Gut Microbial Composition and Function Are Altered in Patients with Early Rheumatoid Arthritis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Research Subjects
2.2. Sample Collection and Next Generation Sequencing
2.3. Bioinformatics Analysis
2.4. Statistical Analysis
3. Results
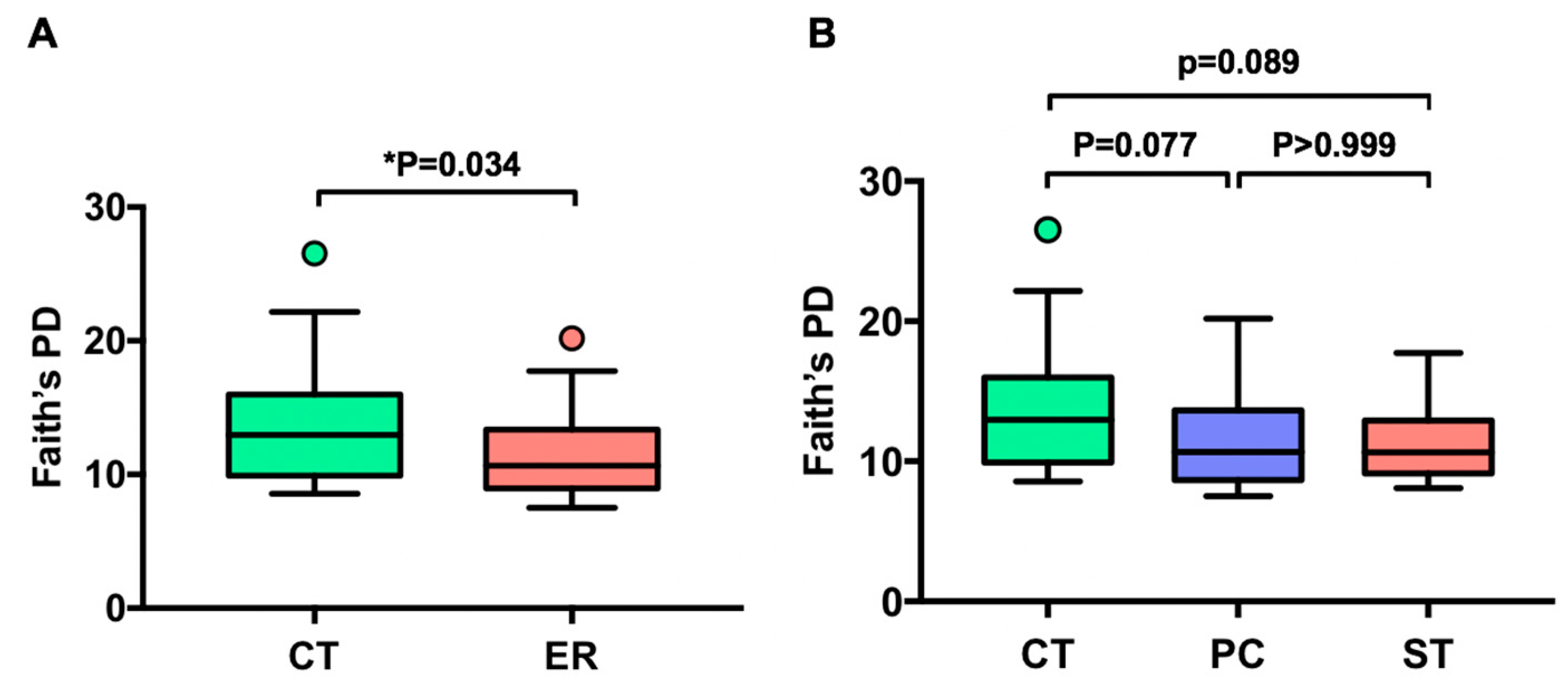
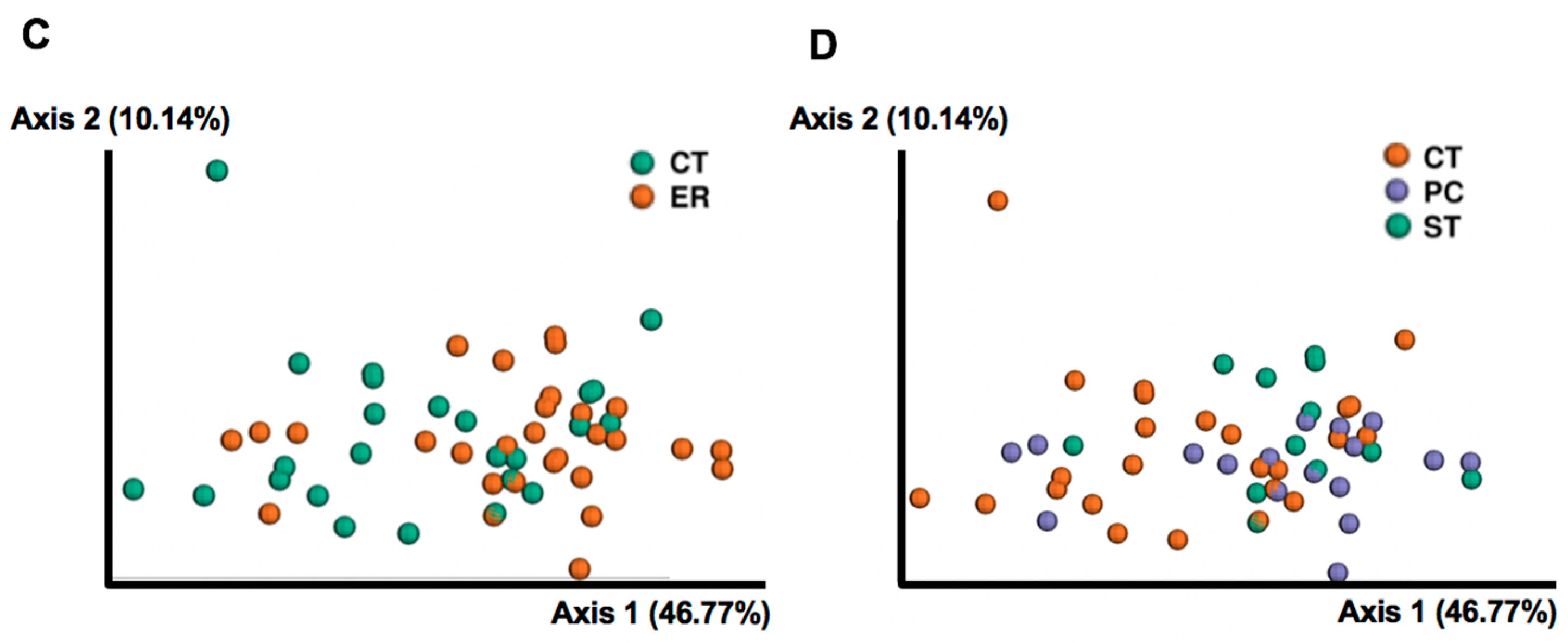
3.1. Differences in Gut Microbial Diversity between RA Patients and Healthy Subjects
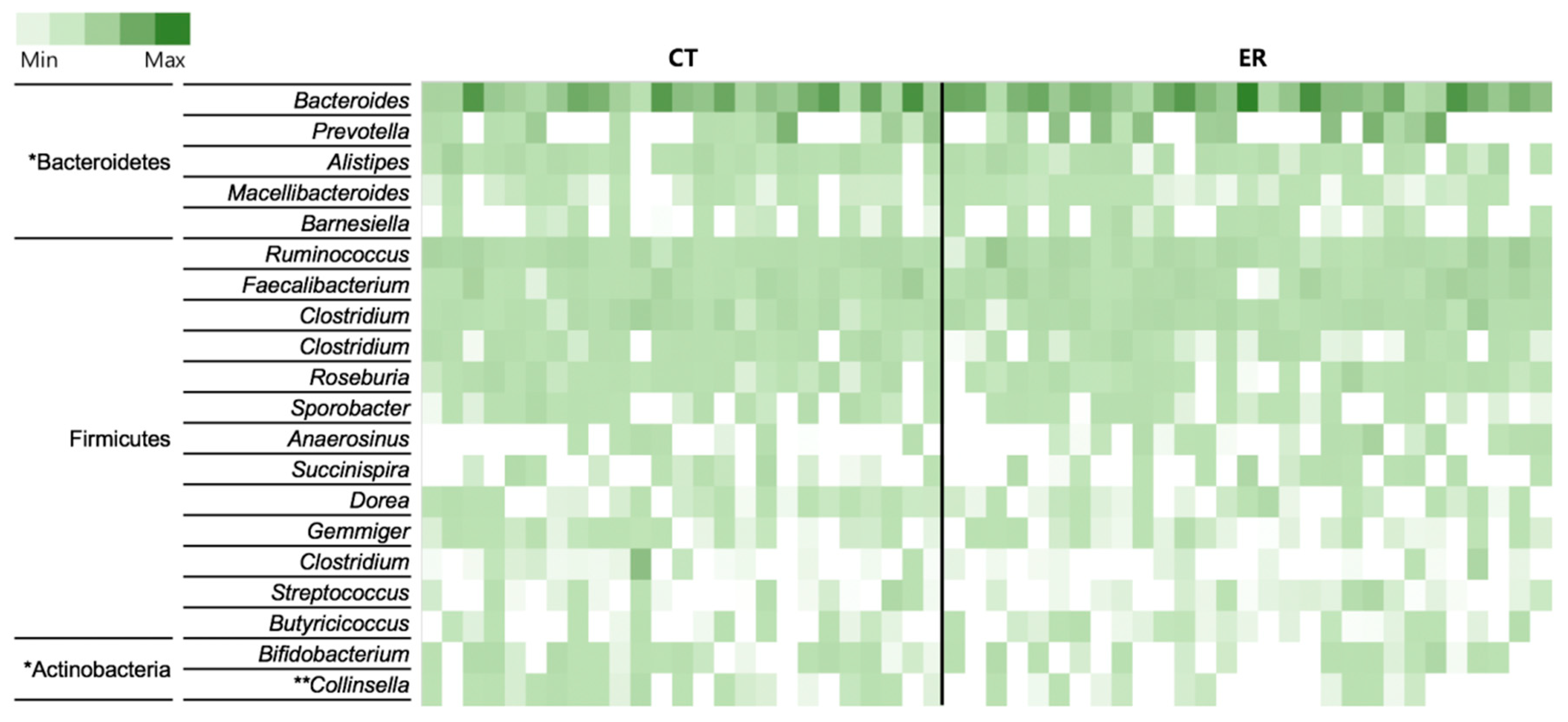
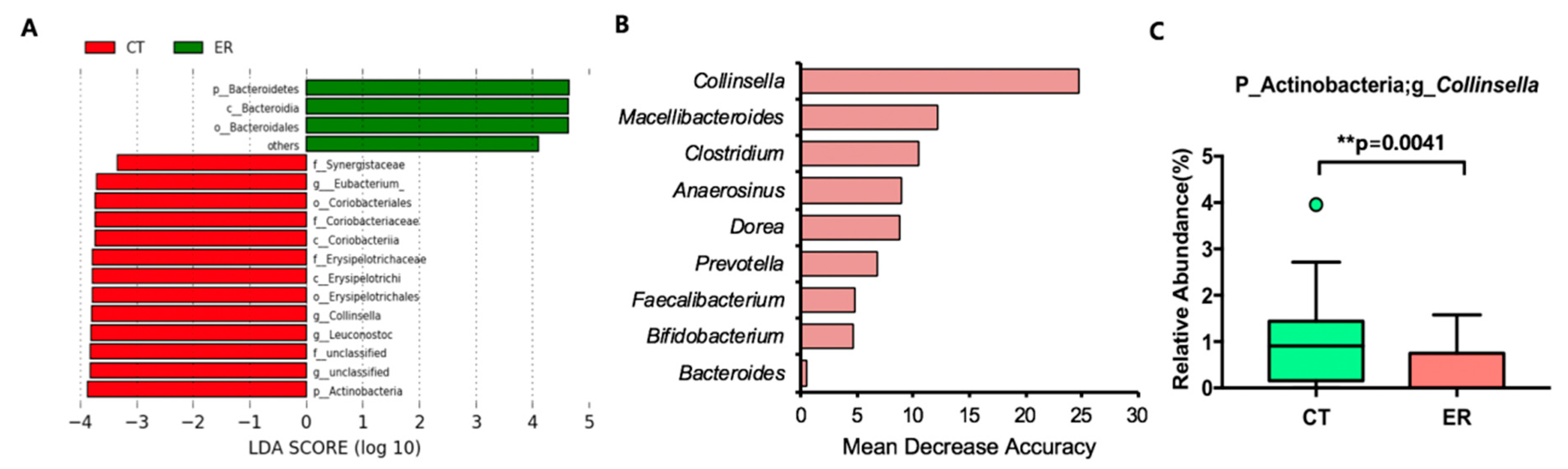
3.2. Featured Microbial Taxa in ER Patients
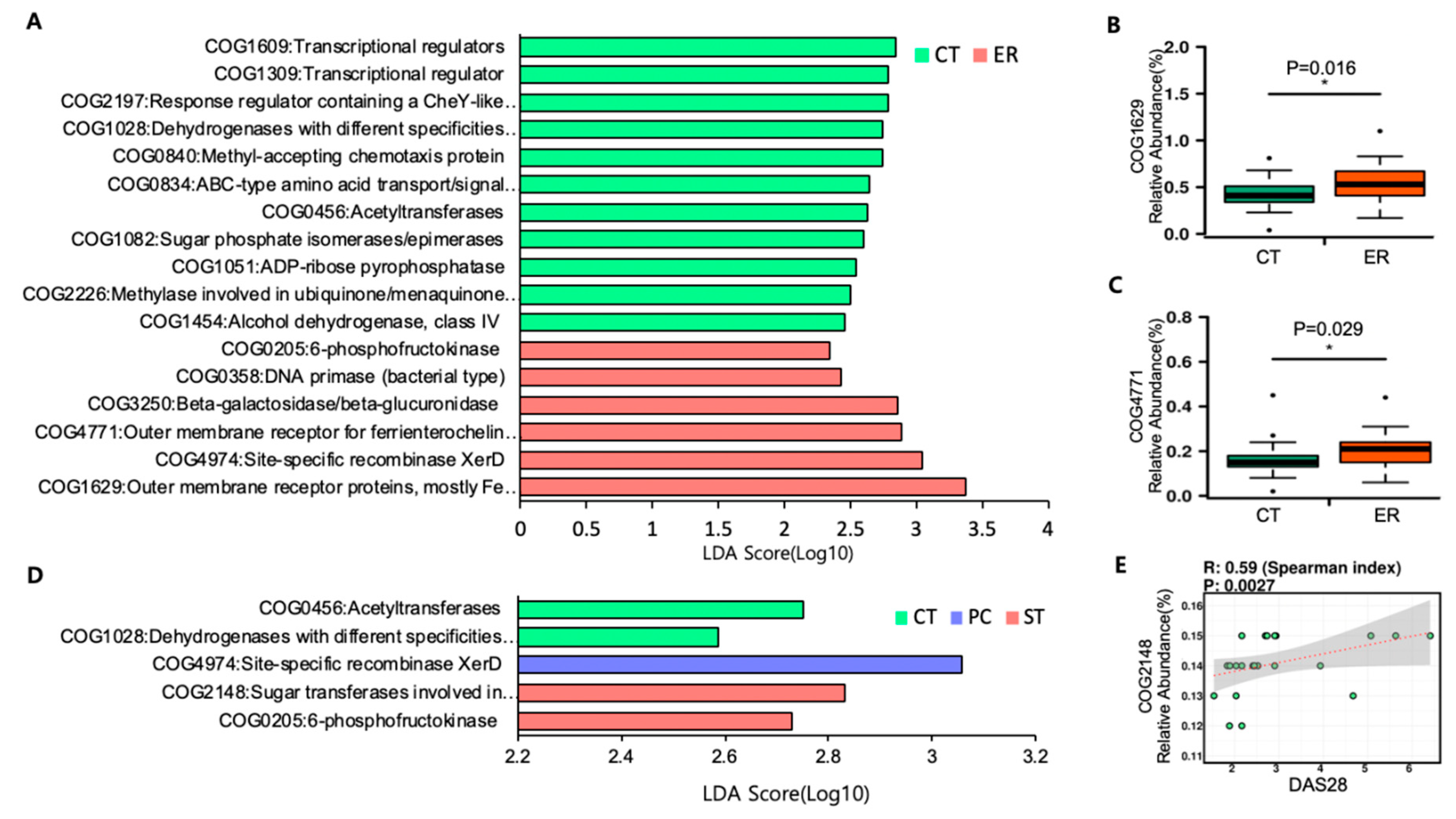
3.3. Microbial Function Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Cojocaru, M.; Cojocaru, I.M.; Silosi, I.; Vrabie, C.D.; Tanasescu, R. Extra-articular Manifestations in Rheumatoid Arthritis. Maedica 2010, 5, 286–291. [Google Scholar]
- Fairweather, D.; Frisancho-Kiss, S.; Rose, N.R. Sex differences in autoimmune disease from a pathological perspective. Am. J. Pathol. 2008, 173, 600–609. [Google Scholar] [CrossRef]
- Tellefsen, S.; Morthen, M.K.; Richards, S.M.; Lieberman, S.M.; Rahimi Darabad, R.; Kam, W.R.; Sullivan, D.A. Sex Effects on Gene Expression in Lacrimal Glands of Mouse Models of Sjogren Syndrome. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5599–5614. [Google Scholar] [CrossRef]
- Maeda, Y.; Takeda, K. Role of Gut Microbiota in Rheumatoid Arthritis. J. Clin. Med. 2017, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Kurakawa, T.; Umemoto, E.; Motooka, D.; Ito, Y.; Gotoh, K.; Hirota, K.; Matsushita, M.; Furuta, Y.; Narazaki, M.; et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol. 2016, 68, 2646–2661. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Picchianti-Diamanti, A.; Panebianco, C.; Salemi, S.; Sorgi, M.L.; Di Rosa, R.; Tropea, A.; Sgrulletti, M.; Salerno, G.; Terracciano, F.; D’Amelio, R.; et al. Analysis of Gut Microbiota in Rheumatoid Arthritis Patients: Disease-Related Dysbiosis and Modifications Induced by Etanercept. Int. J. Mol. Sci. 2018, 19, 2938. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpizar-Rodriguez, D.; Lesker, T.R.; Gronow, A.; Gilbert, B.; Raemy, E.; Lamacchia, C.; Gabay, C.; Finckh, A.; Strowig, T. Prevotella copri in individuals at risk for rheumatoid arthritis. Ann. Rheum. Dis. 2019, 78, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ 2018. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome–environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; Beiko, R.G.; Huttenhower, C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.; Caplan, L.; Yazdany, J.; Robbins, M.L.; Neogi, T.; Michaud, K.; Saag, K.G.; O’Dell, J.R.; Kazi, S. Rheumatoid arthritis disease activity measures: American College of Rheumatology recommendations for use in clinical practice. Arthritis Care Res. 2012, 64, 640–647. [Google Scholar] [CrossRef]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Bag, S.; Ghosh, T.S.; Das, B. Complete Genome Sequence of Collinsella aerofaciens Isolated from the Gut of a Healthy Indian Subject. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Kassinen, A.; Krogius-Kurikka, L.; Makivuokko, H.; Rinttila, T.; Paulin, L.; Corander, J.; Malinen, E.; Apajalahti, J.; Palva, A. The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology 2007, 133, 24–33. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Vyara, M.; Jessica, F.; Riyue, B.; Tara, C.; Yuanyuan, Z.; Maria-Luisa, A.; Jason, J.L.; Thomas, F.G. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vreugdenhil, G.; Swaak, A.J. Anaemia in rheumatoid arthritis: Pathogenesis, diagnosis and treatment. Rheumatol. Int. 1990, 9, 243–257. [Google Scholar] [CrossRef]
- Wahle, M. Anemia in patients with rheumatoid arthritis. Ter Arkh 2012, 84, 64–68. [Google Scholar]
- Kurosu, M.; Begari, E. Vitamin K2 in electron transport system: Are enzymes involved in vitamin K2 biosynthesis promising drug targets? Molecules 2010, 15, 1531–1553. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ER | CT | |||
|---|---|---|---|---|
| PC | ST | Total | ||
| N | 17 | 12 | 29 | 25 |
| Age | 47.71 ± 6.57 | 44.33 ± 14.65 | 46.31 ± 10.58 | 42.32 ± 12.46 |
| Sex (F/M) | 17/0 | 12/0 | 29/0 | 25/0 |
| ** DAS28 | 2.26 ± 0.43 | 3.84 ± 1.52 | 2.92 ± 1.28 | - |
| ** CRP (mg/dL) | 0.11 ± 0.19 | 1.28 ± 1.77 | 0.59 ± 1.26 | - |
| ** ESR (mm/h) | 9.06 ± 5.83 | 27.17 ± 17.78 | 16.55 ± 15.03 | - |
| RF (IU/mL) | 188.20 ± 285.93 | 288.10 ± 245.74 | 203.90 ± 266.85 | - |
| ACCP (CU) | 1048.00 ± 2815.19 | 765.30 ± 705.84 | 942.10 ± 2239.86 | - |
| Taxa | p-value | FDR | CT Mean (%) | ER Mean (%) | Fold Change | CT Prevalence (%) | ER Prevalence (%) | |
|---|---|---|---|---|---|---|---|---|
| Enriched taxa in ER | ||||||||
| Phylum | Bacteroidetes | * 0.011 | 0.091 | 34.78 | 43.07 | 1.24 | 100 | 100 |
| Class | Bacteroidia | * 0.014 | 0.089 | 34.67 | 43.03 | 1.24 | 100 | 100 |
| Order | Bacteroidales | * 0.014 | 0.093 | 34.67 | 43.03 | 1.24 | 100 | 100 |
| Enriched taxa in CT | ||||||||
| Phylum | Actinobacteria | * 0.014 | 0.091 | 2.85 | 1.52 | 0.54 | 92 | 69 |
| Class | Erysipelotrichi | ** 0.0027 | 0.028 | 1.44 | 0.65 | 0.45 | 96 | 72 |
| Class | Coriobacteriia | ** 0.0029 | 0.028 | 1.35 | 0.48 | 0.36 | 80 | 48 |
| Order | Erysipelotrichales | ** 0.0027 | 0.029 | 1.44 | 0.65 | 0.45 | 96 | 72 |
| Order | Coriobacteriales | ** 0.0029 | 0.029 | 1.35 | 0.48 | 0.36 | 80 | 48 |
| Family | Erysipelotrichaceae | ** 0.0027 | 0.029 | 1.44 | 0.65 | 0.45 | 96 | 72 |
| Family | Coriobacteriaceae | ** 0.0029 | 0.029 | 1.35 | 0.48 | 0.36 | 80 | 48 |
| Genus | Collinsella | ** 0.0041 | 0.082 | 1.00 | 0.36 | 0.36 | 76 | 41 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, Y.; Kim, J.-W.; You, H.J.; Park, S.-J.; Lee, J.; Ju, J.H.; Park, M.S.; Jin, H.; Cho, M.-L.; Kwon, B.; et al. Gut Microbial Composition and Function Are Altered in Patients with Early Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 693. https://doi.org/10.3390/jcm8050693
Jeong Y, Kim J-W, You HJ, Park S-J, Lee J, Ju JH, Park MS, Jin H, Cho M-L, Kwon B, et al. Gut Microbial Composition and Function Are Altered in Patients with Early Rheumatoid Arthritis. Journal of Clinical Medicine. 2019; 8(5):693. https://doi.org/10.3390/jcm8050693
Chicago/Turabian StyleJeong, Yunju, Ji-Won Kim, Hyun Ju You, Sang-Jun Park, Jennifer Lee, Ji Hyeon Ju, Myeong Soo Park, Hui Jin, Mi-La Cho, Bin Kwon, and et al. 2019. "Gut Microbial Composition and Function Are Altered in Patients with Early Rheumatoid Arthritis" Journal of Clinical Medicine 8, no. 5: 693. https://doi.org/10.3390/jcm8050693