Autophagy and Inflammasome Activation in Dilated Cardiomyopathy

 , , , , and
, , , , and
Abstract
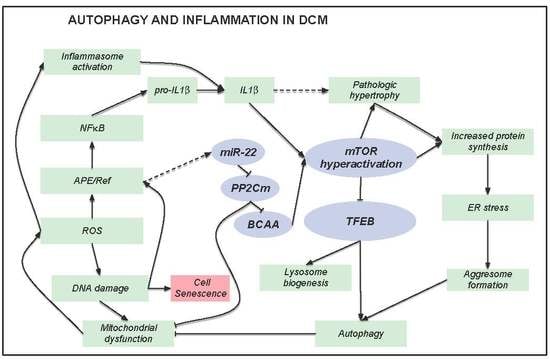
:
1. Introduction
2. Experimental Section
2.1. Patient Enrollment and Ethics
2.2. Tissue Sampling
2.3. Histology, Histochemistry, Immunohistochemistry, and Immunofluorescence Assays
2.4. Western Blotting
2.5. NMR Analysis
2.6. Cardiac Pericyte/Mural Cell Culture and in Vitro Characterization
2.7. Oxygen Consumption Rate (OCR)
2.8. RNA Extraction and Real-Time PCR Analysis of miRNA Expression
2.9. MiRNA Inhibition
2.10. Statistical Analysis
3. Results
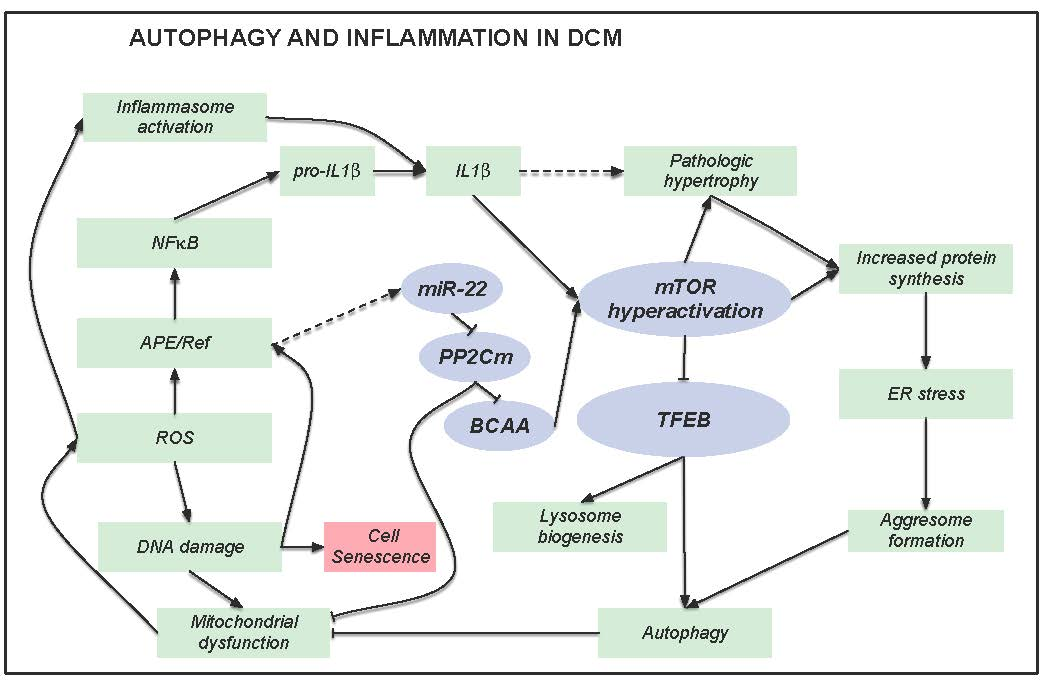
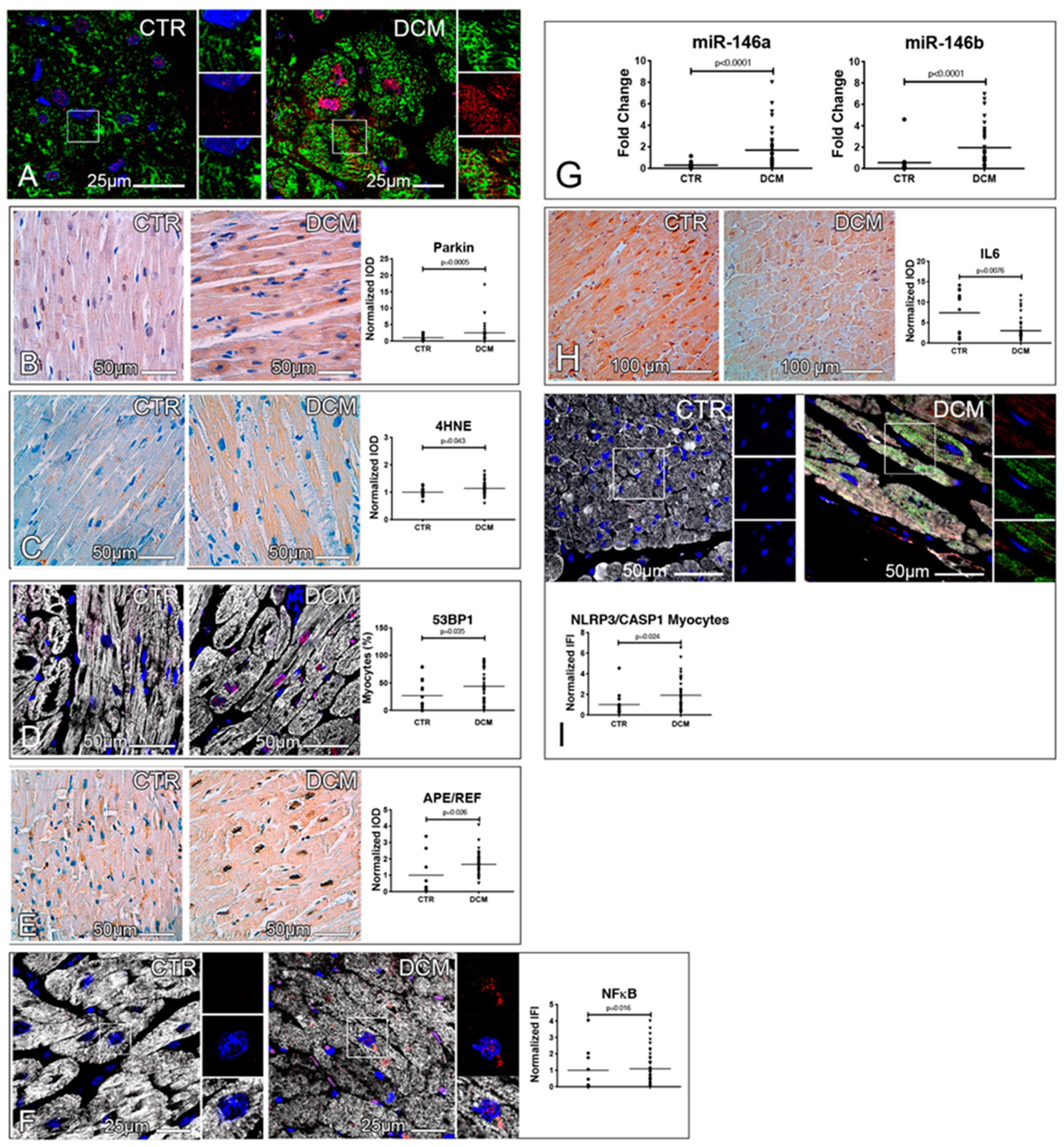
3.1. Failing Hearts of DCM Patients Show Accumulation of Misfolded and Ubiquitinated Proteins
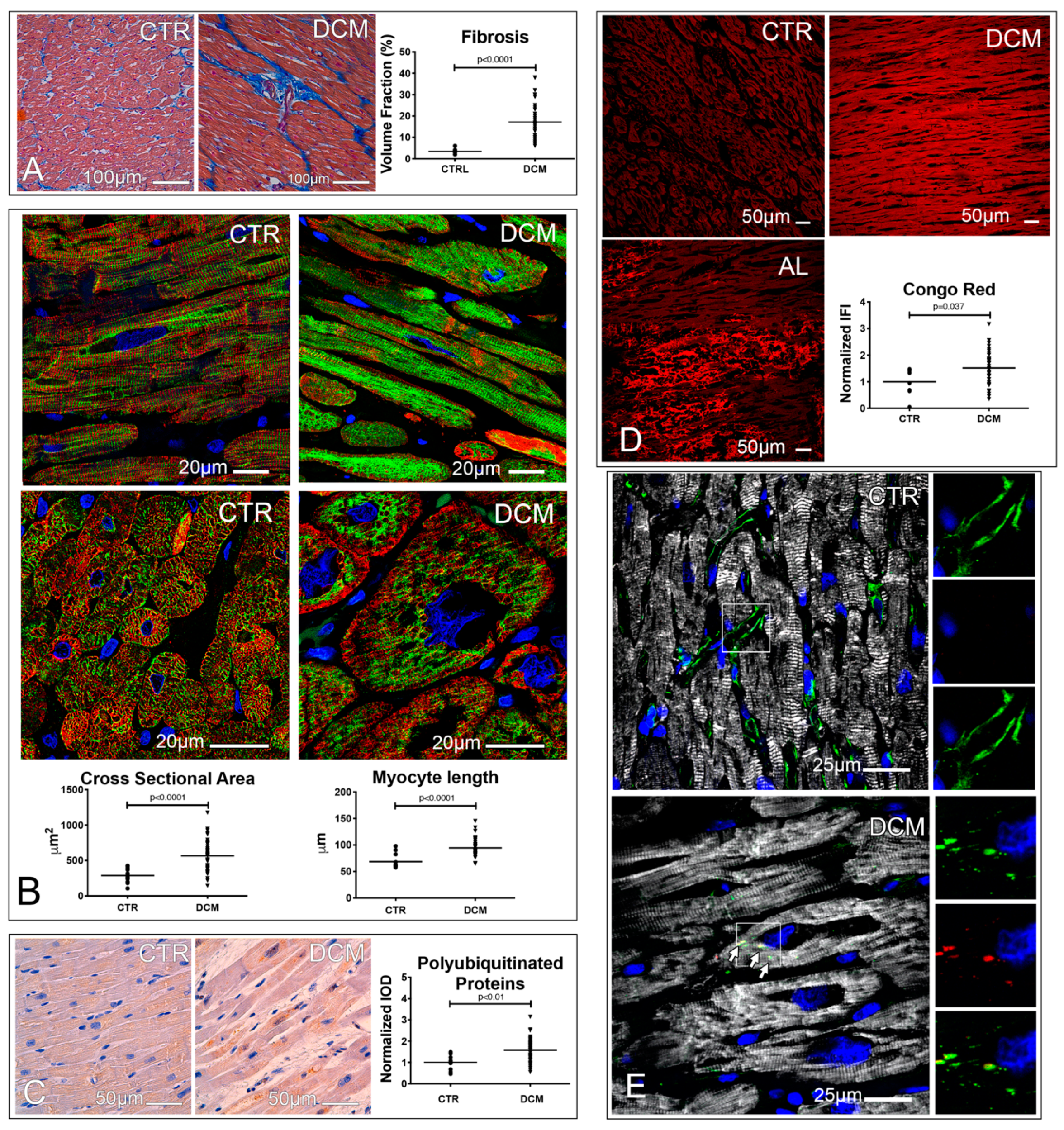
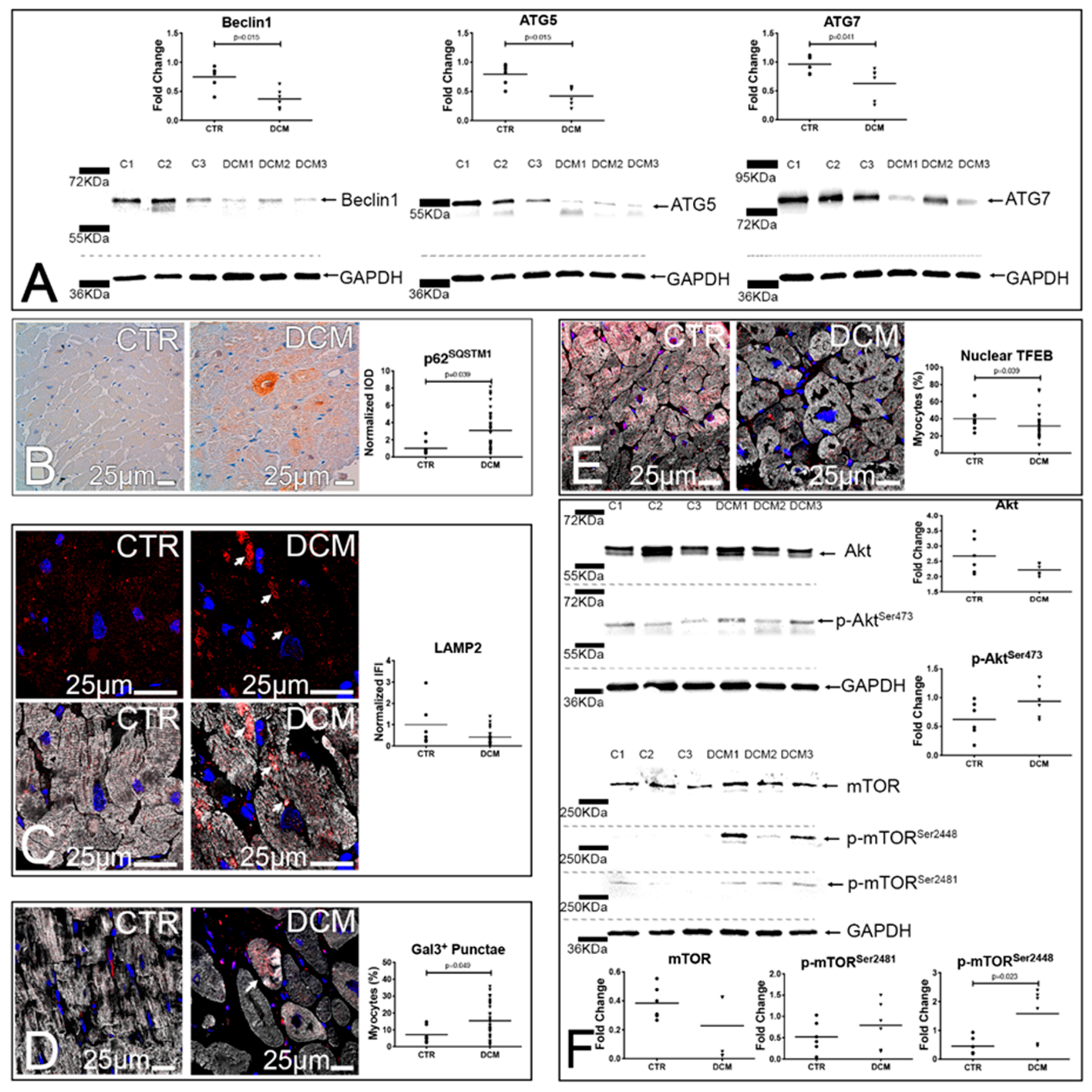
3.2. A Defective Autophagy Lysosomal Pathway Characterizes Failing DCM Hearts
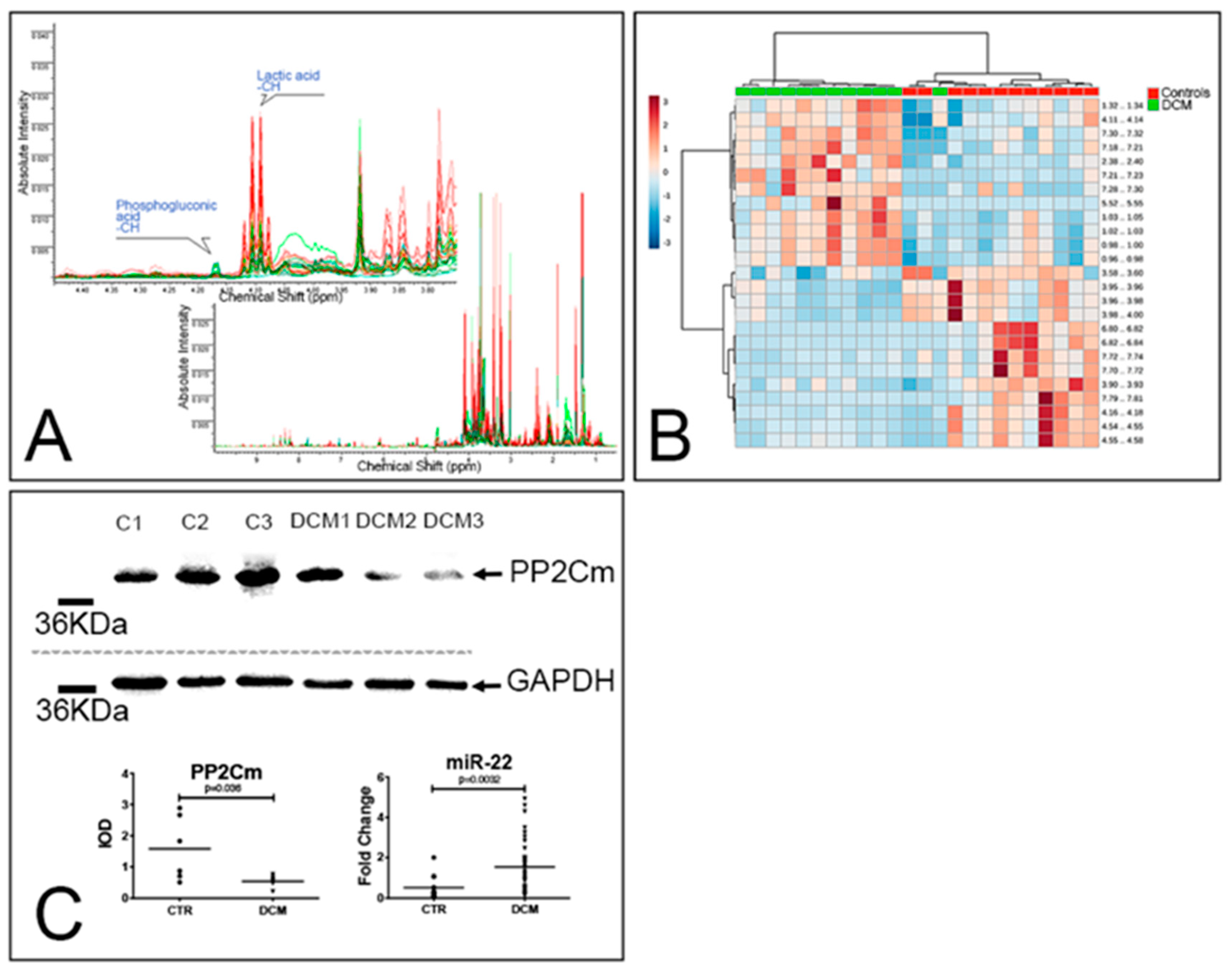
3.3. Accumulation of Dysfunctional Mitochondria, Oxidative Stress and Inflammasome Activation
3.4. Metabolic Alterations in DCM Hearts
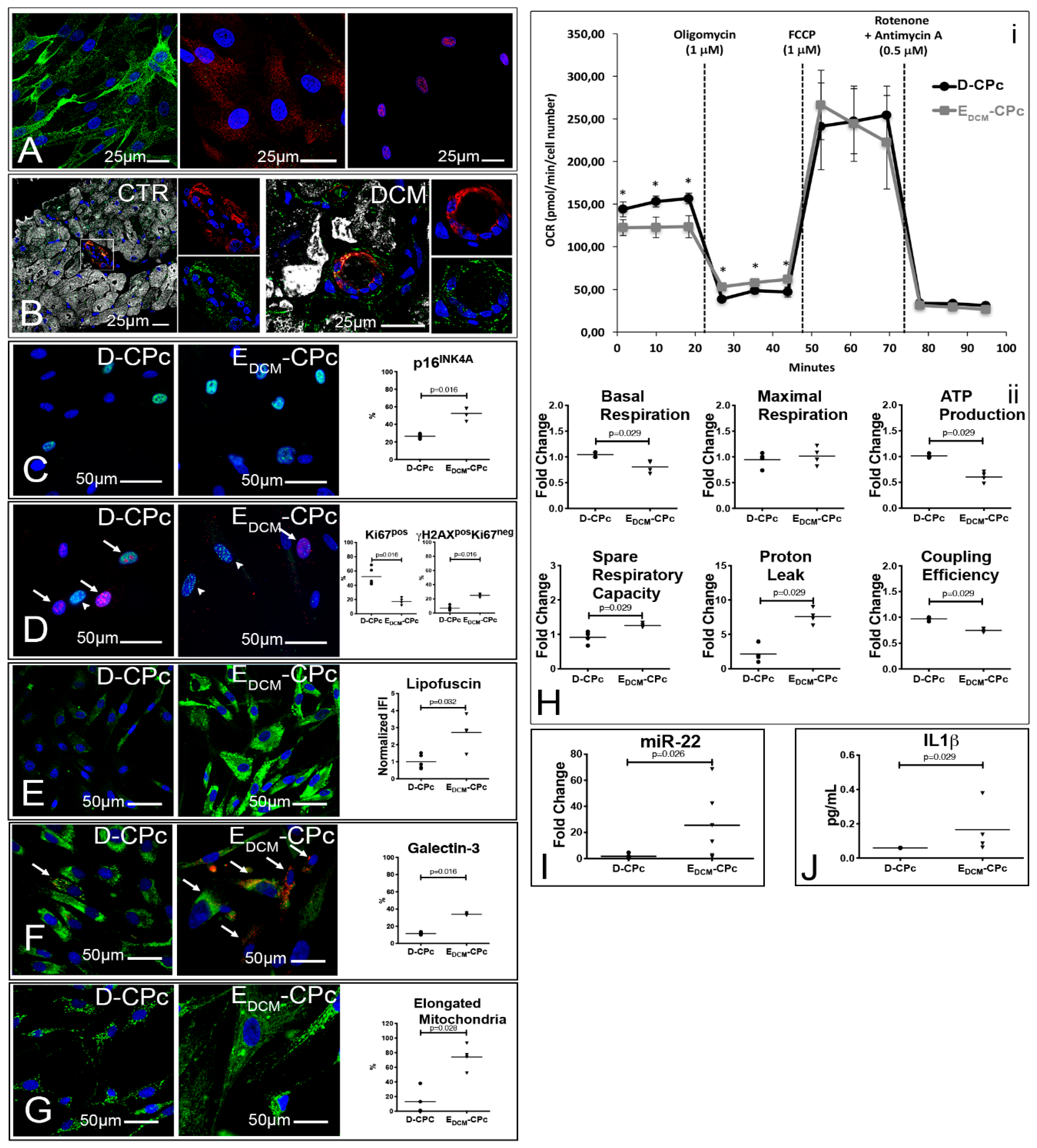
3.5. Alterations of the Autophagic Process and Mitochondrial Function are Coupled with Accelerated Senescence of Cardiac Pericytes in DCM
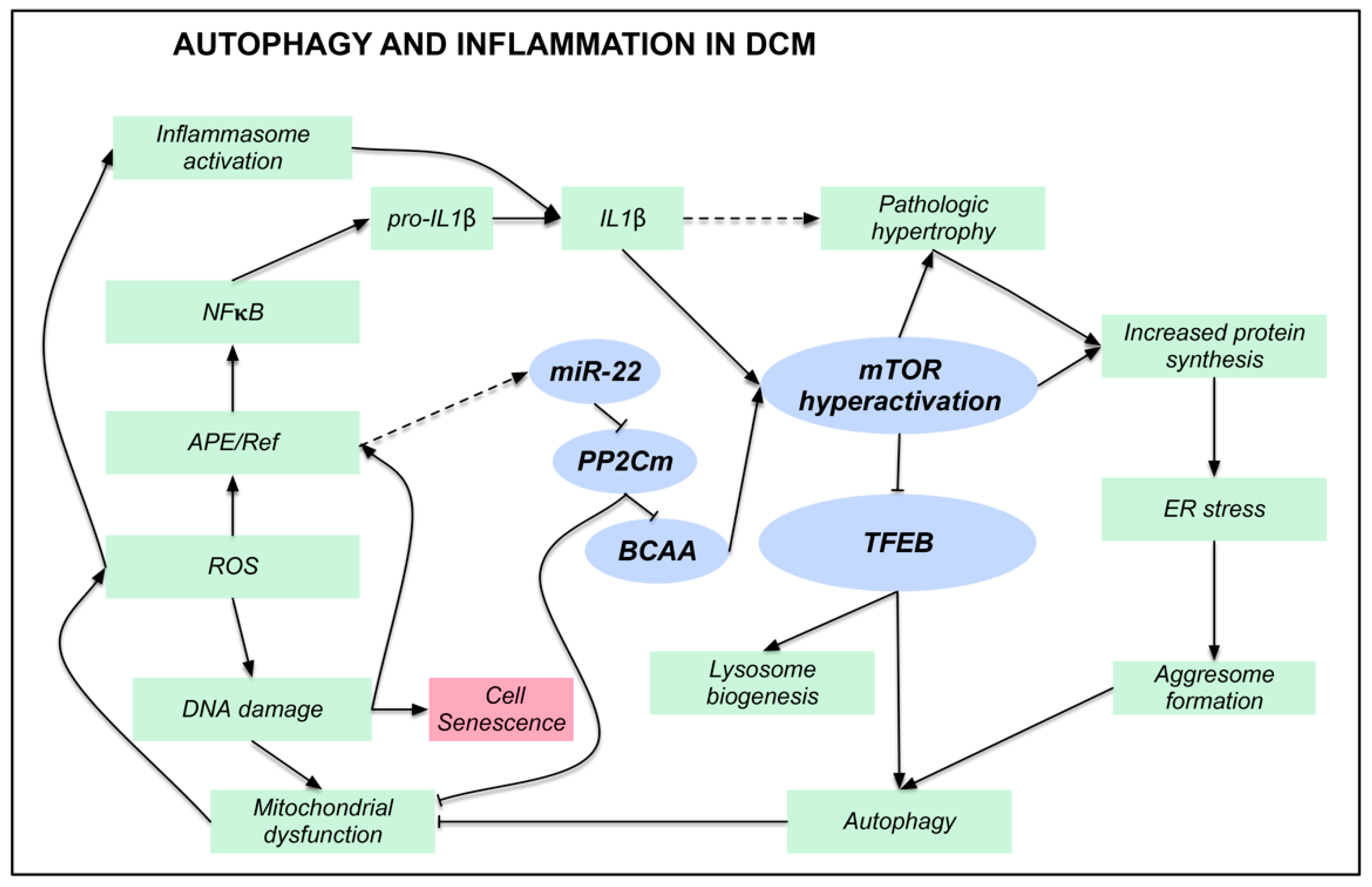
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fatkin, D.; CSANZ Cardiac Genetic Diseases Council Writing Group. Guidelines for the diagnosis and management of familial dilated cardiomyopathy. Heart Lung Circ. 2011, 20, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Siegfried, J.D. Update 2011: Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2011, 57, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. Off. J. Jpn. Circ. Soc. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Fong, L.G.; Muchir, A.; Young, S.G. Laminopathies and the long strange trip from basic cell biology to therapy. J. Clin. Investig. 2009, 119, 1825–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlossarek, S.; Frey, N.; Carrier, L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J. Mol. Cell. Cardiol. 2014, 71, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Lejeune, F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 2017, 50, 175. [Google Scholar] [CrossRef]
- Schafer, S.; de Marvao, A.; Adami, E.; Fiedler, L.R.; Ng, B.; Khin, E.; Rackham, O.J.; van Heesch, S.; Pua, C.J.; Kui, M.; et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat. Genet. 2017, 49, 46–53. [Google Scholar] [CrossRef]
- Ulbricht, A.; Arndt, V.; Hohfeld, J. Chaperone-assisted proteostasis is essential for mechanotransduction in mammalian cells. Commun. Integr. Biol. 2013, 6, e24925. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef]
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.M.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy. Circulation 2010, 121, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Weekes, J.; Morrison, K.; Mullen, A.; Wait, R.; Barton, P.; Dunn, M.J. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 2003, 3, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The role of autophagy in the heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 2012, 4, 166–175. [Google Scholar] [CrossRef]
- Aleksova, A.; Beltrami, A.P.; Carriere, C.; Barbati, G.; Lesizza, P.; Perrieri-Montanino, M.; Isola, M.; Gentile, P.; Salvioni, E.; Not, T.; et al. Interleukin-1β levels predict long-term mortality and need for heart transplantation in ambulatory patients affected by idiopathic dilated cardiomyopathy. Oncotarget 2017. [Google Scholar] [CrossRef]
- Beltrami, C.A.; Finato, N.; Rocco, M.; Feruglio, G.A.; Puricelli, C.; Cigola, E.; Quaini, F.; Sonnenblick, E.H.; Olivetti, G.; Anversa, P. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation 1994, 89, 151–163. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Hendrickx, D.M.; Hoefsloot, H.C.; Hendriks, M.M.; Canelas, A.B.; Smilde, A.K. Global test for metabolic pathway differences between conditions. Anal. Chim. Acta 2012, 719, 8–15. [Google Scholar] [CrossRef]
- Aittokallio, T.; Schwikowski, B. Graph-based methods for analysing networks in cell biology. Brief. Bioinform. 2006, 7, 243–255. [Google Scholar] [CrossRef]
- Cesselli, D.; D’Aurizio, F.; Marcon, P.; Bergamin, N.; Beltrami, C.A.; Beltrami, A.P. Cardiac stem cell senescence. Methods Mol. Biol. 2013, 976, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Aits, S.; Kricker, J.; Liu, B.; Ellegaard, A.M.; Hamalisto, S.; Tvingsholm, S.; Corcelle-Termeau, E.; Hogh, S.; Farkas, T.; Holm Jonassen, A.; et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015, 11, 1408–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardiello, M. Transcription factor EB: From master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann. NY Acad. Sci. 2016, 1371, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Moyzis, A.G.; Sadoshima, J.; Gustafsson, A.B. Mending a broken heart: The role of mitophagy in cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H183–H192. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Porter, N.A.; Schneider, C.; Brash, A.R.; Yin, H. Formation of 4-hydroxynonenal from cardiolipin oxidation: Intramolecular peroxyl radical addition and decomposition. Free Radic. Biol. Med. 2011, 50, 166–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY) 2009, 1, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.A.; Chang, S.; Wakimoto, H.; Gorham, J.M.; Conner, D.A.; Christodoulou, D.C.; Parfenov, M.G.; DePalma, S.R.; Eminaga, S.; Konno, T.; et al. Molecular profiling of dilated cardiomyopathy that progresses to heart failure. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Zhang, Z.; Kolwicz, S.C., Jr.; Abell, L.; Roe, N.D.; Kim, M.; Zhou, B.; Cao, Y.; Ritterhoff, J.; Gu, H.; et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. 2017, 25, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.F.; Gao, C.; Ren, S.X.; Wang, Y.B.; Sun, H.P.; Zhou, M.Y. Regulation of PP2Cm expression by miRNA-204/211 and miRNA-22 in mouse and human cells. Acta Pharmacol. Sin. 2015, 36, 1480–1486. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.P.; Chen, J.; Seok, H.Y.; Zhang, Z.; Kataoka, M.; Hu, X.; Wang, D.Z. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ. Res. 2013, 112, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Foinquinos, A.; Thum, S.; Remke, J.; Zimmer, K.; Bauters, C.; de Groote, P.; Boon, R.A.; de Windt, L.J.; Preissl, S.; et al. Preclinical development of a microRNA-based therapy for elderly patients with myocardial infarction. J. Am. Coll. Cardiol. 2016, 68, 1557–1571. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Del Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Lamming, D.W. The mechanistic target of rapamycin: The grand conducTOR of metabolism and aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Sun, H.; She, P.; Youn, J.Y.; Warburton, S.; Ping, P.; Vondriska, T.M.; Cai, H.; Lynch, C.J.; Wang, Y. Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J. Clin. Investig. 2009, 119, 1678–1687. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.Y.; Ren, S.; Liu, Y.; et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, F.M.; Attwell, D. A role for pericytes in coronary no-reflow. Nat. Rev. Cardiol. 2014, 11, 427–432. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, F.M.; Mastitskaya, S.; Hammond-Haley, M.; Freitas, F.; Wah, W.R.; Attwell, D. Capillary pericytes mediate coronary no-reflow after myocardial ischaemia. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Gulati, A.; Ismail, T.F.; Ali, A.; Hsu, L.Y.; Goncalves, C.; Ismail, N.A.; Krishnathasan, K.; Davendralingam, N.; Ferreira, P.; Halliday, B.P.; et al. Microvascular dysfunction in dilated cardiomyopathy: A quantitative stress perfusion cardiovascular magnetic resonance study. JACC Cardiovasc Imaging 2019. [Google Scholar] [CrossRef]
- Ferland-McCollough, D.; Slater, S.; Richard, J.; Reni, C.; Mangialardi, G. Pericytes, an overlooked player in vascular pathobiology. Pharmacol. Ther. 2017, 171, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Navarro, R.; Compte, M.; Alvarez-Vallina, L.; Sanz, L. Immune regulation by pericytes: Modulating innate and adaptive immunity. Front. Immunol. 2016, 7, 480. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Ren, S.; Korge, P.; Choi, J.; Dong, Y.; Weiss, J.; Koehler, C.; Chen, J.N.; Wang, Y. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 2007, 21, 784–796. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Mosquera, L.; Diogo, C.V.; Yambire, K.F.; Santos, G.L.; Sanchez, M.L.; Benit, P.; Rustin, P.; Lopez, L.C.; Milosevic, I.; Raimundo, N. Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci. Rep. 2017, 7, 45076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konig, J.; Ott, C.; Hugo, M.; Jung, T.; Bulteau, A.L.; Grune, T.; Hohn, A. Mitochondrial contribution to lipofuscin formation. Redox Biol. 2017, 11, 673–681. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiomyopathy (n = 50) | Controls (n = 18) | Normal Values | p | |
|---|---|---|---|---|
| Age (Yr) | 51.6 ± 13.9 | 40.7 ± 12.4 | - | 0.0068 |
| Sex (M/F) | 40/10 | 8/10 | - | 0.0072 |
| Duration of disease (Years) | 8.36 ± 6.54 | - | - | |
| NYHA class (%) | ||||
| II | 9 | - | - | - |
| III | 67 | - | - | - |
| IV | 24 | - | - | - |
| Echocardiography § | ||||
| Left ventricular diameter (mm) | ||||
| Systolic | 62.7 ± 12.1 | - | 21.6–34.8 | - |
| Diastolic | 74.0 ± 11.3 | - | 37.8–52.2 | - |
| Left ventricular Volumes (mL) | ||||
| End Diastolic | 197 ± 75 | - | 46–106 | - |
| End Systolic | 149 ± 60 | - | 14–42 | - |
| LV Ejection Fraction (%) | 23 ± 7 | - | 54–74 | - |
| Hemodynamics ‡ | ||||
| Pulmonary Artery Pressure (mmHg) | ||||
| Systolic | 41.92 ± 13.59 | - | 15–25 | - |
| Diastolic | 22.15 ± 10.31 | - | 8–12 | - |
| Mean | 29.45 ± 11.18 | - | 10–20 | - |
| PCWP (mmHg) | 21.56 ± 10.42 | - | 6–12 | - |
| CI (L⋅min−1⋅m−2) | 2.37 ± 0.73 | - | 2.5–4.0 | - |
| Gross Anatomy † | ||||
| Heart Weight (g) | 494 ± 161 | 334 ± 109 | 196–516 | 0.0011 |
| Transverse diameter (mm) | 129 ± 16 | 100 ± 20 | - | <0.0001 |
| Inner longitudinal diameter (mm) | 94 ± 16 | 73 ± 3 | - | <0.0001 |
| Wall thickness (mm) | ||||
| LV | 9.5 ± 2.1 | 13 ± 1.2 | - | 0.0073 |
| RV | 4.1 ± 1.9 | 3.3 ± 1.5 | - | n.s. |
| Septum | 11.5 ± 2.4 | 12.0 ± 2.0 | - | n.s. |
| Comorbidities | ||||
| BMI>30 (%) | 7.5 | |||
| Arrhythmia (%) | 75.6 | |||
| Mitral Insufficiency (%) | 82.6 | |||
| Chronic Kidney Failure (%) | 4.5 | |||
| BPCO (%) | 6.8 | |||
| Smoke | 4.5 | |||
| Hypothyroidism | 9.1 | |||
| Hyperthyroidism | 2.3 | |||
| Hypertension (%) | 2.3 | |||
| Dyslipidemia (%) | 20.5 | |||
| Diabetes (%) | 11.4 | |||
| Pharmacological therapy | ||||
| ACE-I/ARB (%) | 82.5 | |||
| β-Blockers (%) | 58.5 | |||
| Digitalis (%) | 80.5 | |||
| Dobutamine (%) | 30 | |||
| Amiodarone (%) | 56.1 | |||
| Antialdosteronic (%) | 67.5 | |||
| K Sparing diuretics (%) | 12.5 | |||
| Loop diuretics (%) | 97.5 | |||
| Insulin / Antidiabetics (%) | 7.5 | |||
| Statins (%) | 0 | |||
| Oral Anticoagulants (%) | 57.5 | |||
| Tiroxin (%) | 9.8 | |||
| Cardiomyopathy (n = 12) | Controls (n = 12) | Normal Values | p | |
|---|---|---|---|---|
| Age (Yr) | 57.9 ± 6.1 | 46.9 ± 15.9 | - | 0.04 |
| Sex (M/F) | 11/1 | 6/6 | - | n.s. |
| Duration of disease (Years) | 23.9 ± 32.1 | - | - | |
| NYHA class (%) | ||||
| III | 75 | - | - | - |
| IV | 25 | - | - | - |
| Echocardiography § | ||||
| Left ventricular diameter (mm) | ||||
| Systolic | 61.6 ± 11.9 | 27.1 ± 5.0 | 21.6–34.8 | <0.0001 |
| Diastolic | 71.0 ± 7.9 | 45.4 ± 4.5 | 37.8–52.2 | <0.0001 |
| Left ventricular Volumes (mL) | ||||
| End Diastolic | 220.2 ± 112.4 | 92.0 ± 50.4 | 46–106 | 0.047 |
| End Systolic | 170.6 ± 98.9 | 40.7 ± 23.1 | 14–42 | 0.012 |
| LV Ejection Fraction (%) | 22.8 ± 8.4 | 63.4 ± 5.8 | 54–74 | <0.0001 |
| Hemodynamics ‡ | ||||
| Pulmonary Artery Pressure (mmHg) | ||||
| Systolic | 45.9 ± 15.2 | - | 15–25 | |
| Diastolic | 20.7 ± 11.6 | - | 8–12 | |
| Mean | 30.0 ± 13.8 | - | 10–20 | |
| PCWP (mmHg) | 18.5 ± 11.4 | - | 6–12 | |
| CI (L⋅min−1⋅m−2) | 1.9 ± 0.36 | - | 2.5–4.0 | |
| Gross Anatomy † | ||||
| Heart Weight (g) | 555.4 ± 194.5 | - | 196–516 | |
| Transverse diameter (mm) | 125 ± 12 | - | - | |
| Inner longitudinal diameter (mm) | 93 ± 13 | - | - | |
| Wall thickness (mm) | ||||
| LV | 11.6±1.8 | - | - | |
| RV | 7.2±3.5 | - | - | |
| Septum | 13.9±3.5 | - | - | |
| Comorbidities | ||||
| BMI>30 (%) | 8 | - | ||
| Arrhythmia (%) | 90 | - | ||
| Mitral Insufficiency (%) | 88 | - | ||
| Chronic Kidney Failure | 40 | - | ||
| BPCO (%) | 10 | - | ||
| Smoke | 0 | - | ||
| Hypothyroidism | 10 | - | ||
| Hyperthyroidism | 20 | - | ||
| Hypertension (%) | 30 | - | ||
| Dyslipidemia (%) | 40 | - | ||
| Diabetes (%) | 20 | - | ||
| Pharmacological therapy | ||||
| ACE-I/ARB (%) | 66.7 | - | ||
| β-Blockers (%) | 80 | - | ||
| Digitalis (%) | 60 | - | ||
| Dobutamine (%) | 10 | - | ||
| Amiodarone (%) | 30 | - | ||
| Antialdosteronic (%) | 40 | - | ||
| K Sparing diuretics (%) | 50 | - | ||
| Loop diuretics (%) | 80 | - | ||
| Insulin / Antidiabetics (%) | 10 | - | ||
| Statins (%) | 30 | - | ||
| Oral Anticoagulants (%) | 70 | - | ||
| Tiroxin (%) | 20 | - | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caragnano, A.; Aleksova, A.; Bulfoni, M.; Cervellin, C.; Rolle, I.G.; Veneziano, C.; Barchiesi, A.; Mimmi, M.C.; Vascotto, C.; Finato, N.; et al. Autophagy and Inflammasome Activation in Dilated Cardiomyopathy. J. Clin. Med. 2019, 8, 1519. https://doi.org/10.3390/jcm8101519
Caragnano A, Aleksova A, Bulfoni M, Cervellin C, Rolle IG, Veneziano C, Barchiesi A, Mimmi MC, Vascotto C, Finato N, et al. Autophagy and Inflammasome Activation in Dilated Cardiomyopathy. Journal of Clinical Medicine. 2019; 8(10):1519. https://doi.org/10.3390/jcm8101519
Chicago/Turabian StyleCaragnano, Angela, Aneta Aleksova, Michela Bulfoni, Celeste Cervellin, Irene Giulia Rolle, Claudia Veneziano, Arianna Barchiesi, Maria Chiara Mimmi, Carlo Vascotto, Nicoletta Finato, and et al. 2019. "Autophagy and Inflammasome Activation in Dilated Cardiomyopathy" Journal of Clinical Medicine 8, no. 10: 1519. https://doi.org/10.3390/jcm8101519