Cross-Talk between Lipoproteins and Inflammation: The Role of Microvesicles


1
Cardiovascular Program ICCC, Institut de Recerca Hospital Santa Creu i Sant Pau—IIB Sant Pau, Sant Antoni Maria Claret, 167, 08025 Barcelona, Spain
2
CIBER Enfermedades Cardiovasculares (CIBERCV), Instituto de Salud Carlos III (ISCIII), 28029 Madrid, Spain
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2019, 8(12), 2059; https://doi.org/10.3390/jcm8122059
Submission received: 25 October 2019
/
Revised: 18 November 2019
/
Accepted: 20 November 2019
/
Published: 22 November 2019
(This article belongs to the Special Issue Novel Aspects of Lipoprotein Metabolism with Focus on Systemic Inflammation)
Abstract
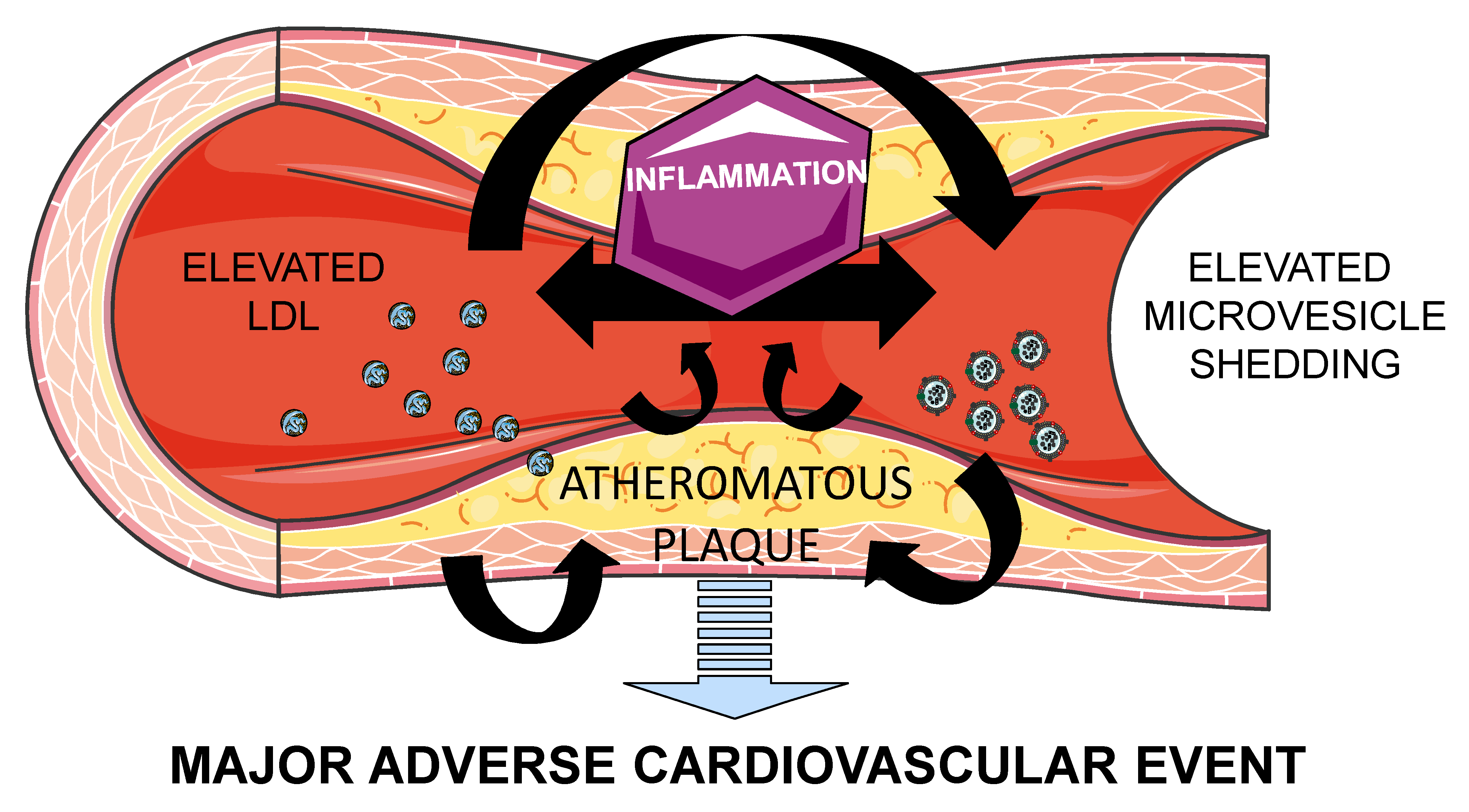
:Atherothrombosis is the principal underlying cause of cardiovascular disease (CVD). Microvesicles (MV) are small blebs originated by an outward budding at the cell plasma membranes, which are released in normal conditions. However, MV release is increased in pathophysiologic conditions such as CVD. Low density lipoprotein (LDL) and MV contribute to atherothrombosis onset and progression by promoting inflammation and leukocyte recruitment to injured endothelium, as well as by increasing thrombosis and plaque vulnerability. Moreover, (oxidized)LDL induces MV release and vice-versa, perpetuating endothelium injury leading to CVD progression. Therefore, MV and lipoproteins exhibit common features, which should be considered in the interpretation of their respective roles in the pathophysiology of CVD. Understanding the pathways implicated in this process will aid in developing novel therapeutic approaches against atherothrombosis.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Atherothrombosis is the principal underlying cause of cardiovascular disease (CVD). Atherosclerosis is caused by lipid accumulation associated with endothelial dysfunction and chronic low-grade inflammation and oxidative stress. Innate immunity cells and vascular smooth muscle cells respond to these perturbations by initiating interactions and gene programs that contribute to vascular dysfunction and atherosclerotic plaque formation. Advanced atherosclerotic plaques, with a lipid-rich atheroma, show inward remodeling encroaching in the arterial lumen, which decrease blood flow, thus leading to tissue ischemia. Eventually, atherosclerotic plaques can rupture and provoke thrombus formation that may occlude the lumen interrupting oxygen supply [1].
High plasma concentrations of low density lipoprotein (LDL) cholesterol induce atherosclerosis, while decreasing LDL cholesterol levels associates with a reduced incidence of major CV events [2]. In fact, the life-long exposure of an artery to LDL cholesterol remains a principal determinant of atherosclerotic progression [3]. Recently, enhanced perivascular adipose tissue mass and local inflammation has been associated with increased atherosclerotic plaque burden [4], and immunity cells and inflammation have a causal role in atherosclerosis progression by modulating the resident cells in the artery wall [3].
Microvesicles (MV) derived from blood and vascular cells seem also being able to participate in the initiation, progression and complications of atherothrombosis by a direct and a paracrine regulation of target cells. Therefore, this review is aimed at summarizing the crosstalk between lipoproteins, inflammation, and microvesicles in the pathophysiology of atherothrombosis.
2. Atherosclerosis: Lipids and Inflammation
Atherosclerosis is considered a lipid-initiated, chronic and progressive inflammatory systemic disease of large and medium arteries and characterized by inflammatory and immune responses contributing to the onset and evolution of the disease and to plaque instability and rupture. It is triggered by the presence of elevated levels of cholesterol in the vessel wall. On the other hand, high density lipoprotein (HDL) particles, when functionally capable, can exert the opposite effect by removing cholesterol from the circulation through the induction of reverse cholesterol transport, by protecting LDL and other proteins from oxidative damage [5], and by inhibiting monocyte production of inflammatory cytokines [6,7] and monocyte differentiation [8]. However, alterations in the HDL structure and function can render dysfunctional HDL particles that may exert deleterious effects in the CV system [9,10]. Inflammation plays a pivotal role in atherosclerotic plaque formation and instability by promoting endothelial cell activation, endothelial dysfunction, loss of integrity and lipid deposition in the intima and by impairing endothelial-repairing capacity [11].
The involvement of inflammation besides LDL in atherosclerosis has been recently proven in the proof-of-concept CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcome Study) trial, showing that interleukin 1β inhibition resulted in a 15% reduction in cardiovascular events [12]. Supporting these findings, it has been recently observed that interleukin 6 trans-signaling increases the risk of cardiovascular events [13]. Interleukin 1β/ interleukin 6 signaling is activated by the nucleotide-binding leucine-rich repeat-containing pyrine receptor 3 (NLRP3) inflammasome platform [14]. However, (minimally modified) LDL activates the inflammasome [15,16,17], in its turn activating the Interleukin 1β/ interleukin 6 signaling pathway, and thus hampering the dissection of the separate roles of lipids and inflammation in atherosclerosis progression.
Atherosclerosis initiates with increases in endothelial permeability to circulating LDL and its accumulation in the intima. Activated endothelial cells then release cytokines, chemokines, and adhesion molecules attracting circulating leukocytes, and more specifically monocytes, into the atherosclerotic lesion, inducing the differentiation of monocytes into proinflammatory macrophages and finally foam cells [18]. Although the primary origin of foam cells in atherosclerotic lesions are leukocytes [19], smooth muscle cells have been shown to differentiate to foam cells [20]. The rupture of the atherosclerotic plaque is the most common trigger of thrombosis, leading to an infarction.
Therefore, atherosclerosis is driven by LDL and inflammation mediated by immune cells, leading to thrombosis and a major CV event. In this process, MV act as a paracrine complementary system ensuing atherothrombosis as will be further discussed.
3. Microvesicles
MV belong to the family of extracellular vesicles. Extracellular vesicles are literally defined by the International Society for Extracellular Vesicles as “the generic term for particles naturally released from the cell that are delimited by a lipid bilayer and cannot replicate, i.e. do not contain a functional nucleus” [21]. MV are medium/large extracellular vesicles (0.1–1 µm) originated by an outward budding at the plasma membrane from almost all cell types [21,22,23,24], and their release is increased when activated, injured or undergoing apoptosis. The biogenesis of MV requires a structured rearrangement of the plasma membrane which provokes physical bending of the membrane and restructuring of the underlying actin cytoskeleton, inducing membrane budding, exposure of phosphatidylserine (PS) from the inner leaflet to the cell surface and formation of membrane blebs. However, MV biogenesis also occurs even if membrane lipid asymmetry is maintained [25]. Given their biogenesis, they contain bioactive molecules from their parental cells, and lipids are essential components of MV. Cholesterol is a structural component of the plasma membrane, which regulates cell functionality and subcellular compartmentalization, and is a precursor or cofactor for several signaling molecules/pathways. In fact, MV are enriched in cholesterol compared to plasma membranes [26,27], and cholesterol mediates their formation and release, ensures membrane stability, and it is necessary for their uptake by target cells. Although MV are known to carry biomolecules such as proteins, RNAs and even DNA, their complete molecular fingerprint is largely unexplored [28], probably because the molecular composition of MV is largely influenced by the stimulus originating them [29,30].
Once released from the cell, MV reach target cells and transfer their molecular cargo, triggering functional responses and phenotypic changes affecting cell functionality, both at the local and systemic environments. The net effect of MV in target cells involves signaling at multiple levels including their interaction with cell surface receptors, and transference of genetic material, metabolites and cytosolic enzymes.
Although the current review is focused in the lipid-mediated role of MV in the pathogenesis of atherothrombosis, it is worth mentioning that there are smaller extracellular vesicles sized ≤150 nm called exosomes, formed by the fusion of intracellular multivesicular bodies with the plasma membrane [23]. Exosomes are rich in biomolecules such as protein, mRNA and non-coding RNA such as miRNA, and have also emerged as both regulators and biomarkers of CVD progression [31]. For further information please refer to reviews [32,33,34,35,36,37].
4. Role of Microvesicles in Atherosclerosis
As previously mentioned, MV are released from all cell types, including platelets, endothelial cells, smooth muscle cells, leukocytes and erythrocytes. Therefore, MV are released in the bloodstream affecting local and distal vulnerable zones.
MV are involved in CVD progression by supporting cellular cross-talk leading to vascular inflammation and dysfunction, leukocyte adhesion and tissue remodeling, thus creating an inflammatory and prothrombotic milieu. In parallel to LDL, MV accumulate and promote the progression of atherosclerotic plaques [38]. Given the externalization of PS during their biogenesis, and, in some cases, tissue factor on their surfaces, MV are potent inducers of coagulation [39], and within the atherosclerotic plaque, retained MV account for the procoagulant activity of the lipid core. MV are the main reservoir of tissue factor activity, thus MV increase plaque vulnerability by promoting coagulation after erosion or rupture of the atherosclerotic plaque. In fact, MV exposing PS in their surface enhance clot propagation, cause apoptosis of nearby cells, and modulate gene expression via a variety of pathways [25], for instance, by transferring monomeric C-reactive protein (CRP) to the cell surface and generating pro-inflammatory signals [40]. Endothelial release of MV is associated to increased cytokine production, and in its turn, increased cytokine production increases MV release. In fact, it has been observed that pharmacological attenuation of inflammation induces a decreased shedding of endothelial-derived MV [41]. Monocyte-derived MV increase endothelial thrombogenicity and apoptosis in vitro by increasing endothelial tissue factor expression, also by the adherence of tissue factor derived from MV to the endothelial cell membrane, by decreasing levels of tissue factor pathway inhibitor (TFPI) and thrombomodulin, and by disrupting endothelial integrity [42]. In addition, MV can transfer cluster of differentiation 40 ligand (CD40L) which binds to monocyte CD40 triggering the activation of tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) downstream signaling, activating in its turn nuclear factor (NF)-kB, thus inducing monocyte release of inflammatory mediators [43]. Besides their prothrombotic activity [44], platelet-derived MV enhance the expression of adhesion molecules and trigger the production of interleukins and TNF-α [45]. In addition, platelet MV recruit activated platelets to the endothelial injury area and can also activate platelets; and platelet-derived MV also contribute to atherogeneis by inducing smooth muscle cell proliferation [46]. Finally, it has been shown that erythrocytes can induce the vulnerability and rupture of plaques in a dose-dependant manner [47]. This effect has been proposed to be indirectly mediated by erythrocyte MV by activating the endothelium, attracting leukocytes and platelets, and enhancing the whole inflammatory pathway [46], although scarce research has been done in this direction.
Therefore, MV contribute to atherothrombosis progression by indirectly elevating the thrombotic risk through the induction of an inflammatory response and directly activating platelets and coagulation. Although up to 200-fold elevated concentrations of MV have been found at the local area of atherosclerotic plaques compared to plasma circulating levels [48], circulating MV from CVD patients [49] and healthy subjects [44] have shown significant prothrombotic activity per se, a role that should not be underestimated in atherothrombotic progression.
Crosstalk between Lipoproteins and MV
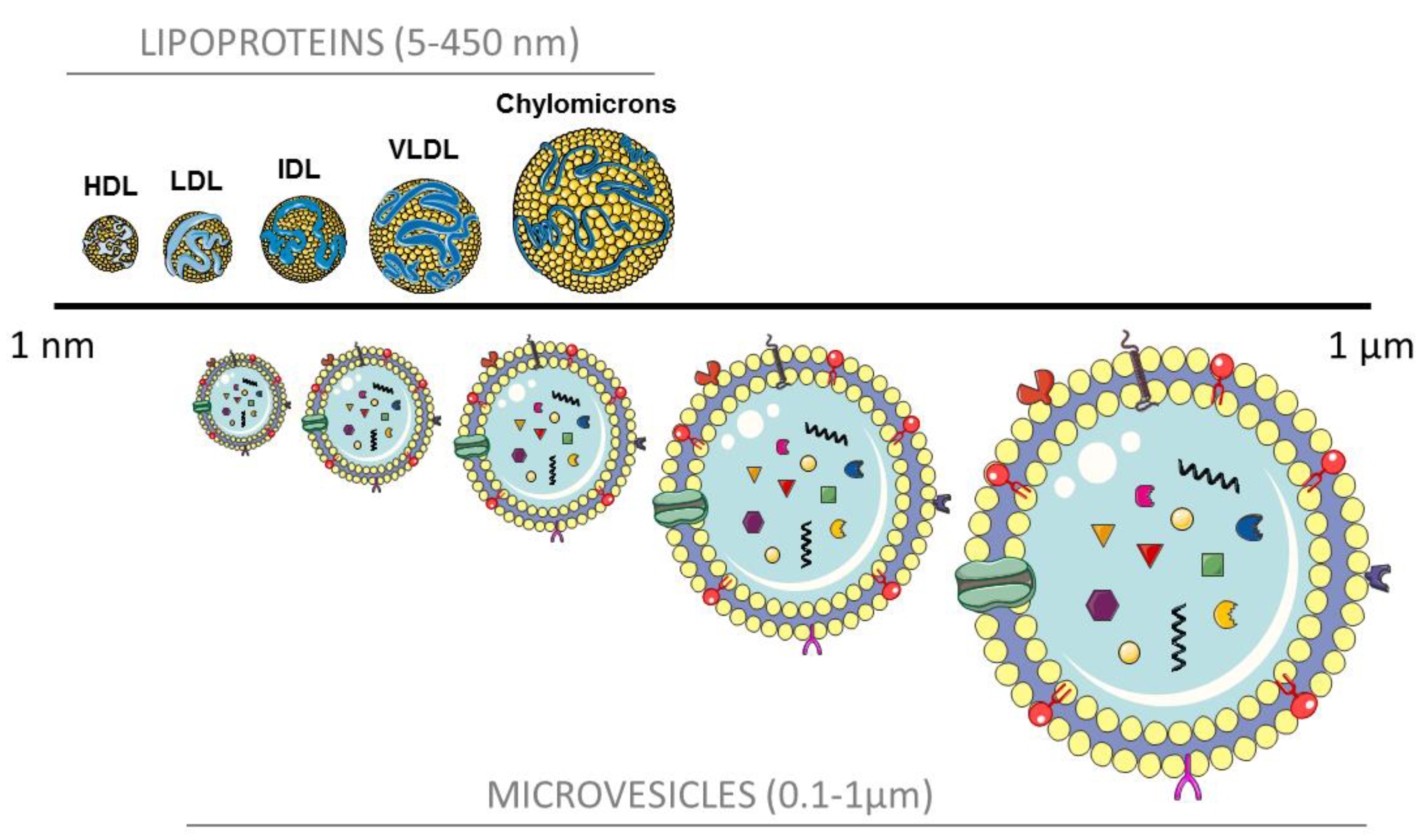
Recent findings pinpoint some similarities between MV and lipoproteins: both MV and HDL and LDL contain and transfer miRNA to target cells [50,51]; MV extracts have been shown to carry ApoE, ApoB, ApoC-II, among others; and the size and density of small- and middle-sized MV closely overlap with lipoproteins, as can be observed in Figure 1. As a matter of fact, some isolation techniques result in co-purification of MV and lipoprotein particles [22,23,52].
Given this size overlap, it is of extreme importance that blood extraction for MV isolation and characterization is being performed in fasting conditions. Depletion of lipoproteins for MV analyses and vice-versa should be carefully considered in biomarker and functional studies. However, the half-life of lipoproteins and MV differs significantly. In physiological conditions, remnant chylomicrons, LDL and HDL, are readily metabolized and stored as intracellular lipid droplets in the liver and adipose tissue. Oppositely, MV rapidly accumulate in resident macrophages of the liver, lungs, and spleen [53]. During atherosclerosis progression, oxidized lipoproteins accumulate in macrophages and other subendothelial cells of the vascular wall provoking an inflammatory response and the progression of the plaque formation. The contribution of circulating (not local) MV in atherothrombosis needs further research.
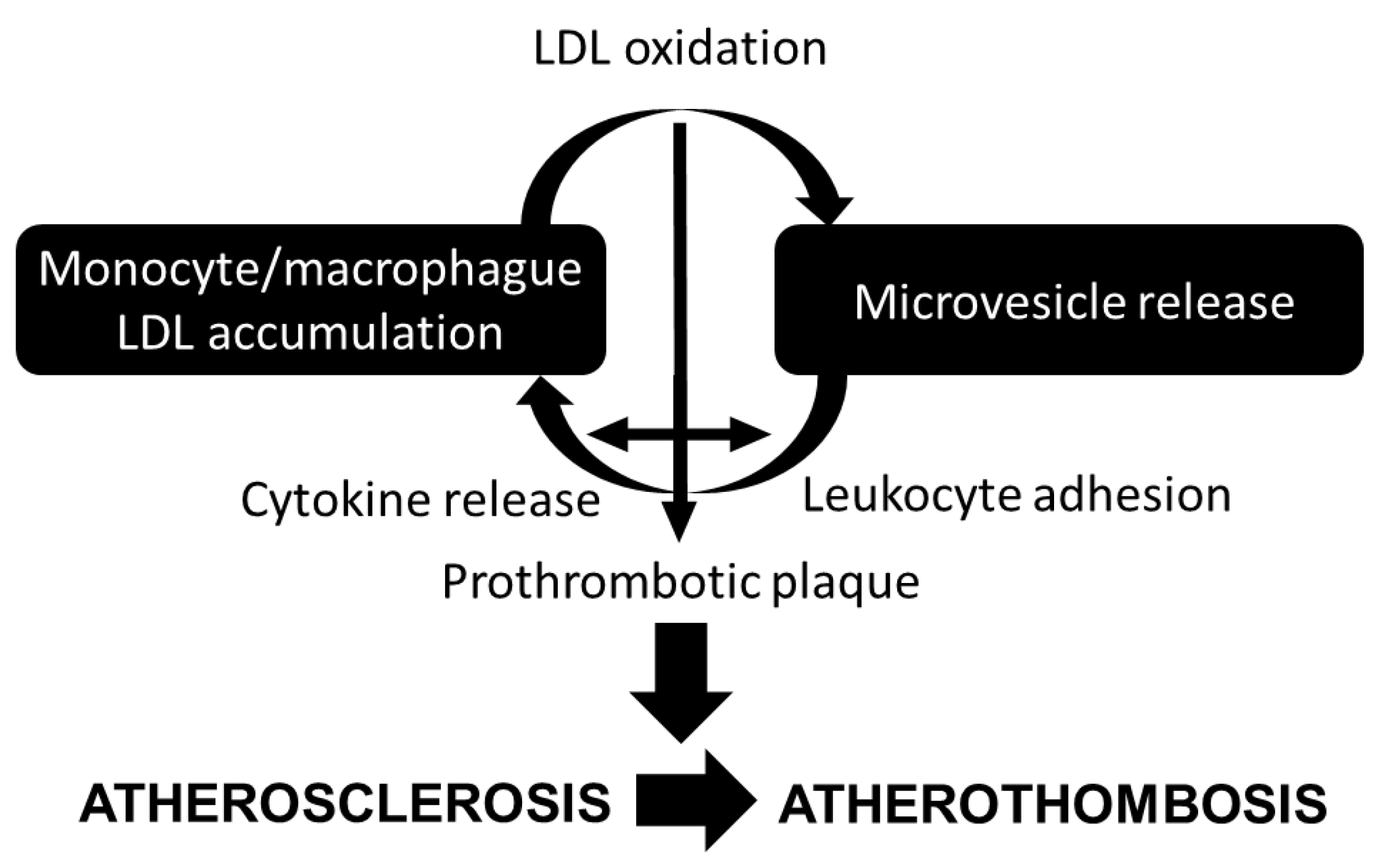
As previously mentioned and summarized in Figure 2, monocyte-derived macrophages accumulate cholesterol modified within the arterial wall during atherogenesis [54]. In its turn, cholesterol accumulation in macrophages and foam cells enhance AV+ and tissue factor+ MV release [55], potentially contributing to the prothrombotic state in hypercholesterolemia [56], and to the prothrombotic core of the lipid-rich vulnerable plaque [57].
It is known that oxidized low-density lipoprotein (oxLDL) activates platelets [58,59,60], thus inducing a prothrombotic state [39]. In addition, oxLDL induces the cellular release of MV [33,39]. In fact, total, AV+ and CD41a+ MV release was shown increased after challenging platelets with oxLDL (but not native LDL) [61], and this effect was comparable to that of ADP. However, Nielsen et al. [62] observed that in vitro incubation of platelets with oxLDL (or native LDL as well) did not significantly stimulate CD41+ and CD41+/CD36+ MV release, suggesting that MV release induced by oxLDL may not be mediated by the interaction with CD36 on platelets.
It has been shown that oxLDL, in a time- and dose-dependent manner, promotes the in vitro release of endothelial MV rich in intercellular adhesion molecule 1 (ICAM-1) [63], which can be transferred from MV to endothelial cells increasing monocyte adhesion to endothelial cells [64], further propagating the atheroprone effects of oxLDL even in its absence. Moreover, enrichment of THP-1 monocytic cells with unsterilized cholesterol resulted in increased MV production [65], and these MV induced extensive leukocyte rolling and adherence to the endothelium. In the presence of oxLDL, high shear stress-induced platelet-derived MV were able to activate THP-1 monocytes and induce them to generate tissue factor-rich MV in vitro [66].
Elevated concentrations of circulating autoantibodies for oxLDL, a surrogate biomarker for LDL oxidation in vivo, have been associated with increased levels of platelet- and monocyte-derived circulating MV in acute coronary syndrome patients [66]. As previously stated, oxLDL induces the cellular release of tissue factor-exposing MV, promoting coagulation and thrombosis, and also disseminating the inflammatory response [39]. Although surface molecules of parental cells are transferred to MV, the interactions between of oxLDL and MV are not entirely elucidated and the consequent effects still remain unknown.
In addition to oxidized LDL, aggregated LDL but not native LDL increases tissue factor-loaded MV from smooth muscle cells [67]. In their turn, oxidized MV have been shown to stimulate monocyte adhesion to endothelial cells through oxidized membrane phospholipids, thus also contributing to atherosclerosis progression [65,68,69].
Given the heterogenic composition of MV, they may have cardioprotective functions as well. As recently reviewed, some MV carry antioxidant enzymes, conferring antioxidant activity at MV under specific stimuli [70]. In its turn, MV have also been shown to both stimulate or inhibit angiogenesis by several mechanisms of action, again depending on the cellular origin and molecular composition derived from the trigger or condition originating their release [71].
5. Role of Microvesicles in Dyslipidemia
The pathophysiological link between postprandial hypertriglyceridemia, inflammation and endothelial injury may be provoked by an excessive retention of lipoproteins in the extracellular matrix and increased uptake by macrophages, thus initiating the atherogenic process. A large body of evidence indicates a direct relationship between postprandial hypertriglyceridemia and CVD risk [72,73]. Postprandial dyslipidemia, independently of the caloric intake or the postprandial state itself, is associated with increased endothelial-derived CD31+/CD42− [74], and total circulating MV levels in healthy subjects [75], and to increased platelet-derived MV in men with different CV risk burden [76], in parallel to increased markers of oxidative stress such as oxLDL and impaired flow-mediated dilation [75]. The effects of postprandial hypertriglyceridemia on MV release in subjects with metabolic dysregulation are quite unexplored and deserve further research, as the reported results are controversial. Type 2 diabetic patients, who show exacerbated postprandial dyslipidemia, show around 3.5 fold increased concentration of endothelial-derived CD144 circulating MV after a meal [77]. However, patients with carotid atherosclerosis show a similar postprandial elevation of circulating platelet-derived MV, despite having higher postprandial hypertriglyceridemia than control subjects free of atherosclerosis [78].
Patients with hypercholesterolemia show higher levels of monocyte- and platelet-derived MV than healthy subjects [79,80,81]. Familial hypercholesterolemia (FH) is an autosomal dominant genetic disorder associated with elevated LDL cholesterol levels and deposition in tendons (xanthomas), and premature heart disease [82]. MV have been associated with atherosclerosis progression and with a higher risk of atherothrombosis in FH patients. In fact, elevated concentrations of total, endothelial cell-derived, erythrocyte-derived, monocyte-derived, tissue factor-loaded MV [83], and platelet-derived MV [84], have been found in FH patients compared to healthy controls [85], and identify subclinical atherosclerosis [80]. In addition, circulating CD36+ MV derived from endothelial cells and monocytes were significantly higher in FH patients compared to healthy controls [85], and monocyte-derived circulating MV in FH patients directly correlated with oxLDL plasma concentrations [79]. Moreover, these patients showed increased concentrations of circulating MV derived from leukocytes, and lymphocyte-derived CD3+/CD45+ circulating MV have been shown elevated in FH patients with lipid-rich atherosclerotic plaques [86]. In addition, platelet- (CD41a+/AV+, CD31+/AV+, CD41a+/CD31+/AV+), granulocyte- (CD66+/AV+), neutrophil- (CD11b+/CD66+/AV+), and endothelial cell-derived (CD62E+/AV+/−) circulating MV discriminate and map coronary atherosclerotic plaque and calcification [84], and leukocyte- and activated platelet-derived circulating MV predict a major cardiovascular event in these patients three years before it takes place [87].
In FH patients, plasma apheresis reduces circulating MV concentration, mainly platelet-derived MV AV+ [88].
Pharmacological Treatment of Dyslipidemia and Circulating MV
Given the crosstalk between lipids and MV, as discussed in a previous section, it appears plausible that lipid lowering therapy may elicit a pleiotropic effect by (partially) inhibiting MV release, contributing to explain their beneficial effects in atherothrombotic complications. We have observed that statins reduce MV shedding from platelets, endothelial cells and leukocytes carrying markers of cell activation [89], and this effect was found to be accumulative throughout years of treatment. Other researchers have also observed that statins decrease MV release from several cell origins [90,91,92], and modify their molecular fingerprint by decreasing their cargo of cell activation markers [93,94]. In stroke patients with hyperlipidemia, simvastatin treatment for 6 months reduced the percentage of CD61+ platelet-derived MV to similar levels to those of age- and sex-matched controls [95].
6. Conclusions and Future Perspectives
The studies summarized in this review evidence that MV are settled in the tandem lipids inflammation, contributing to the progression of atherothrombosis leading to a major CV event. Despite a huge body of research towards the understanding of the exact contribution of MV in this process, several questions remain unanswered, such as the specific interaction of MV with native and modified LDL, the molecular fingerprint of MV, how do they qualitatively (by which pathways) and quantitatively (in what amounts) contribute in the presentation of major CV events, and if they can be considered real candidates as drug delivery vectors in the clinical setting. Given the central role of MV in systemic inflammation and endothelial injury within the pathophysiology of atherothrombosis, their link with lipids and lipoproteins are of particular interest. Understanding the pathways implicated in this process will aid in developing novel therapeutic approaches against early atherosclerosis. In this setting, MV and lipoproteins exhibit common features, which should be considered in the interpretation of their respective roles in the pathophysiology of CVD.
Author Contributions
Conceptualization, G.C.-B. and L.B.; writing—original draft preparation, G.C.-B. and L.B.; writing—review and editing, G.C.-B. and L.B.; funding acquisition, L.B.
Funding
This research was funded by the Spanish Ministry of Economy and Competitiveness of Science, SAF2016-76819-R; the Institute of Health Carlos III (ISCIII), TERCEL—RD16/0011/0018 and CIBERCV CB16/11/00411; FEDER “Una Manera de Hacer Europa”, and by the Agency for Management of University and Research Grants, 2017SGR1480. The APC was funded by SAF2016-76819-R.
Acknowledgments
We thank the Fundació d’Investigació Cardiovascular (FIC)-Fundación Jesús Serra, Barcelona, Spain, for their continuous support. Figures have been created with templates of Servier Medical Art.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-analysis. JAMA 2016, 316, 1289–1297. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. [Google Scholar] [CrossRef]
- Vilahur, G.; Ben-Aicha, S.; Badimon, L. New insights into the role of adipose tissue in thrombosis. Cardiovasc. Res. 2017, 113, 1046–1054. [Google Scholar] [CrossRef]
- Zerrad-Saadi, A.; Therond, P.; Chantepie, S.; Couturier, M.; Rye, K.-A.; Chapman, M.J.; Kontush, A. HDL3-Mediated Inactivation of LDL-Associated Phospholipid Hydroperoxides Is Determined by the Redox Status of Apolipoprotein A-I and HDL Particle Surface Lipid Rigidity. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. Ability of reconstituted high density lipoproteins to inhibit cytokine-induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J. Lipid Res. 1999, 40, 345–353. [Google Scholar] [PubMed]
- Jonas, K.; Kopeć, G. HDL Cholesterol as a Marker of Disease Severity and Prognosis in Patients with Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2019, 20, 3514. [Google Scholar] [CrossRef] [PubMed]
- Grün, J.L.; Manjarrez-Reyna, A.N.; Gómez-Arauz, A.Y.; Leon-Cabrera, S.; Rückert, F.; Fragoso, J.M.; Bueno-Hernández, N.; Islas-Andrade, S.; Meléndez-Mier, G.; Escobedo, G. High-Density Lipoprotein Reduction Differentially Modulates to Classical and Nonclassical Monocyte Subpopulations in Metabolic Syndrome Patients and in LPS-Stimulated Primary Human Monocytes In Vitro. J. Immunol. Res. 2018, 2018, 2737040. [Google Scholar] [CrossRef]
- Padró, T.; Cubedo, J.; Camino, S.; Béjar, M.T.; Ben-Aicha, S.; Mendieta, G.; Escolà-Gil, J.C.; Escate, R.; Gutiérrez, M.; Casani, L.; et al. Detrimental Effect of Hypercholesterolemia on High-Density Lipoprotein Particle Remodeling in Pigs. J. Am. Coll. Cardiol. 2017, 70, 165–178. [Google Scholar] [CrossRef]
- Ben-Aicha, S.; Escate, R.; Casaní, L.; Padró, T.; Peña, E.; Arderiu, G.; Mendieta, G.; Badimón, L.; Vilahur, G. High-density lipoprotein remodelled in hypercholesterolaemic blood induce epigenetically driven down-regulation of endothelial HIF-1α expression in a preclinical animal model. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, L.; Frumento, P.; Wallén, H.; de Faire, U.; Gigante, B. The predictive role of interleukin 6 trans-signalling in middle-aged men and women at low-intermediate risk of cardiovascular events. Eur. J. Prev. Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Von Moltke, J.; Trinidad, N.J.; Moayeri, M.; Kintzer, A.F.; Wang, S.B.; Van Rooijen, N.; Brown, C.R.; Krantz, B.A.; Leppla, S.H.; Gronert, K.; et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 2012, 490, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Jukema, R.A.; Ahmed, T.A.N.; Tardif, J.-C. Does low-density lipoprotein cholesterol induce inflammation? If so, does it matter? Current insights and future perspectives for novel therapies. BMC Med. 2019, 17, 197. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nũez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Varghese, J.F.; Patel, R.; Yadav, U.C.S. Sterol regulatory element binding protein (SREBP) -1 mediates oxidized low-density lipoprotein (oxLDL) induced macrophage foam cell formation through NLRP3 inflammasome activation. Cell. Signal. 2019, 53, 316–326. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Obama, T.; Ohinata, H.; Takaki, T.; Iwamoto, S.; Sawada, N.; Aiuchi, T.; Kato, R.; Itabe, H. Cooperative Action of Oxidized Low-Density Lipoproteins and Neutrophils on Endothelial Inflammatory Responses Through Neutrophil Extracellular Trap Formation. Front. Immunol. 2019, 10, 1899. [Google Scholar] [CrossRef]
- Llorente-Cortés, V.; Royo, T.; Juan-Babot, O.; Badimon, L. Adipocyte differentiation-related protein is induced by LRP1-mediated aggregated LDL internalization in human vascular smooth muscle cells and macrophages. J. Lipid Res. 2007, 48, 2133–2140. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Suades, R.; Arderiu, G.; Peña, E.; Chiva-Blanch, G.; Padró, T. Microvesicles in Atherosclerosis and Angiogenesis: From Bench to Bedside and Reverse. Front. Cardiovasc. Med. 2017, 4, 77. [Google Scholar] [CrossRef]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; López, J.A. Tissue-factor–bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Vitale, N. Cholesterol and the journey of extracellular vesicles. J. Lipid Res. 2018, 59, 2255–2261. [Google Scholar] [CrossRef]
- BIRO, E.; Akkerman, J.W.N.; Hoek, F.J.; Gorter, G.; Pronk, L.M.; Sturk, A.; Nieuwland, R. The phospholipid composition and cholesterol content of platelet-derived microparticles: A comparison with platelet membrane fractions. J. Thromb. Haemost. 2005, 3, 2754–2763. [Google Scholar] [CrossRef]
- Gudbergsson, J.M.; Jønsson, K.; Simonsen, J.B.; Johnsen, K.B. Systematic review of targeted extracellular vesicles for drug delivery—Considerations on methodological and biological heterogeneity. J. Control. Release 2019, 306, 108–120. [Google Scholar] [CrossRef]
- Williams, C.; Palviainen, M.; Reichardt, N.-C.; Siljander, P.R.-M.; Falcón-Pérez, J.M. Metabolomics Applied to the Study of Extracellular Vesicles. Metabolites 2019, 9, 276. [Google Scholar] [CrossRef]
- Turchinovich, A.; Drapkina, O.; Tonevitsky, A. Transcriptome of extracellular vesicles: State-of-the-art. Front. Immunol. 2019, 10, 202. [Google Scholar] [CrossRef]
- De Giusti, C.J.; Santalla, M.; Das, S. Exosomal non-coding RNAs (Exo-ncRNAs) in cardiovascular health. J. Mol. Cell. Cardiol. 2019, 137, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wang, M. New insights into the immunomodulatory role of exosomes in cardiovascular disease. Rev. Cardiovasc. Med. 2019, 20, 153–160. [Google Scholar] [PubMed]
- Yang, W.; Zou, B.; Hou, Y.; Yan, W.; Chen, T.; Qu, S. Extracellular vesicles in vascular calcification. Clin. Chim. Acta 2019, 499, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Kita, S.; Maeda, N.; Shimomura, I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J. Clin. Investig. 2019, 129, 4041–4049. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wang, Z. Cardiomyocyte-Derived Exosomes: Biological Functions and Potential Therapeutic Implications. Front. Physiol. 2019, 10, 1049. [Google Scholar] [CrossRef]
- Jafarzadeh-Esfehani, R.; Soudyab, M.; Parizadeh, S.M.; Jaripoor, M.E.; Nejad, P.S.; Shariati, M.; Nabavi, A.S. Circulating exosomes and their role in stroke. Curr. Drug Targets 2019, 20. [Google Scholar] [CrossRef]
- Aghabozorgi, A.S.; Ahangari, N.; Eftekhaari, T.E.; Torbati, P.N.; Bahiraee, A.; Ebrahimi, R.; Pasdar, A. Circulating exosomal miRNAs in cardiovascular disease pathogenesis: New emerging hopes. J. Cell. Physiol. 2019, 234, 21796–21809. [Google Scholar] [CrossRef]
- Owens, A.P.; Mackman, N. Microparticles in Hemostasis and Thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef]
- Obermayer, G.; Afonyushkin, T.; Binder, C.J. Oxidized low-density lipoprotein in inflammation-driven thrombosis. J. Thromb. Haemost. 2018, 16, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Habersberger, J.; Strang, F.; Scheichl, A.; Htun, N.; Bassler, N.; Merivirta, R.-M.; Diehl, P.; Krippner, G.; Meikle, P.; Eisenhardt, S.U.; et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc. Res. 2012, 96, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Pirro, M.; Bianconi, V.; Paciullo, F.; Mannarino, M.R.; Bagaglia, F.; Sahebkar, A. Lipoprotein(a) and inflammation: A dangerous duet leading to endothelial loss of integrity. Pharmacol. Res. 2017, 119, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Tsimerman, G.; Roguin, A.; Bachar, A.; Melamed, E.; Brenner, B.; Aharon, A. Involvement of microparticles in diabetic vascular complications. Thromb. Haemost. 2011, 106, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Bei, J.-J.; Liu, C.; Peng, S.; Liu, C.-H.; Zhao, W.-B.; Qu, X.-L.; Chen, Q.; Zhou, Z.; Yu, Z.-P.; Peter, K.; et al. Staphylococcal SSL5-induced platelet microparticles provoke proinflammatory responses via the CD40/TRAF6/NFKB signalling pathway in monocytes. Thromb. Haemost. 2016, 115, 632–645. [Google Scholar] [CrossRef] [PubMed]
- Suades, R.; Padró, T.; Vilahur, G.; Badimon, L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb. Haemost. 2012, 108, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Hugel, B.; Martínez, M.C.; Kunzelmann, C.; Freyssinet, J.-M. Membrane Microparticles: Two Sides of the Coin. Physiology 2005, 20, 22–27. [Google Scholar] [CrossRef]
- Blum, A. The possible role of red blood cell microvesicles in atherosclerosis. Eur. J. Intern. Med. 2009, 20, 101–105. [Google Scholar] [CrossRef]
- Lin, H.-L.; Xu, X.-S.; Lu, H.-X.; Zhang, L.; Li, C.-J.; Tang, M.-X.; Sun, H.-W.; Liu, Y.; Zhang, Y. Pathological mechanisms and dose dependency of erythrocyte-induced vulnerability of atherosclerotic plaques. J. Mol. Cell. Cardiol. 2007, 43, 272–280. [Google Scholar] [CrossRef]
- Leroyer, A.S.; Isobe, H.; Lesèche, G.; Castier, Y.; Wassef, M.; Mallat, Z.; Binder, B.R.; Tedgui, A.; Boulanger, C.M. Cellular Origins and Thrombogenic Activity of Microparticles Isolated from Human Atherosclerotic Plaques. J. Am. Coll. Cardiol. 2007, 49, 772–777. [Google Scholar] [CrossRef] [Green Version]
- Mallat, Z.; Benamer, H.; Hugel, B.; Benessiano, J.; Steg, P.G.; Freyssinet, J.M.; Tedgui, A. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation 2000, 101, 841–843. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Zhang, D.; Zhu, Z.; Dela Cruz, C.S.; Jin, Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci. Rep. 2016, 6, 35250. [Google Scholar] [CrossRef] [Green Version]
- Shu, Z.; Tan, J.; Miao, Y.; Zhang, Q. The role of microvesicles containing microRNAs in vascular endothelial dysfunction. J. Cell. Mol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Mahmood Hussain, M. A proposed model for the assembly of chylomicrons. Atherosclerosis 2000, 148, 1–15. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skålén, K.; Gustafsson, M.; Rydberg, E.K.; Hultén, L.M.; Wiklund, O.; Innerarity, T.L.; Borén, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef]
- Liu, M.-L.; Reilly, M.P.; Casasanto, P.; McKenzie, S.E.; Williams, K.J. Cholesterol enrichment of human monocyte/macrophages induces surface exposure of phosphatidylserine and the release of biologically-active tissue factor-positive microvesicles. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 430–435. [Google Scholar] [CrossRef]
- Cipollone, F.; Mezzetti, A.; Porreca, E.; Di Febbo, C.; Nutini, M.; Fazia, M.; Falco, A.; Cuccurullo, F.; Davì, G. Association Between Enhanced Soluble CD40L and Prothrombotic State in Hypercholesterolemia. Circulation 2002, 106, 399–402. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.J.; Tabas, I. Lipoprotein retention—And clues for atheroma regression. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1536–1540. [Google Scholar] [CrossRef] [Green Version]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Febbraio, M.; Li, W.; Silverstein, R.L. A Specific CD36-Dependent Signaling Pathway Is Required for Platelet Activation by Oxidized Low-Density Lipoprotein. Circ. Res. 2008, 102, 1512–1519. [Google Scholar] [CrossRef] [Green Version]
- Wraith, K.S.; Magwenzi, S.; Aburima, A.; Wen, Y.; Leake, D.; Naseem, K.M. Oxidized low-density lipoproteins induce rapid platelet activation and shape change through tyrosine kinase and Rho kinase-signaling pathways. Blood 2013, 122, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, Z.-H.; Kong, J.; Yang, M.-Y.; Jiang, G.-H.; Wang, X.-P.; Zhong, M.; Zhang, Y.; Deng, J.-T.; Zhang, W. Oxidized Low-Density Lipoprotein-Dependent Platelet-Derived Microvesicles Trigger Procoagulant Effects and Amplify Oxidative Stress. Mol. Med. 2012, 18, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.B.; Nielsen, M.H.; Handberg, A. In vitro Incubation of Platelets with oxLDL Does Not Induce Microvesicle Release When Measured by Sensitive Flow Cytometry. Front. Cardiovasc. Med. 2015, 2, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Zhou, E.; Wang, X.; Tian, M.; Kong, J.; Li, J.; Ji, L.; Niu, C.; Shen, H.; Dong, S.; et al. Oxidized low-density lipoprotein-induced microparticles promote endothelial monocyte adhesion via intercellular adhesion molecule 1. Am. J. Physiol. Physiol. 2017, 313, C567–C574. [Google Scholar] [CrossRef] [PubMed]
- Rautou, P.-E.; Leroyer, A.S.; Ramkhelawon, B.; Devue, C.; Duflaut, D.; Vion, A.-C.; Nalbone, G.; Castier, Y.; Leseche, G.; Lehoux, S.; et al. Microparticles From Human Atherosclerotic Plaques Promote Endothelial ICAM-1–Dependent Monocyte Adhesion and Transendothelial Migration. Circ. Res. 2011, 108, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.L.; Scalia, R.; Mehta, J.L.; Williams, K.J. Cholesterol-induced membrane microvesicles as novel carriers of damage-associated molecular patterns: Mechanisms of formation, action, and detoxification. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2113–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, N.; Nomura, S.; Kamihata, H.; Kimura, Y.; Iwasaka, T. Increased level of oxidized LDL-dependent monocytederived microparticles in acute coronary syndrome. Thromb. Haemost. 2004, 91, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Llorente-Cortés, V.; Otero-Viñas, M.; Camino-López, S.; Llampayas, O.; Badimon, L. Aggregated low-density lipoprotein uptake induces membrane tissue factor procoagulant activity and microparticle release in human vascular smooth muscle cells. Circulation 2004, 110, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Huber, J.; Vales, A.; Mitulovic, G.; Blumer, M.; Schmid, R.; Witztum, J.L.; Binder, B.R.; Leitinger, N. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Papac-Milicevic, N.; Busch, C.J.L.; Binder, C.J. Malondialdehyde Epitopes as Targets of Immunity and the Implications for Atherosclerosis. In Advances in Immunology; Academic Press Inc.: Cambridge, MA, USA, 2016; Volume 131, pp. 1–59. [Google Scholar]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef] [Green Version]
- Todorova, D.; Simoncini, S.; Lacroix, R.; Sabatier, F.; Dignat-George, F. Extracellular vesicles in angiogenesis. Circ. Res. 2017, 120, 1658–1673. [Google Scholar] [CrossRef]
- Karpe, F. Postprandial lipoprotein metabolism and atherosclerosis. J. Intern. Med. 1999, 246, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nappo, F.; Esposito, K.; Cioffi, M.; Giugliano, G.; Molinari, A.M.; Paolisso, G.; Marfella, R.; Giugliano, D. Postprandial endothelial activation in healthy subjects and in type 2 diabetic patients: Role of fat and carbohydrate meals. J. Am. Coll. Cardiol. 2002, 39, 1145–1150. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.C.; Peter, A.A.; Mendez, A.J.; Jimenez, J.J.; Mauro, L.M.; Chirinos, J.A.; Ghany, R.; Virani, S.; Garcia, S.; Horstman, L.L.; et al. Postprandial Hypertriglyceridemia Increases Circulating Levels of Endothelial Cell Microparticles. Circulation 2004, 110, 3599–3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tushuizen, M.E.; Nieuwland, R.; Scheffer, P.G.; Sturk, A.; Heine, R.J.; Diamant, M. Two consecutive high-fat meals affect endothelial-dependent vasodilation, oxidative stress and cellular microparticles in healthy men. J. Thromb. Haemost. 2006, 4, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Tamburrelli, C.; Gianfagna, F.; D’imperio, M.; De Curtis, A.; Rotilio, D.; Iacoviello, L.; De Gaetano, G.; Donati, M.B.; Cerletti, C. Postprandial cell inflammatory response to a standardised fatty meal in subjects at different degree of cardiovascular risk. Thromb. Haemost. 2012, 107, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tushuizen, M.E.; Nieuwland, R.; Rustemeijer, C.; Hensgens, B.E.; Sturk, A.; Heine, R.J.; Diamant, M. Elevated endothelial microparticles following consecutive meals are associated with vascular endothelial dysfunction in type 2 diabetes. Diabetes Care 2007, 30, 728–730. [Google Scholar] [CrossRef] [Green Version]
- Elevated Levels of Platelet Microparticles in Carotid Atherosclerosis and during the Postprandial State- ClinicalKey. Available online: https://www.clinicalkey.es/#!/content/playContent/1-s2.0-S0049384808005082?scrollTo=%23hl0000494 (accessed on 14 November 2019).
- Hjuler Nielsen, M.; Irvine, H.; Vedel, S.; Raungaard, B.; Beck-Nielsen, H.; Handberg, A. Elevated Atherosclerosis-Related Gene Expression, Monocyte Activation and Microparticle-Release Are Related to Increased Lipoprotein-Associated Oxidative Stress in Familial Hypercholesterolemia. PLoS ONE 2015, 10, e0121516. [Google Scholar] [CrossRef]
- Suades, R.; Padró, T.; Alonso, R.; Mata, P.; Badimon, L. High levels of TSP1+/CD142+ platelet-derived microparticles characterise young patients with high cardiovascular risk and subclinical atherosclerosis. Thromb. Haemost. 2015, 114, 1310–1321. [Google Scholar] [CrossRef]
- Pirro, M.; Schillaci, G.; Paltriccia, R.; Bagaglia, F.; Menecali, C.; Mannarino, M.R.; Capanni, M.; Velardi, A.; Mannarino, E. Increased ratio of CD31+/CD42− microparticles to endothelial progenitors as a novel marker of atherosclerosis in hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2530–2535. [Google Scholar] [CrossRef] [Green Version]
- Badimon, L.; Chiva-Blanch, G. Lipid Metabolism in Dyslipidemia and Familial Hypercholesterolemia. Mol. Nutr. Fats 2019, 307–322. [Google Scholar]
- Owens, A.P.; Passam, F.H.; Antoniak, S.; Marshall, S.M.; McDaniel, A.L.; Rudel, L.; Williams, J.C.; Hubbard, B.K.; Dutton, J.-A.; Wang, J.; et al. Monocyte tissue factor-dependent activation of coagulation in hypercholesterolemic mice and monkeys is inhibited by simvastatin. J. Clin. Investig. 2012, 122, 558–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiva-Blanch, G.; Padró, T.; Alonso, R.; Crespo, J.; Perez de Isla, L.; Mata, P.; Badimon, L. Liquid biopsy of extracellular microvesicles maps coronary calcification and atherosclerotic plaque in asymptomatic patients with familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 945–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.H.; Irvine, H.; Vedel, S.; Raungaard, B.; Beck-Nielsen, H.; Handberg, A. The Impact of Lipoprotein-Associated Oxidative Stress on Cell-Specific Microvesicle Release in Patients with Familial Hypercholesterolemia. Oxid. Med. Cell. Longev. 2016, 2016, 2492858. [Google Scholar] [CrossRef] [PubMed]
- Suades, R.; Padró, T.; Alonso, R.; López-Miranda, J.; Mata, P.; Badimon, L. Circulating CD45+/CD3+ lymphocyte-derived microparticles map lipid-rich atherosclerotic plaques in familial hypercholesterolaemia patients. Thromb. Haemost. 2013, 111, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Suades, R.; Padró, T.; Crespo, J.; Sionis, A.; Alonso, R.; Mata, P.; Badimon, L. Liquid Biopsy of Extracellular Microvesicles Predicts Future Major Ischemic Events in Genetically Characterized Familial Hypercholesterolemia Patients. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1172–1181. [Google Scholar] [CrossRef] [Green Version]
- Connolly, K.D.; Willis, G.R.; Datta, D.B.N.; Ellins, E.A.; Ladell, K.; Price, D.A.; Guschina, I.A.; Rees, D.A.; James, P.E. Lipoprotein-apheresis reduces circulating microparticles in individuals with familial hypercholesterolemia. J. Lipid Res. 2014, 55, 2064–2072. [Google Scholar] [CrossRef] [Green Version]
- Suades, R.; Padró, T.; Alonso, R.; Mata, P.; Badimon, L. Lipid-lowering therapy with statins reduces microparticle shedding from endothelium, platelets and inflammatory cells. Thromb. Haemost. 2013, 110, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Nomura, S.; Inami, N.; Shouzu, A.; Omoto, S.; Kimura, Y.; Takahashi, N.; Tanaka, A.; Urase, F.; Maeda, Y.; Ohtani, H.; et al. The effects of pitavastatin, eicosapentaenoic acid and combined therapy on platelet-derived microparticles and adiponectin in hyperlipidemic, diabetic patients. Platelets 2009, 20, 16–22. [Google Scholar] [CrossRef]
- Nomura, S.; Shouzu, A.; Omoto, S.; Inami, N.; Ueba, T.; Urase, F.; Maeda, Y. Effects of eicosapentaenoic acid on endothelial cell-derived microparticles, angiopoietins and adiponectin in patients with type 2 diabetes. J. Atheroscler. Thromb. 2009, 16, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Tramontano, A.F.; O’Leary, J.; Black, A.D.; Muniyappa, R.; Cutaia, M.V.; El-Sherif, N. Statin decreases endothelial microparticle release from human coronary artery endothelial cells: Implication for the Rho-kinase pathway. Biochem. Biophys. Res. Commun. 2004, 320, 34–38. [Google Scholar] [CrossRef]
- Sommeijer, D.W.; Joop, K.; Leyte, A.; Reitsma, P.H.; Cate, H.T. Pravastatin reduces fibrinogen receptor gpIIIa on platelet-derived microparticles in patients with type 2 diabetes. J. Thromb. Haemost. 2005, 3, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Mobarrez, F.; He, S.; Bröijersen, A.; Wiklund, B.; Antovic, A.; Antovic, J.; Egberg, N.; Jörneskog, G.; Wallén, H. Atorvastatin reduces thrombin generation and expression of tissue factor, P-selectin and GPIIIa on platelet-derived microparticles in patients with peripheral arterial occlusive disease. Thromb. Haemost. 2011, 106, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Pawelczyk, M.; Chmielewski, H.; Kaczorowska, B.; Przybyła, M.; Baj, Z. The influence of statin therapy on platelet activity markers in hyperlipidemic patients after ischemic stroke. Arch. Med. Sci. 2015, 1, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo, L.M.; França, C.N.; Izar, M.C.; Bianco, H.T.; Lins, L.S.; Barbosa, S.P.; Pinheiro, L.F.; Fonseca, F.A.H. Effects of simvastatin/ezetimibe on microparticles, endothelial progenitor cells and platelet aggregation in subjects with coronary heart disease under antiplatelet therapy. Braz. J. Med. Biol. Res. 2014, 47, 432–437. [Google Scholar] [CrossRef]
- Lins, L.C.A.; França, C.N.; Fonseca, F.A.H.; Barbosa, S.P.M.; Matos, L.N.; Aguirre, A.C.; Bianco, H.T.; do Amaral, J.B.; Izar, M.C. Effects of Ezetimibe on Endothelial Progenitor Cells and Microparticles in High-Risk Patients. Cell Biochem. Biophys. 2014, 70, 687–696. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2019. [Google Scholar] [CrossRef]
Figure 1.
Particle size of lipoproteins and microvesicles. HDL, high density lipoprotein; LDL, low density lipoprotein; IDL, intermediate density lipoprotein; VLDL, very low density lipoprotein; and MV, microvesicles.
Figure 1.
Particle size of lipoproteins and microvesicles. HDL, high density lipoprotein; LDL, low density lipoprotein; IDL, intermediate density lipoprotein; VLDL, very low density lipoprotein; and MV, microvesicles.

Figure 2.
Lipid and microvesicle crosstalk contributing to atherosclerosis progression.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chiva-Blanch, G.; Badimon, L. Cross-Talk between Lipoproteins and Inflammation: The Role of Microvesicles. J. Clin. Med. 2019, 8, 2059. https://doi.org/10.3390/jcm8122059
AMA Style
Chiva-Blanch G, Badimon L. Cross-Talk between Lipoproteins and Inflammation: The Role of Microvesicles. Journal of Clinical Medicine. 2019; 8(12):2059. https://doi.org/10.3390/jcm8122059
Chicago/Turabian StyleChiva-Blanch, Gemma, and Lina Badimon. 2019. "Cross-Talk between Lipoproteins and Inflammation: The Role of Microvesicles" Journal of Clinical Medicine 8, no. 12: 2059. https://doi.org/10.3390/jcm8122059
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.