka, O.; Mekalanos, J.J. Vibrio cholerae T3SS effector VopE modulates mitochondrial dynamics and innate immune signaling by targeting Miro GTPases. Cell Host Microbe 2014, 16, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czyz, D.M.; Willett, J.W.; Crosson, S. Brucella abortus Induces a Warburg Shift in Host Metabolism That Is Linked to Enhanced Intracellular Survival of the Pathogen. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobet, E.; Willemart, K.; Ninane, N.; Demazy, C.; Sedzicki, J.; Lelubre, C.; De Bolle, X.; Renard, P.; Raes, M.; Dehio, C.; et al. Mitochondrial fragmentation affects neither the sensitivity to TNFalpha-induced apoptosis of Brucella-infected cells nor the intracellular replication of the bacteria. Sci. Rep. 2018, 8, 5173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavru, F.; Bouillaud, F.; Sartori, A.; Ricquier, D.; Cossart, P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA 2011, 108, 3612–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escoll, P.; Song, O.R.; Viana, F.; Steiner, B.; Lagache, T.; Olivo-Marin, J.C.; Impens, F.; Brodin, P.; Hilbi, H.; Buchrieser, C. Legionella pneumophila Modulates Mitochondrial Dynamics to Trigger Metabolic Repurposing of Infected Macrophages. Cell Host Microbe 2017, 22, 302–316. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Reimer, A.; Sharan, M.; Kozjak-Pavlovic, V.; Eulalio, A.; Prusty, B.K.; Fraunholz, M.; Karunakaran, K.; Rudel, T. Chlamydia preserves the mitochondrial network necessary for replication via microRNA-dependent inhibition of fission. J. Cell Biol. 2017, 216, 1071–1089. [Google Scholar] [CrossRef]
- Liang, P.; Rosas-Lemus, M.; Patel, D.; Fang, X.; Tuz, K.; Juarez, O. Dynamic energy dependency of Chlamydia trachomatis on host cell metabolism during intracellular growth: Role of sodium-based energetics in chlamydial ATP generation. J. Biol. Chem. 2018, 293, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Zhang, Z.; Tian, L.; Wang, S.; Hu, S.; Qiao, J.J. The Salmonella effector SopB prevents ROS-induced apoptosis of epithelial cells by retarding TRAF6 recruitment to mitochondria. Biochem. Biophys. Res. Commun. 2016, 478, 618–623. [Google Scholar] [CrossRef]
- Abuaita, B.H.; Schultz, T.L.; O’Riordan, M.X. Mitochondria-Derived Vesicles Deliver Antimicrobial Reactive Oxygen Species to Control Phagosome-Localized Staphylococcus aureus. Cell Host Microbe 2018, 24, 625–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [Green Version]
- Kapetanovic, R.; Bokil, N.J.; Sweet, M.J. Innate immune perturbations, accumulating DAMPs and inflammasome dysregulation: A ticking time bomb in ageing. Ageing Res. Rev. 2015, 24, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Licastro, F.; Pedrini, S.; Caputo, L.; Annoni, G.; Davis, L.J.; Ferri, C.; Casadei, V.; Grimaldi, L.M. Increased plasma levels of interleukin-1, interleukin-6 and alpha-1-antichymotrypsin in patients with Alzheimer’s disease: Peripheral inflammation or signals from the brain? J. Neuroimmunol. 2000, 103, 97–102. [Google Scholar] [CrossRef]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spielman, L.J.; Little, J.P.; Klegeris, A. Inflammation and insulin/IGF-1 resistance as the possible link between obesity and neurodegeneration. J. Neuroimmunol. 2014, 273, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Lastres-Becker, I.; Cuadrado, A. Role of microglial redox balance in modulation of neuroinflammation. Curr. Opin. Neurol. 2009, 22, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Prinz, M. Factors regulating microglia activation. Front. Cell Neurosci. 2013, 7, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; Lopez, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid Redox Signal 2014, 21, 1766–1801. [Google Scholar] [CrossRef] [Green Version]
- Dheen, S.T.; Kaur, C.; Ling, E.A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Franco, R.; Vargas, M.R. Redox Biology in Neurological Function, Dysfunction, and Aging. Antioxid Redox Signal 2018, 28, 1583–1586. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid Redox Signal 2012, 16, 1323–1367. [Google Scholar] [CrossRef]
- Rossi, S.; Zanier, E.R.; Mauri, I.; Columbo, A.; Stocchetti, N. Brain temperature, body core temperature, and intracranial pressure in acute cerebral damage. J. Neurol. Neurosurg. Psychiatry 2001, 71, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 2017, 391, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero-Mendez, A.; Almeida, A.; Fernandez, E.; Maestre, C.; Moncada, S.; Bolanos, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Zhang, H.; Sheu, K.F.; Bogdanovich, N.; Lindsay, J.G.; Lannfelt, L.; Vestling, M.; Cowburn, R.F. Alpha-ketoglutarate dehydrogenase in Alzheimer brains bearing the APP670/671 mutation. Ann. Neurol. 1998, 44, 676–681. [Google Scholar] [CrossRef]
- Mastrogiacoma, F.; Lindsay, J.G.; Bettendorff, L.; Rice, J.; Kish, S.J. Brain protein and alpha-ketoglutarate dehydrogenase complex activity in Alzheimer’s disease. Ann. Neurol. 1996, 39, 592–598. [Google Scholar] [CrossRef]
- Nijland, P.G.; Molenaar, R.J.; van der Pol, S.M.; van der Valk, P.; van Noorden, C.J.; de Vries, H.E.; van Horssen, J. Differential expression of glucose-metabolizing enzymes in multiple sclerosis lesions. Acta Neuropathol. Commun. 2015, 3, 79. [Google Scholar] [CrossRef] [Green Version]
- Nagra, R.M.; Becher, B.; Tourtellotte, W.W.; Antel, J.P.; Gold, D.; Paladino, T.; Smith, R.A.; Nelson, J.R.; Reynolds, W.F. Immunohistochemical and genetic evidence of myeloperoxidase involvement in multiple sclerosis. J. Neuroimmunol. 1997, 78, 97–107. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Rhees, J.; Maciejewski, D.; Paladino, T.; Sieburg, H.; Maki, R.A.; Masliah, E. Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp. Neurol. 1999, 155, 31–41. [Google Scholar] [CrossRef]
- Shi, Q.; Xu, H.; Yu, H.; Zhang, N.; Ye, Y.; Estevez, A.G.; Deng, H.; Gibson, G.E. Inactivation and reactivation of the mitochondrial alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem. 2011, 286, 17640–17648. [Google Scholar] [CrossRef] [Green Version]
- Lunnon, K.; Ibrahim, Z.; Proitsi, P.; Lourdusamy, A.; Newhouse, S.; Sattlecker, M.; Furney, S.; Saleem, M.; Soininen, H.; Kloszewska, I.; et al. Mitochondrial dysfunction and immune activation are detectable in early Alzheimer’s disease blood. J. Alzheimers Dis. 2012, 30, 685–710. [Google Scholar] [CrossRef] [Green Version]
- Doll, D.N.; Rellick, S.L.; Barr, T.L.; Ren, X.; Simpkins, J.W. Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J. Neurochem. 2015, 132, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Kastl, L.; Sauer, S.W.; Ruppert, T.; Beissbarth, T.; Becker, M.S.; Suss, D.; Krammer, P.H.; Gulow, K. TNF-alpha mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-kappaB activation in liver cells. FEBS Lett. 2014, 588, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.; Huttemann, M. Energy crisis: The role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Biophys. Acta 2014, 1842, 1579–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samavati, L.; Lee, I.; Mathes, I.; Lottspeich, F.; Huttemann, M. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J. Biol. Chem. 2008, 283, 21134–21144. [Google Scholar] [CrossRef] [Green Version]
- Suliman, H.B.; Welty-Wolf, K.E.; Carraway, M.; Tatro, L.; Piantadosi, C.A. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc. Res. 2004, 64, 279–288. [Google Scholar] [CrossRef]
- Moe, G.W.; Marin-Garcia, J.; Konig, A.; Goldenthal, M.; Lu, X.; Feng, Q. In vivo TNF-alpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1813–H1820. [Google Scholar] [CrossRef] [Green Version]
- Venters, H.D.; Dantzer, R.; Kelley, K.W. A new concept in neurodegeneration: TNFalpha is a silencer of survival signals. Trends Neurosci. 2000, 23, 175–180. [Google Scholar] [CrossRef]
- Prajapati, P.; Sripada, L.; Singh, K.; Bhatelia, K.; Singh, R.; Singh, R. TNF-alpha regulates miRNA targeting mitochondrial complex-I and induces cell death in dopaminergic cells. Biochim. Biophys. Acta 2015, 1852, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Palomer, X.; Alvarez-Guardia, D.; Rodriguez-Calvo, R.; Coll, T.; Laguna, J.C.; Davidson, M.M.; Chan, T.O.; Feldman, A.M.; Vazquez-Carrera, M. TNF-alpha reduces PGC-1alpha expression through NF-kappaB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovasc. Res. 2009, 81, 703–712. [Google Scholar] [CrossRef]
- Hahn, W.S.; Kuzmicic, J.; Burrill, J.S.; Donoghue, M.A.; Foncea, R.; Jensen, M.D.; Lavandero, S.; Arriaga, E.A.; Bernlohr, D.A. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1033–E1045. [Google Scholar] [CrossRef] [Green Version]
- Motori, E.; Puyal, J.; Toni, N.; Ghanem, A.; Angeloni, C.; Malaguti, M.; Cantelli-Forti, G.; Berninger, B.; Conzelmann, K.K.; Gotz, M.; et al. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 2013, 18, 844–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Daniels, B.P.; Snyder, A.G.; Olsen, T.M.; Orozco, S.; Oguin, T.H., 3rd; Tait, S.W.G.; Martinez, J.; Gale, M., Jr.; Loo, Y.M.; Oberst, A. RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell 2017, 169, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Tang, M.B.; Luo, H.Y.; Shi, C.H.; Xu, Y.M. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef] [Green Version]
- Iliev, A.I.; Stringaris, A.K.; Nau, R.; Neumann, H. Neuronal injury mediated via stimulation of microglial toll-like receptor-9 (TLR9). FASEB J. 2004, 18, 412–414. [Google Scholar] [CrossRef]
- Ajmone-Cat, M.A.; Bernardo, A.; Greco, A.; Minghetti, L. Non-Steroidal Anti-Inflammatory Drugs and Brain Inflammation: Effects on Microglial Functions. Pharmaceuticals 2010, 3, 1949–1965. [Google Scholar] [CrossRef]
- Koch, J.C.; Tatenhorst, L.; Roser, A.E.; Saal, K.A.; Tonges, L.; Lingor, P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol. Ther. 2018, 189, 1–21. [Google Scholar] [CrossRef]
- Yu, Y.; Shen, Q.; Lai, Y.; Park, S.Y.; Ou, X.; Lin, D.; Jin, M.; Zhang, W. Anti-inflammatory Effects of Curcumin in Microglial Cells. Front. Pharmacol. 2018, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Gan, P.; Zhang, L.; Chen, Y.; Zhang, Y.; Zhang, F.; Zhou, X.; Zhang, X.; Gao, B.; Zhen, X.; Zhang, J.; et al. Anti-inflammatory effects of glaucocalyxin B in microglia cells. J. Pharmacol. Sci. 2015, 128, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, M.; Al-Suhaimi, E.A.; Wahid, F.; Shehzad, O.; Shehzad, A. Therapeutic potential of curcumin for multiple sclerosis. Neurol. Sci. 2018, 39, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Liang, A.; Rangel, A.; Gyengesi, E.; Niedermayer, G.; Munch, G. High bioavailability curcumin: An anti-inflammatory and neurosupportive bioactive nutrient for neurodegenerative diseases characterized by chronic neuroinflammation. Arch. Toxicol. 2017, 91, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Jun-Long, H.; Yi, L.; Bao-Lian, Z.; Jia-Si, L.; Ning, Z.; Zhou-Heng, Y.; Xue-Jun, S.; Wen-Wu, L. Necroptosis Signaling Pathways in Stroke: From Mechanisms to Therapies. Curr. Neuropharmacol. 2018, 16, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.Z.; Cao, Q.; Liu, C. Targeting NLRP3 Inflammasome in the Treatment of CNS Diseases. Front. Mol. Neurosci. 2018, 11, 320. [Google Scholar] [CrossRef]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2011, 2, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Vringer, E.; Tait, S.W.G. Mitochondria and Inflammation: Cell Death Heats Up. Front. Cell Dev. Biol. 2019, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Kim, D. Cancer Metabolism: Fueling More than Just Growth. Mol. Cells 2016, 39, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-Garcia, C.; Valcarcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Maximo, V.; Botelho, T.; Capela, J.; Soares, P.; Lima, J.; Taveira, A.; Amaro, T.; Barbosa, A.P.; Preto, A.; Harach, H.R.; et al. Somatic and germline mutation in GRIM-19, a dual function gene involved in mitochondrial metabolism and cell death, is linked to mitochondrion-rich (Hurthle cell) tumours of the thyroid. Br. J. Cancer 2005, 92, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Putignani, L.; Raffa, S.; Pescosolido, R.; Aimati, L.; Signore, F.; Torrisi, M.R.; Grammatico, P. Alteration of expression levels of the oxidative phosphorylation system (OXPHOS) in breast cancer cell mitochondria. Breast Cancer Res. Treat. 2008, 110, 439–452. [Google Scholar] [CrossRef]
- Bonora, E.; Porcelli, A.M.; Gasparre, G.; Biondi, A.; Ghelli, A.; Carelli, V.; Baracca, A.; Tallini, G.; Martinuzzi, A.; Lenaz, G.; et al. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial DNA mutations affecting complexes I and III. Cancer Res. 2006, 66, 6087–6096. [Google Scholar] [CrossRef] [Green Version]
- Stankov, K.; Biondi, A.; D’Aurelio, M.; Gasparre, G.; Falasca, A.; Romeo, G.; Lenaz, G. Mitochondrial activities of a cell line derived from thyroid Hurthle cell tumors. Thyroid 2006, 16, 325–331. [Google Scholar] [CrossRef]
- Simonnet, H.; Alazard, N.; Pfeiffer, K.; Gallou, C.; Beroud, C.; Demont, J.; Bouvier, R.; Schagger, H.; Godinot, C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Modica-Napolitano, J.S.; Singh, K.K. Mitochondrial dysfunction in cancer. Mitochondrion 2004, 4, 755–762. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hold, G.L.; El-Omar, E.M. Genetic aspects of inflammation and cancer. Biochem. J. 2008, 410, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Khansari, N.; Shakiba, Y.; Mahmoudi, M. Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2009, 3, 73–80. [Google Scholar] [CrossRef]
- Segal, A.W. How superoxide production by neutrophil leukocytes kills microbes. Novartis Found. Symp. 2006, 279, 92–98, discussion 98-100, 216-109. [Google Scholar]
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef] [Green Version]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, T. Metabolism: The mitochondria that wag the dog. Nat. Rev. Cancer 2011, 11, 155. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.V.; Dong, L.; Neuzil, J. Mitochondrial DNA in Tumor Initiation, Progression, and Metastasis: Role of Horizontal mtDNA Transfer. Cancer Res. 2015, 75, 3203–3208. [Google Scholar] [CrossRef] [Green Version]
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernandez-Delgado, I.; Latorre-Pellicer, A.; Acin-Perez, R.; Martin-Cofreces, N.B.; Jaso-Tamame, A.L.; Iborra, S.; Jorge, I.; et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun. 2018, 9, 2658. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Feng, M.; Guan, W. Mitochondrial DNA sensing by STING signaling participates in inflammation, cancer and beyond. Int. J. Cancer 2016, 139, 736–741. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013, 20, 1631–1643. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca(2+)-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafa, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Choi, S.C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of CD4+ T cell metabolism reverses lupus. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wen, H.; Zhou, C.; Su, Q.; Lin, Y.; Xie, Y.; Huang, Y.; Qiu, Q.; Lin, J.; Huang, X.; et al. TNF-alpha derived from M2 tumor-associated macrophages promotes epithelial-mesenchymal transition and cancer stemness through the Wnt/beta-catenin pathway in SMMC-7721 hepatocellular carcinoma cells. Exp. Cell Res. 2019, 378, 41–50. [Google Scholar] [CrossRef]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bahre, H.; et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 2014, 20, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.; Constantin, C.; Popescu, I.D.; Zipeto, D.; Tzanakakis, G.; Nikitovic, D.; Fenga, C.; Stratakis, C.A.; Spandidos, D.A.; Tsatsakis, A.M. Inflammation and Metabolism in Cancer Cell-Mitochondria Key Player. Front. Oncol. 2019, 9, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Hahn, T.; Polanczyk, M.J.; Borodovsky, A.; Ramanathapuram, L.V.; Akporiaye, E.T.; Ralph, S.J. Use of anti-cancer drugs, mitocans, to enhance the immune responses against tumors. Curr. Pharm. Biotechnol. 2013, 14, 357–376. [Google Scholar] [CrossRef] [Green Version]
- Gillies, R.J.; Gatenby, R.A. Metabolism and its sequelae in cancer evolution and therapy. Cancer J. 2015, 21, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Hartman, M.L.; Czyz, M. Pro-apoptotic activity of BH3-only proteins and BH3 mimetics: From theory to potential cancer therapy. Anticancer Agents Med. Chem. 2012, 12, 966–981. [Google Scholar] [CrossRef]
- Manole, E.; Ceafalan, L.C.; Popescu, B.O.; Dumitru, C.; Bastian, A.E. Myokines as Possible Therapeutic Targets in Cancer Cachexia. J. Immunol. Res. 2018, 2018, 8260742. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Selivanov, V.A.; Votyakova, T.V.; Pivtoraiko, V.N.; Zeak, J.; Sukhomlin, T.; Trucco, M.; Roca, J.; Cascante, M. Reactive oxygen species production by forward and reverse electron fluxes in the mitochondrial respiratory chain. PLoS Comput. Biol. 2011, 7, e1001115. [Google Scholar] [CrossRef] [Green Version]
- Egawa, Y.; Saigo, C.; Kito, Y.; Moriki, T.; Takeuchi, T. Therapeutic potential of CPI-613 for targeting tumorous mitochondrial energy metabolism and inhibiting autophagy in clear cell sarcoma. PLoS ONE 2018, 13, e0198940. [Google Scholar] [CrossRef]
- Lycan, T.W.; Pardee, T.S.; Petty, W.J.; Bonomi, M.; Alistar, A.; Lamar, Z.S.; Isom, S.; Chan, M.D.; Miller, A.A.; Ruiz, J. A Phase II Clinical Trial of CPI-613 in Patients with Relapsed or Refractory Small Cell Lung Carcinoma. PLoS ONE 2016, 11, e0164244. [Google Scholar] [CrossRef] [Green Version]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R., Jr.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. New Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Ogasawara, M.A.; Huang, P. Small mitochondria-targeting molecules as anti-cancer agents. Mol. Asp. Med. 2010, 31, 75–92. [Google Scholar] [CrossRef] [Green Version]
- Young, K.E.; Flaherty, S.; Woodman, K.M.; Sharma-Walia, N.; Reynolds, J.M. Fatty acid synthase regulates the pathogenicity of Th17 cells. J. Leukoc. Biol. 2017, 102, 1229–1235. [Google Scholar] [CrossRef]
- Engin, A.B.; Engin, A.; Gonul, I.I. The effect of adipocyte-macrophage crosstalk in obesity-related breast cancer. J. Mol. Endocrinol. 2019, 62, R201–R222. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Rimessi, A.; Bezzerri, V.; Patergnani, S.; Marchi, S.; Cabrini, G.; Pinton, P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 2015, 6, 6201. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Missiroli, S.; Genovese, I.; Perrone, M.; Vezzani, B.; Vitto, V.A.M.; Giorgi, C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. J. Clin. Med. 2020, 9, 740. https://doi.org/10.3390/jcm9030740
Missiroli S, Genovese I, Perrone M, Vezzani B, Vitto VAM, Giorgi C. The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders. Journal of Clinical Medicine. 2020; 9(3):740. https://doi.org/10.3390/jcm9030740
Chicago/Turabian StyleMissiroli, Sonia, Ilaria Genovese, Mariasole Perrone, Bianca Vezzani, Veronica A. M. Vitto, and Carlotta Giorgi. 2020. "The Role of Mitochondria in Inflammation: From Cancer to Neurodegenerative Disorders" Journal of Clinical Medicine 9, no. 3: 740. https://doi.org/10.3390/jcm9030740