
Epithelial-Mesenchymal Transition and Breast Cancer

Abstract
:1. Introduction
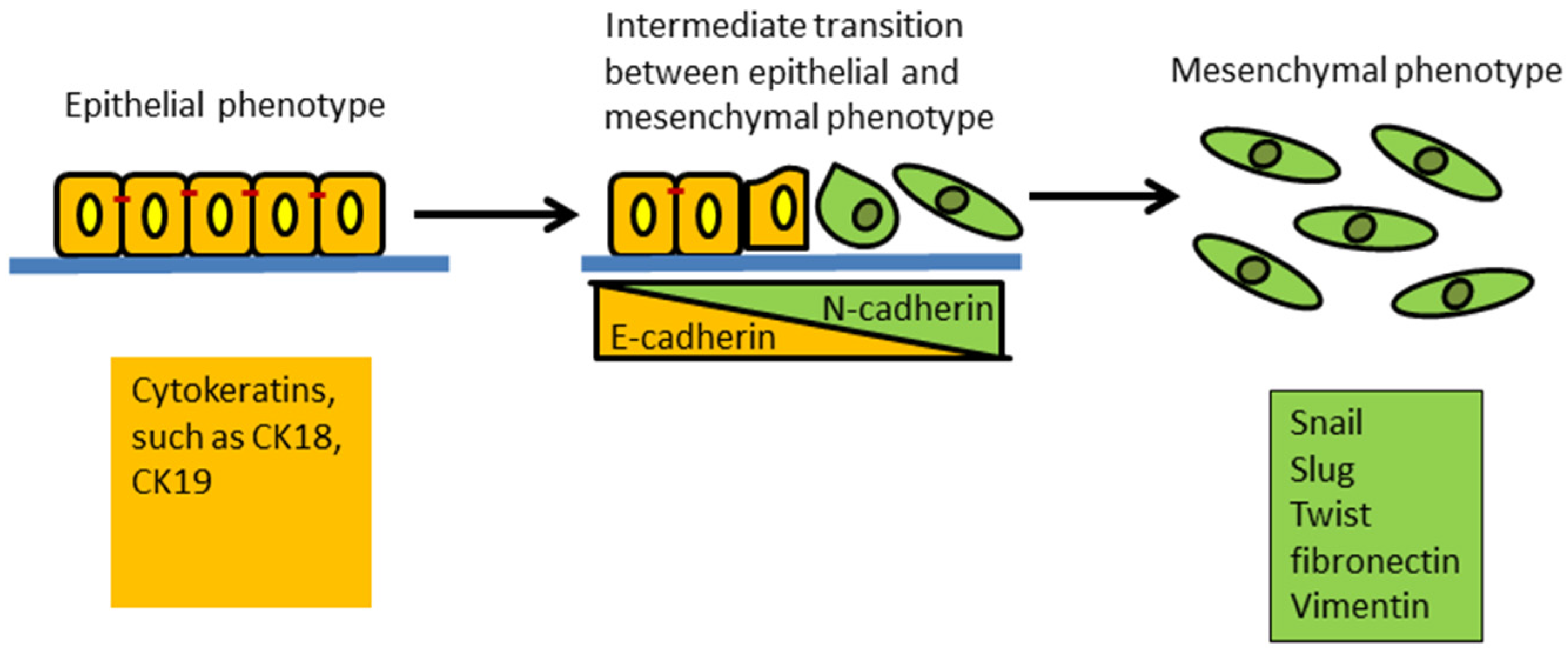
2. EMT and Its Plasticity Features

3. EMT and Breast Cancer Metastasis
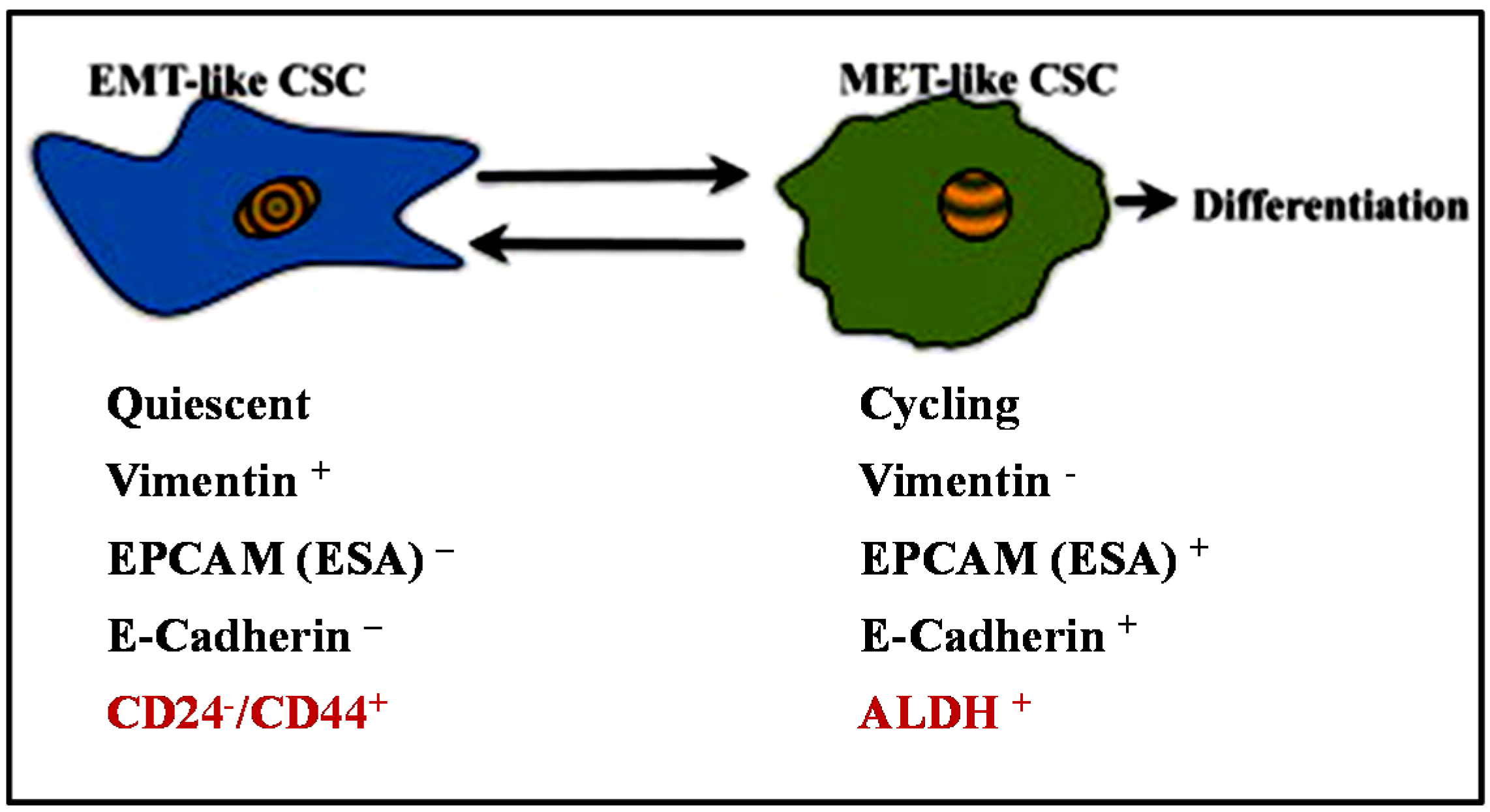
4. EMT and Cancer Stem-Like Cell (CSC)

5. EMT and Resistance to Treatment
6. Molecular Mechanisms of EMT
6.1. Signaling Pathways Mediate EMT
6.1.1. TGF-β Signaling
6.1.2. Wnt Signaling
6.1.3. Notch Signaling
6.1.4. Interaction of Signaling Pathways on EMT
6.2. Epigenetics Regulation of EMT
6.2.1. DNA Methylation and Histone Modification in Regulation of EMT
6.2.2. MicroRNAs in Regulation of EMT
6.2.3. Epigenetic Therapies and EMT
7. Conclusions

{kind=link}
{kind=link}
| Study | N | Method | Markers | Clinical Outcome | p-Value |
|---|---|---|---|---|---|
| Lin et al. [26] | 441 | IHC | Low E-cadherin, High Slug, High Vimentin | Associated with Low DFS and Low OS | <0.01 |
| Aleskandarany et al. [27] | 1035 | IHC, RPPA | Low E-cadhirin and High N-cadherin | Associated with Low DFS and Low OS | <0.001 |
| Wu et al. [37] | 126 | IHC | CD44+/CD24− vs. CD44−/CD24−; | Associated with Low DFS | 0.05 |
| CD24+/CD44− vs. CD44−/CD24− | 0.016 | ||||
| Lin et al. [40] | 147 | IHC | CD44high/CD24low | Associated with Low DFS and Low OS | <0.05 |
| Ma et al. [98] | 45 | RT-qPCR | miR9 | Associated with Metastasis | <0.01 |
| Bonnie et al. [127] | 492 | IHC | Low E-cadherin | Increased HR of all-cause mortality | <0.05 |
| Khramtsov et al. [128] | 117 | IHC | High cytosolic β-catenin | Associated with Low OS | 0.0005 |
| Or High or High nuclear β-catenin | 0.039 | ||||
| Martin et al. [129] | 190 | RT-qPCR | High Twist | High mortality | n.s. |
| High Snail | High mortality | n.s. | |||
| High Slug | High metastasis | 0.05 | |||
| Mylona et al. [130] | 155 | IHC | CD44+/CD24−; CD24+/CD44− | No association with DFS and OS; | n.s. |
| Associated with low DFS and OS | <0.05 | ||||
| Gwak et al. [131] | 295 | IHC, RT-qPCR | miR9 | Associated with low DFS and OS | <0.05 |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumors. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Neo, S.Y.; McShane, L.M.; Korn, E.L.; Long, P.M.; Jazaeri, A.; Martiat, P.; Fox, S.B.; Harris, A.L.; Liu, E.T. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl. Acad. Sci. USA 2003, 100, 10393–10398. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Sarkar, F.H.; Ahmed, Q.; Bandyopadhyay, S.; Nahleh, Z.A.; Semaan, A.; Sarkr, W.; Munkarah, A.; Ali-Fehmi, R. Molecular markers of epithelial-to mesenchymal transition are associated with tumor aggressiveness in breast carcinoma. Translational. Oncol. 2011, 4, 222–226. [Google Scholar] [CrossRef]
- Blick, T.; Widodo, E.; Hugo, H.; Waltham, M.; Lenburg, M.E.; Neve, R.M.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin. Exp. Metastasis. 2008, 25, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Montagna, E.; Cancello, G.; D’Agostino, D.; Lauria, R.; Forestieri, V.; Esposito, A.; Silvestro, L.; Accurso, A.; de Placido, S.; de Laurentiis, M. Central nervous system metastases in a cohort of metastatic breast cancer patients treated with trastuzumab. Cancer Chemother. Pharmacol. 2009, 63, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.J.; Danson, S.; Jolly, S.; Ryder, W.D.J.; Burt, P.A.; Stewart, A.L.; Wilkinson, P.M.; Welch, R.S.; Magee, B.; Wilson, G.; et al. Incidence of cerebral metastases in patients treated with trastuzumab for metastatic breast cancer. Br. J. Cancer 2004, 91, 639–643. [Google Scholar] [CrossRef]
- Thiery, J.P.; Lim, C.T. Tumor dissemination: An EMT affair. Cancer Cell 2013, 23. [Google Scholar] [CrossRef] [PubMed]
- Christofori, G.; Bill, R. The relevance of EMT in breast cancer metastasis: Correlation or causality. FEBS 2015, 589, 1577–1587. [Google Scholar]
- Fidler, I.J.; Kripke, M.L. Metastasis results from preexisting variant cells within a malignant tumor. Science 1977, 197, 893–895. [Google Scholar] [CrossRef] [PubMed]
- Daleba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Blick, T.; Hugo, H.; Widodo, E.; Waltham, M.; Pinto, C.; Mani, S.A.; Weinberg, R.A.; Neve, R.M.; Lenburg, M.E.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44hi/CD24lo/− stemcell phenotype in human breast cancer. J. Mammary Gland Biol. Neoplasia. 2010, 15, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, M.; Mimori, K.; Yokobori, T.; Ishi, H.; Beppu, T.; Nakamori, S.; Baba, H.; Mori, M. Epithelia-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010, 101, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Foroni, C.; Broggini, M.; Generali, D.; Damia, G. Epithelial-mesenchymal transition and breast cancer: Role, molecular mechanisms and clinical impact. Cancer Treat. Rev. 2012, 38, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Mallini, P.; Lennard, T.; Kriby, J.; Meeson, A. Epithelial-to-mesenchymal transition: What is the impact on breast cancer stem cells and drug resistance. Cancer Treat. Rev. 2014, 40, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Lien, H.C.; Hsiao, Y.H.; Lin, Y.S.; Yao, Y.T.; Juan, H.F.; Kuo, W.H.; Hung, M.C.; Chang, K.J.; Hsieh, F.J. Molecular signatures of metaplastic carcinoma of the breast by large-scale transcriptional profiling: Identification of genes potentially related to epithelial-mesenchymal transition. Oncogene 2007, 26, 7859–7871. [Google Scholar] [CrossRef] [PubMed]
- Bebee, T.W.; Cieply, B.W.; Carstens, R.P. Genome-wide activities of RNA binding proteins that regulate cellular changes in the epithelial to mesenchymal transition (EMT). Adv. Exp. Med. Biol. 2014, 825, 267–302. [Google Scholar] [PubMed]
- Cheng, Q.; Chang, J.T.; Gwin, W.R.; Zhu, J.; Ambs, S.; Geradts, J.; Lyerly, H.K. A signature of epithelial-mesenchymal plasticity and stromal activation in primary tumor modulates late recurrence in breast cancer independent of disease subtype. Breast Cancer Res. 2014, 16, 407. [Google Scholar] [CrossRef] [PubMed]
- Hazan, R.B.; Qiao, R.; Keren, R.; Badano, I.; Suyama, K. Cadherin switch in tumor progression. Ann. N. Y. Acad. Sci. 2004, 1014, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Johnson, K.R.; Wheelock, M.J. Cadherin switching: Essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J. Cell Sci. 2005, 118, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Sarrió, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, X.; Shang, M.; Zhang, Y.; Xia, B.; Niu, M.; Liu, Y.; Pang, D. Dysregulated expression of Slug, Vimentin, and E-cadherin correlates with poor clinical outcome in patients with basal-like breast cancer. J. Surg. Oncol. 2013, 107, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Aleskandarany, M.A.; Negm, O.H.; Green, A.R.; Ahmed, M.A.H.; Nolan, C.C.; Tighe, P.J.; Ellis, I.; Rakha, E.A. Epithelial mesenchymal transition in early invasive breast cancer: An immunohistochemical and reverse phase protein array study. Breast Cancer Res. Treat. 2014, 145, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, H.J.; Jang, M.H.; Gwak, J.M.; Lee, K.S.; Kim, E.J.; Kim, H.J.; Lee, H.E.; Park, S.Y. Epithelial-mesenchymal transition increases during the progression of in situ to invasive basal-like breast cancer. Hum. Pathol. 2013, 44, 2581–2589. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.; Au, S.K.; Ni, Y.B.; Shao, M.M.; Siu, W.M.; Hui, S.W.; Chan, S.K.; Chan, K.W.; Kwok, Y.K.; Chan, K.F.; et al. P-cadherin and Vimentin are useful basal markers in breast cancers. Hum. Pathol. 2013, 44, 2782–2791. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.A.; Kallergi, G.; Zafeiriou, Z.; Manouras, L.; Theodoropoulos, P.A.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Co-expression of putative stemness and epithelial-to-mesenchymal transition markers on single circulating tumour cells from patients with early and metastatic breast cancer. BMC Cancer 2014, 14, 651. [Google Scholar] [CrossRef] [PubMed]
- Kallergi, G.; Papadaki, M.A.; Politaki, E.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res. 2011, 13, R59. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; LaBarge, M.A. Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell 2008, 2, 511–512. [Google Scholar] [PubMed]
- AL-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. PNAS 2003, 100, 3983–3988. [Google Scholar] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2013, 2, 78–91. [Google Scholar] [PubMed]
- Wu, Y.; Sarkissyan, M.; Elshimali, Y.; Vadgama, J.V. Triple negative breast tumors in African-American and Hispanic/Latina women are high in CD44+, low in CD24+, and have loss of PTEN. PLoS ONE 2013, 8, e78259. [Google Scholar] [CrossRef] [PubMed]
- Honeth, G.; Bendahl, P.O.; Ringnér, M.; Saal, L.H.; Gruvberger-Saal, S.K.; Lövgren, K.; Grabau, D.; Fernö, M.; Borg, A.; et al. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10, R53. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Sivridis, E.; Fiska, A.; Koukourakis, M.I. The CD44+/CD24− phenotype relates to “triple-negative” state and unfavorable prognosis in breast cancer patients. Med. Oncol. 2011, 28, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhong, Y.; Guan, H.; Zhang, X.; Sun, Q. CD44+/CD24− phenotype contributes to malignant relapse following surgical resection and chemotherapy in patients with invasive ductal carcinoma. J. Exp. Clin. Cancer Res. 2012, 31, 31–59. [Google Scholar]
- Perrone, G.; Gaeta, L.M.; Zagami, M.; Nasorri, F.; Coppola, R.; Borzomati, D.; Bartolozzi, F.; Altomare, V.; Trodella, L.; Tonini, G.; et al. In situ identification of CD44+/CD24− cancer cells in primary human breast carcinomas. PLOS ONE 2012, 7, e43110. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes ro chemoresistance. Nature 2015, 527. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xu, J.; Wang, W.; Cao, X.; Chen, Q.; Tang, F.; Chen, Z.Q.; Liu, X.P.; Xu, Z.D. Twist1-mediated Adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer Res. 2009, 15, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Huang, J.; Wu, Q.; Cai, Y.; Zhu, L.; Lu, X.; Chen, S.; Chen, C.; Wang, Z. Acquisition of epithelial-mesenchymal transition is associated with Skp2 expression in paclitaxel-resistant breast cancer cells. Br. J. Cancer 2014, 110, 1958–1967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Feng, M.; Zheng, G.; Chen, Y.; Wang, X.; Pen, B.; Yin, J.; Yu, Y.; He, Z. Chemoresistance to 5-fluorouracil induces epithelial-mesenchymal transition via up-regulation of Snail in MCF7 human breast cancer cells. Biochem. Biophys. Res. Commun. 2012, 417, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Işeri, Ö.D.; Kars, M.D.; Arpaci, F.; Atalay, C.; Pak, I.; Gündüz, U. Drug resistant MCF-7 cells exhibit epithelial-mesenchymal transition gene expression pattern. Biomed. Phamacother. 2011, 65, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, S.; Jiang, W.G.; Obermeier, K.; Taylor, K.; Morgan, L.; Burmi, R.; Barrow, D.; Nicholson, R.I. Tamoxifen resistance in MCF7 cells promotes EMT-like behavior and involves modulation of β-catenin phosphorylation. Int. J. Cancer 2006, 118, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.W.; Sun, X.F.; Guo, X.H.; He, Y.N.; Cui, S.D.; Fan, Q.X. Tamoxifen-resistant breast cancer cells possess cancer stem-like cell properties. Chin. Med. J. 2013, 126, 3030–3034. [Google Scholar] [PubMed]
- Oliveras-Ferraros, C.; Corominas-Faja, B.; Cufi, S.; Vazquez-Martin, A.; Martin-Castillo, B.; Iglesias, J.M.; López-Bonet, E.; Martin, Á.G.; Menendez, J.A. Epithelial-to mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin). Cell Cycle 2012, 11, 4020–4032. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 activates Wnt/β-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol. Cancer Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Mego, M.; Mani, S.A.; Lee, B.N.; Li, C.; Evans, K.W.; Cohen, E.N.; Gao, H.; Jackson, S.A.; Giordano, A.; Hortobagyi, G.N.; et al. Expression of epithelial-mesenchymal transition-inducing transcription factors in primary breast cancer: The effect of neoadjuvant therapy. Int. J. Cancer 2012, 130, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Derynck, R. Specificity and versatility in TGF-β signaling though Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signaling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.; Moustakas, A. Role of Smads in TGFβ signaling. Cell Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Lin, X.; Chiu, W.T.; Chen, Y.H.; Yu, G.; Liu, M.; Feng, X.H.; Sawaya, R.; Medema, R.H.; Hung, M.C.; et al. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-β-dependent cancer metastasis. J. Clin. Invest. 2014, 124, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Chen, C.R.; Massagué, J. A self-enabling TGFβ response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol. Cell 2003, 11, 915–926. [Google Scholar] [CrossRef]
- Wang, D.; Lu, P.; Zhang, H.; Luo, M.; Zhang, X.; Wei, X.; Gao, J.; Zhao, Z.; Liu, C. Oct-4 and Nanog promote the epithelial-mesenchymal transition of breast cancer stem cells and are associated with poor prognosis in breast cancer patients. Oncotarget 2014, 5, 10803–10815. [Google Scholar] [CrossRef] [PubMed]
- Yook, J.I.; Li, X.Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A Wnt-Axin2-GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Gilles, C.; Polette, M.; Mestdagt, M.; Nawrocki-Raby, B.; Ruggeri, P.; Birembaut, P.; Foidart, J.M. Transactivation of Vimentin by β-catenin in human breast cancer cells. Cancer Res. 2003, 63, 2658–2664. [Google Scholar] [PubMed]
- Li, Y.; Ma, C.; Shi, X.; Wen, Z.; Li, D.; Sun, M.; Ding, H. Effect of nitric oxide synthase on multiple drug resistance is related to Wnt signaling in non-small cell lung cancer. Oncol. Rep. 2014, 32, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Gheidari, F.; Bakhshandeh, B.; Teimoori-Toolabi, L.; Mehrtash, A.; Ghadir, M.; Zeinali, S. TCF4 silencing sensitizes the colon cancer cell line to oxaliplatin as a common chemotherapeutic drug. Anticancer Drugs 2014, 25, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.N.; Hedditch, E.L.; Baker, L.A.; Jary, E.; Ward, R.L.; Ford, C.E. The Wnt signaling pathway is upregulated in an in vitro model of acquired tamoxifen resistant breast cancer. BMC Cancer 2013, 13, 1471–2407. [Google Scholar]
- Reedijk, M.; Odorcic, S.; Chang, L.; Zhang, H.; Miller, N.; McCready, D.R.; Lockwood, G.; Egan, S.E. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005, 65, 8530–8537. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The Role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Das, T.P.; Damodaran, C. Silencing NOTCH signaling causes growth arrest in both breast cancer stem cells and breast cancer cells. Br. J. Cancer 2013, 109, 2587–2596. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Bio. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Emergence of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin axis in transforming growth factor-β-induced epithelial-mesenchymal transition. Cells Tissues Organs 2011, 193, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-β—Induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Lamouille1, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk pathway is required for TGF-β-induced EMT in vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-β. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landström, M. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–11207. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Giehl, N.; Wu, Y.; Vadgama, J.V. STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. Int. J. Oncol. 2014, 44, 403–411. [Google Scholar] [PubMed]
- Wendt, M.K.; Smith, J.A.; Schiemann, W.P. Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene 2010, 29, 6485–6498. [Google Scholar] [CrossRef] [PubMed]
- Shirakihara, T.; Horiguchi, K.; Miyazawa, K.; Ehata, S.; Shibata, T.; Morita, I.; Miyazono, K.; Saitoh, M. TGF-β regulates isoform switching of FGF receptors and epithelial-mesenchymal transition. EMBO J. 2011, 30, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Hay, E.D.; Goodenough, D.A. Cooperation between snail and LEF-1 transcription factors is essential for TGF-β1-induced epithelial-mesenchymal transition. Mol. Biol Cell 2006, 17, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J. Clin. Invest. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.; Stadler, S.C. Role of epigenetic mechanisms in epithelial-to-mesenchymal transition of breast cancer cells. Transl. Res. 2015, 165, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Esteller, M. How epigenetics can explain human metastasis: A new role for microRNAs. Cell Cycle 2009, 8, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shang, Y. Epigenetic control of epithelial-to-mesenchymal transition and cancer metastasis. Exp. Cell Res. 2013, 319, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Herman, J.G.; Lapidus, R.G.; Chopra, H.; Xu, R.; Jarrard, D.F.; Isaacs, W.B.; Pitha, P.M.; Davidson, N.E.; Baylin, S.B. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995, 55, 5195–5199. [Google Scholar] [PubMed]
- Yoshiura, K.; Kanai, Y.; Ochiai, A.; Shimoyama, Y.; Sugimura, T.; Hirohashi, S. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc. Natl. Acad. Sci. USA 1995, 92, 7416–7419. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Gabrielson, E.; Fujii, H.; Baylin, S.B.; Herman, J.G. Methylation patterns of the E-cadherin 5′-CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J. Biol. Chem. 2000, 275, 2727–2732. [Google Scholar] [CrossRef] [PubMed]
- Ulirsch, J.; Fan, C.; Knafl, G.; Wu, M.J.; Coleman, B.; Perou, C.M.; Swift-Scanlan, T. Vimentin DNA methylation predicts survival in breast cancer. Breast Cancer Res. Treat. 2013, 137, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.M.; Chen, M.W.; Chen, C.A.; Chien, M.H.; Hua, K.T.; Hsiao, M.; Kuo, M.L.; Wei, L.H. The H3K9 Methyltransferase G9a Represses E-cadherin and is Associated with Myometrial Invasion in Endometrial Cancer. Ann. Surg. Oncol. 2015, 3, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Invest. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Bolós, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Hajra, K.M.; Chen, D.Y.; Fearon, E.R. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002, 62, 1613–1618. [Google Scholar] [PubMed]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Bullock, M.D.; Sayan, A.E.; Packham, G.K.; Mirnezami, A.H. MicroRNAs: Critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression. Biol. Cell 2012, 104, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.A.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A.; et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009, 23, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumor invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Deng, H.; Bi, F.; Liu, J.; Bemis, L.T.; Norris, D.; Wang, X.J.; Zhang, Q. MicroRNA-137 targets carboxyl-terminal binding protein 1 in melanoma cell lines. Int. J. Biol. Sci. 2011, 7, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.D.; Lv, M.M.; Chen, W.X.; Zhong, S.L.; Zhang, X.H.; Chen, L.; Ma, T.F.; Tang, J.H.; Zhao, J.H. Role of miR-155 in drug resistance of breast cancer. Tumor Biol. 2015, 36, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Mattiske, S.; Suetani, R.J.; Neilsen, P.M.; Callen, D.F. The oncogenic role of miR-155 in breast cancer. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Neilsen, P.M.; Noll, J.E.; Mattiske, S.; Bracken, C.P.; Gregory, P.A.; Schulz, R.B.; Lim, S.P.; Kumar, R.; Suetani, R.J.; Goodall, G.J. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene 2013, 32, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Yang, H.; He, L.; Zhao, J.J.; Coppola, D.; Dalton, W.S.; Cheng, J.Q. MicroRNA-155 is regulated by the transforming growth factor β/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol. Cell Biol. 2008, 28, 6773–6784. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Berg, T.; Kurzejamska, E.; Pang, M.F.; Tabor, V.; Jansson, M.; Roswall, P.; Pietras, K.; Sund, M.; Religa, P. MiR-155-mediated loss of C/EBPβ shifts the TGF-β response from growth inhibition to epithelial-mesenchymal transition, invasion and metastasis in breast cancer. Oncogene 2013, 32, 5614–5624. [Google Scholar] [CrossRef] [PubMed]
- Eades, G.; Yao, Y.; Yang, M.; Zhang, Y.; Chumsri, S.; Zhou, Q. miR-200a regulates SIRT1 expression and epithelial to mesenchymal transition (EMT)-like transformation in mammary epithelial cells. J. Biol. Chem. 2011, 286, 25992–26002. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.S. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 2007, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E. MALAT1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.H.; Spieker, T.; Koschmieder, S.; Schäffers, S.; Humberg, J.; Jungen, D.; Bulk, E.; Hascher, A.; Wittmer, D.; Marra, A.; et al. The long noncoding MALAT1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J. Thorac. Oncol. 2011, 6, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, Y.; Nie, L.; Gui, Y.; Cai, Z. Inducing cell proliferation inhibition, apoptosis, and motility reduction by silencing long noncoding ribonucleic acid metastasis-associated lung adenocarcinoma transcript 1 in urothelial carcinoma of the bladder. Urology 2013, 81, e201–e207. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Chen, Q.; Wang, Y.; Zhou, Z.; Huang, Y.; Qiu, F. Upregulated MALAT1 contributes to bladder cancer cell migration by inducing epithelial-to-mesenchymal transition. Mol. Biosyst. 2012, 8, 2289–2294. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yang, M.; Tian, J.; Wang, X.; Li, Z. MALAT1: A long non-coding RNA and its important 3′ end functional motif in colorectal cancer metastasis. Int. J. Oncol. 2011, 39, 169–175. [Google Scholar] [PubMed]
- Gutschner, T.; Hämmerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. 2013, 91, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Guffanti, A.; Iacono, M.; Pelucchi, P.; Kim, N.; Solda, G.; Croft, L.J.; Taft, R.J.; Rizzi, E.; Askarian-Amiri, M.; Bonnal, R.J. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics 2009, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.S.; Ino, Y.; Kanai, Y.; Sakamoto, M.; Hirohashi, S. 5-aza-2′-deoxycytidine restores the E-cadherin system in E-cadherin-silenced cancer cells and reduces cancer metastasis. Clin. Exp. Metastasis. 2004, 21, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar]
- Kelly, T.K.; de Carvalho, D.D.; Jones, P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Braiteh, F.; Soriano, A.O.; Garcia-Manero, G.; Hong, D.; Johnson, M.M.; de Padua Silva, L.; Yang, H.; Alexander, S.; Wolff, J.; Kurzrock, R. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin. Cancer Res. 2008, 14, 6296–6301. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Gilbert, J.; Rudek, M.A.; Zwiebel, J.A.; Gore, S.; Jiemjit, A.; Zhao, M.; Baker, S.D.; Ambinder, R.F.; Herman, J.G.; et al. A phase I dose-finding study of 5-azacytidine in combination with sodium phenylbutyrate in patients with refractory solid tumors. Clin. Cancer Res. 2009, 15, 6241–6249. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Marchion, D.; Thomas, S.; Egorin, M.; Minton, S. Phase I trial of vorinostat and doxorubicin in solid tumours: Histone deacetylase 2 expression as a predictive marker. Br. J. Cancer. 2009, 10, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Bonnie, E.; Rothberg, G.; Bracken, M.B. E-cadherin immunohistochemical expression as a prognostic factor in infiltrating ductal carcinoma of the breast: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2006, 100, 139–148. [Google Scholar]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/β-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Goyalm, A.; ChB, M.B.; Watkins, G.; Kiang, W.G. Expression of the transcription factors Snail, Slug, and Twist and their clinical significance in human breast cancer. Ann. Surg. Oncol. 2005, 12, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Mylona, E.; Giannopoulou, I.; Fasomytakis, E.; Nomikos, A.; Magkou, C.; Bakarakos, P.; Nakopoulou, L. The clinicopathologic and prognostic significance of CD44+/CD24−/low and CD44−/CD24+ tumor cells in invasive breast carcinomas. Hum. Pathol. 2008, 39, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Gwak, J.M.; Kim, H.J.; Kim, E.J.; Chung, Y.R.; Yun, S.; Seo, A.N.; Lee, H.J.; Park, S.Y. MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast cancer. Breast Cancer Res. Treat. 2014, 147, 39–49. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epithelial-Mesenchymal Transition and Breast Cancer. J. Clin. Med. 2016, 5, 13. https://doi.org/10.3390/jcm5020013
Wu Y, Sarkissyan M, Vadgama JV. Epithelial-Mesenchymal Transition and Breast Cancer. Journal of Clinical Medicine. 2016; 5(2):13. https://doi.org/10.3390/jcm5020013
Chicago/Turabian StyleWu, Yanyuan, Marianna Sarkissyan, and Jaydutt V. Vadgama. 2016. "Epithelial-Mesenchymal Transition and Breast Cancer" Journal of Clinical Medicine 5, no. 2: 13. https://doi.org/10.3390/jcm5020013