
Helicobacter pylori Infection and Chronic Immune Thrombocytopenia


Department of Medical Laboratory Sciences, Health and Sciences, International University of Health and Welfare Graduate School, 4-3 Kouzunomori, Narita-city 286-8686, Japan
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2022, 11(16), 4822; https://doi.org/10.3390/jcm11164822
Submission received: 11 July 2022
/
Revised: 15 August 2022
/
Accepted: 15 August 2022
/
Published: 17 August 2022
(This article belongs to the Special Issue New Approaches to the Diagnosis and Management of Anemia and Malaria)
{kind=link}
Abstract
:Approximately half of the world’s population is infected with Helicobacter pylori, which causes gastric disease. Recent systematic reviews and meta-analyses have reported that H. pylori may also have extragastric manifestations such as hematologic diseases, including chronic immune thrombocytopenia (cITP). However, the molecular mechanisms by which H. pylori induces cITP remain unclear, and may involve the host immune response, bacterial strain diversity, and delivery of bacterial molecules to the host blood vessels. This review discusses the important pathophysiological mechanisms by which H. pylori potentially contributes to the development of cITP in infected patients.
1. Introduction
Helicobacter pylori is a gram-negative spiral pathogenic bacterium that mainly causes upper gastrointestinal diseases such as inflammation, hyperplastic polyp, ulcer, and cancer [1,2]. Approximately half of the world’s population is estimated to have been infected with H. pylori at some point; typically, the infection occurs in childhood, and persistent, lifelong infection of the stomach can occur if left untreated [3]. Epidemiological research has documented that bacterial virulence factors differ between Western and East Asian strains of H. pylori, and the East Asian strains are thought to be more highly pathogenic [4]. Pathogenic H. pylori is highly genetically diverse and adapts to survive in the microenvironments/conditions present in different anatomical parts of individual patients’ stomachs [5]. Furthermore, H. pylori bacteriophages (phages) also promote the development of a flexible H. pylori community with variable characteristics [6]. Thus, the H. pylori population within the stomach is continuously changing in response to changes in conditions, which contributes to persistent infection and the development of considerable biological polymorphism [5].
According to many reports, H. pylori also causes extragastric disease, including blood diseases such as iron deficiency anemia, vitamin B12 deficiency, and chronic immune thrombocytopenia (cITP), as well as other diseases such as metabolic syndrome, diabetes, non-alcoholic fatty liver disease, Alzheimer’s disease, neurologic disease, skin disease, and cardiovascular disease [7,8,9,10]. The cardiovascular diseases caused by H. pylori, including atherosclerosis and acute coronary syndrome, develop because of vessel damage and the appearance of plaques due to inflammatory cytokine production and platelet activation following infection [9,11]. Eradication therapy for H. pylori infection has been covered by insurance since 2013 in Japan, which was the first country in the world to do this [12], and since then the insurance coverage has been expanded to include treatment of H. pylori-induced extragastric diseases such as cITP. This review focuses on the relationship between H. pylori infection and cITP. Relevant papers were retrieved by searching the MEDLINE/PubMed database using the terms “Helicobacter”, “platelet”, and “ITP/thrombocytopenia.”
2. Immune Thrombocytopenia and H. pylori Infection
Immune thrombocytopenia (ITP), an acquired autoimmune disease, is characterized by the presence of antiplatelet antibodies that lead to platelet destruction and a reduced platelet count (thrombocytopenia, <100 × 109/L). ITP can be a primary disease or can develop as a secondary etiology, such as infection with microorganisms including H. pylori and others [13,14]. Thrombocytopenia that is continuously observed for at least 12 months is referred to as chronic ITP (cITP). cITP is typically treated with immunosuppressant therapy, and splenectomy can be performed if medical treatment is unsuccessful. However, how antiplatelet antibodies are produced and whether the antibodies trigger the development of cITP remain unclear. Gasbarrini et al. first reported that H. pylori eradication therapy improved the platelet count in H. pylori-positive cITP patients (H. pylori-associated cITP), suggesting a pathophysiological relationship between H. pylori infection and cITP [15]. Since then, studies have shown that eradication therapy partially or completely restores the platelet count in more than 50% of H. pylori-infected patients with cITP, with some geographical/regional variability [16,17,18]. Especially, the rate of effective response to eradication therapy on cITP is approximately more than 80% in Middle East region/Mediterranean [19]. In H. pylori-infected cITP patients, the effective response to eradication therapy is relatively high in Middle East, Asia, and Europe, and low in North America. A study involving North American cITP patients demonstrated that H. pylori-infected Hispanic cITP patients responded more effectively to H. pylori eradication than did white, non-Hispanic cITP patients. Current guidelines in multiple countries recommend H. pylori eradication in H. pylori-infected cITP patients who are unresponsive to traditional treatment for cITP. However, more pathophysiological research is needed to elucidate the etiology and pathophysiological mechanisms by which H. pylori infection influences the development of cITP.
3. Mechanistic Pathway of ITP Development
3.1. Molecular Mimicry and Cross-Reaction
Human immuno-complexity, which involves the interactions among infectious agents, environmental and hormonal factors, inflammation, and the host immune response, is well known to trigger autoimmunity by different pathways. Previous reports have shown that various autoimmune disorders are most likely associated with H. pylori, including cITP [8,20,21,22,23]. The mechanistic pathways by which H. pylori promotes cITP development are postulated to include molecular mimicry, increased plasmacytoid dendritic cell count, and the host immune response to virulence factors such as vacuolating-associated cytotoxin gene A (VacA) and cytotoxin-associated gene A (CagA). Michel et al. performed antibody profiling with platelets from three H. pylori-infected ITP patients and found no H. pylori-specific antibodies. However, they did find autoantibodies to platelet surface glycoproteins (IIb/IIIa or Ib) that did not directly react with H. pylori molecules such as CagA, VacA, UreB, HspA, FsB, FlaA, and UreA [24]. Since then, additional studies have shown that the molecular mimicry between H. pylori-derived molecules as CagA and VacA, and platelet surface glycoproteins (IIb/IIIa or Ib) is responsible for cITP induction [25,26,27,28,29]. The anti-CagA and/or anti-VacA antibodies react to platelets (cross-reaction), leading to platelet aggregation and destruction [26,28,29]. These platelet-associated IgG (PAIgG) antibodies derived from H. pylori infection are predictive of platelet recovery after H. pylori eradication, suggesting that antibody titers could be used as a marker to determine the effectiveness of H. pylori eradication in H. pylori-infected cITP patients [27]. In particular, H. pylori CagA was identified an effector molecule that translocates into host gastric epithelial cells via a type IV secretion system, and has also been discovered in exosomes isolated from blood samples [30], indicating that it can directly affect organs (tissues and cells), including platelets. Future studies should focus on bacterial components encapsulated not only in exosomes but also in outer membrane vesicles (OMVs) released from bacteria to clarify the pathophysiological mechanisms of H. pylori-associated disorders including cITP.
The evidence for cross-reaction based on molecular mimicry described above is not sufficient to exactly explain the pathophysiological pathway by which H. pylori causes cITP. Studies have shown that the titers of antibodies such as anti-CagA antibodies in H. pylori-associated cITP patients who are responsive to eradication therapy (responders) decrease, but do not reach the negative level, within the few weeks following therapy. In addition, decreased antibody titers are frequently observed even in non-responders. Interestingly, in our cases and in previous reports, an increased platelet count is observed in responders in the first few weeks following eradication therapy [31]. In addition, even in responders with unsuccessful eradication therapy, the platelet count temporarily increases and then returns to its original level, most likely because of numbers of bacteria grown again in the stomach. These clinical manifestations are difficult to explain based on antibody cross-reaction pathways. Thus, the platelet response should be monitored on a weekly basis following eradication therapy to quantitatively determine temporal fluctuations in platelet count and antigen and antibody levels.
Two recent studies proposed an alternative pathway for cITP induction by H. pylori in which an immunocomplex forms comprising the H. pylori antigen Lpp20, H. pylori-specific antibodies, and platelets [32,33]. These studies demonstrated that the H. pylori-specific antibodies (anti-Lpp20 antibodies) were present at significantly higher levels in responders compared with in non-responders and in H. pylori-uninfected cITP patients. The anti-Lpp20 antibodies reacted to H. pylori Lpp20 but not platelets; however, Lpp20 can bind to platelets, resulting in immunocomplex formation and platelet destruction. This alternative pathway could be responsible for the clinical manifestations mentioned above, as H. pylori-specific platelet-binding antigens could decrease quickly and temporally even in responders with unsuccessful eradication therapy. Lpp20 is released from H. pylori and may have high antigenicity [34,35]. It is typically contained in membranous vesicles or masked by the bacterial outer membrane, so anti-Lpp20 antibodies are not always produced in H. pylori-infected patients [36]. However, significant levels of anti-Lpp20 antibodies are detected in patients with H. pylori-associated cITP, implying that Lpp20 triggers an immune response in certain H. pylori-infected patients. This indicates that an individual’s immune response to H. pylori Lpp20 may be involved in the development of H. pylori-associated cITP. Thus, future studies should focus on genetic and strain diversities, as well as the variation in H. pylori communities within individual stomachs, and microparticles (exosomes and OMVs) should be analyzed to elucidate the pathophysiological etiologies of H. pylori-associated cITP (Figure 1).
3.2. Platelet Activation and Aggregation
Lpp20 directly binds to platelets, eventually leading to platelet activation and aggregation. Some H. pylori strains induce platelet activation and aggregation, resulting in platelet destruction [23,31,37]. The platelet activation/aggregation pathway is thought to involve interactions between anti-H. pylori IgG and IgG receptors (FcgRIIA) on platelets, as well as between von Willebrand factor (vWf) on H. pylori and platelet surface glycoproteins Ib/IX (gp-Ib/IX (CD42)). Consequently, the interaction between vWf and gp-Ib causes activation of gp-IIb/IIIa (CD41/CD61) and irreversible binding of vWf to platelets [37]. The occurrence of this type of H. pylori-induced platelet aggregation in the gastric microvasculature and/or blood vessel may lead to the development of systemic disorders such as cardiovascular diseases. Previous reports have demonstrated a correlation between H. pylori infection and coronary artery diseases including atherosclerosis, dyslipidemia, and thrombosis [8,22,38]. Platelet aggregation prompts the formation of atherosclerotic lesions, which is accompanied by multiple events such as plaque formation, inflammatory cytokine release, damaged endothelial cells, fibrous cap formation, and thrombosis [9,38,39]. H. pylori-specific DNA was observed in atherosclerotic plaques in severe cardiovascular disease patients [40], and H. pylori CagA encapsulated in exosomes was discovered in blood samples [30]. In addition, H. pylori infection is associated with increased levels of the exosome-associated miRNA miR-25, even in peripheral blood, and this miRNA is known to regulate the nuclear factor (NF)-κB signaling pathway [41]. Taken together, these findings indicate that H. pylori infection may be involved in the development of cardiovascular diseases and propose a pathway by which delivery of as yet unidentified H. pylori molecules to the blood vessels trigger the induction of these diseases though platelet aggregation and dysfunction, endothelial damage, and other processes [41].
Interestingly, the mechanical pathway described, based on platelet activation and aggregation, does not involve CagA or VacA [42]. So far, the exact H. pylori molecules that are responsible for platelet activation and aggregation have not been identified. Lpp20 was recently discovered to directly bind to platelets and eventually lead to platelet activation and aggregation, suggesting that it may promote thrombosis in H. pylori-infected patients with the absence of anti-Lpp20 antibodies. More research is needed to evaluate whether and how this molecule triggers and/or enhances the induction of H. pylori-associated disorders.
3.3. Host Immune Response to H. pylori Infection
Another potential mechanism for H. pylori-associated cITP is inhibition of Fcγ receptors on monocytes, which eventually results in increased antiplatelet antibodies and platelet turnover by reducing FcγRIIB levels. Reduced FcγRIIB production and the presence of autoreactive B cells increase the phagocytic activity of monocytes, leading to decreased platelet counts [43,44]. In addition, the number of plasmacytoid dendritic cells, a type of antigen-presenting cells, was found to be increased in patients with H. pylori-associated cITP [45,46]. In gastric epithelium infected with H. pylori, dendritic cells enhance host immune response with an influx of T cells and induce IL-12 and IL-10 release. Furthermore, H. pylori outer membrane proteins (Omp) such as HpaA and Omp 18 proteins stimulate the production of IL-12 and IL-10 by dendritic cells [46,47]. In addition to CagA and VacA, other H. pylori molecules such as outer inflammatory protein A (OipA), blood group antigen-binding adhesion A (BabA), sialic acid-binding adhesion (SabA), and picB have also been reported to be involved in H. pylori-associated disorders based on the host immune response to H. pylori infection [23,48]. Tyrosine phosphorylation of the Glu-Pro-Ile-Tyr-Ala (EPIYA) motif in CagA is a strong antigenic factor that induces IL-8- and NF-κB-mediated inflammation, as well as the production of anti-CagA antibodies, which cross-react with platelets, as described above [49]. VacA suppresses helper T-cells via interruption of the T-cell receptor IL-2 pathway [50]. VacA also binds to multimerin-1 and elastin microfibril interfacer 4 on megakaryocytes and platelets, respectively, and enhances platelet activation and clearance [28]. Taken together, these findings suggest that these host immune response pathways play a role in the pathophysiological progression of H. pylori-associated cITP.
4. Conclusions
Even though the exact mechanism remains unclear, multiple reviews and meta-analyses regarding H. pylori-associated extragastric disorders, including cITP, have outlined the broad pathophysiological mechanisms by which H. pylori directly and/or indirectly contributes to the development of cITP. Guidelines from different countries around the world, including Japan, agree that cITP is an extragastric manifestation of H. pylori infection, and that eradication therapy is recommended [51,52,53,54]. In fact, many cases of platelet count recovery after eradication therapy (responders) have been reported in different countries. On the other hand, some patients do not experience platelet recovery after successful eradication therapy (non-responders). At present, little information is available regarding how to predict which patients will be responders and which will be non-responders. The H. pylori protein Lpp20 and anti-Lpp20 antibodies may be useful markers in this context. More research is needed to identify predictive markers by obtaining a more accurate understanding of the biological and immunological events that occur in the H. pylori-infected host. Furthermore, the clinical manifestations of cITP in H. pylori-infected patients, including platelet activation/aggregation, appear to be dependent on H. pylori strain variation. Thus, more attention should be paid to the diversity of H. pylori strains in individual patients’ stomachs, as well as ongoing changes in the composition of the H. pylori population in the stomach. Finally, H. pylori molecules have been detected encapsulated in serum-derived extracellular vesicles such as exosomes and OMVs, indicating that they can directly and functionally interact with multiple organs/tissues/cells outside the stomach. Thus, there are multiple promising avenues for future research regarding the pathophysiological etiologies of H. pylori-associated cITP.
Author Contributions
Conceptualization, writing—original draft preparation, writing—review and editing, funding acquisition, H.T.; data curation, visualization, A.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by JSPS KAKENHI, grant number 20K07857.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fischbach, W.; Malfertheiner, P. Helicobacter pylori Infection. Dtsch. Arztebl. Int. 2018, 115, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Zhang, W.; Huang, Y.; Wang, Y.; Shao, Q.; Wu, X.; Lu, N.; Xie, C. Effect of Helicobacter pylori eradication on hyperplastic gastric polyps: A systematic review and meta-analysis. Helicobacter 2021, 26, e12838. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Lee, H.; Kang, M.; Kim, J.E.; Choi, Y.-H.; Min, Y.W.; Min, B.-H.; Lee, J.H.; Son, H.J.; Rhee, P.-L.; et al. Helicobacter pylori is associated with dyslipidemia but not with other risk factors of cardiovascular disease. Sci. Rep. 2016, 6, 38015. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Fujita, A.; Yamazaki, S.; Asaka, M.; Azuma, T.; Hatakeyama, M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc. Natl. Acad. Sci. USA 2002, 99, 14428–14433. [Google Scholar] [CrossRef]
- Ailloud, F.; Didelot, X.; Woltemate, S.; Pfaffinger, G.; Overmann, J.; Bader, R.C.; Schulz, C.; Malfertheiner, P.; Suerbaum, S. Within-host evolution of Helicobacter pylori shaped by niche-specific adaptation, intragastric migrations and selective sweeps. Nat. Commun. 2019, 10, 2273. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kira, M.; Konishi, S.; Uchiyama, J.; Matsuzaki, S.; Matsumura, Y. Polymorphisms in the Helicobacter pylori NY43 strain and its prophage-cured derivatives. Microbiology 2018, 164, 877–882. [Google Scholar] [CrossRef]
- De Korwin, J.-D.; Ianiro, G.; Gibiino, G.; Gasbarrini, A. Helicobacter pylori infection and extragastric diseases in 2017. Helicobacter 2017, 22 (Suppl. S1), e12411. [Google Scholar] [CrossRef]
- Pellicano, R.; Ianiro, G.; Fagoonee, S.; Settanni, C.R.; Gasbarrini, A. Review: Extragastric diseases and Helicobacter pylori. Helicobacter 2020, 25 (Suppl. S1), e12741. [Google Scholar] [CrossRef]
- Qiang, L.; Hu, J.; Tian, M.; Li, Y.; Ren, C.; Deng, Y.; Jiang, Y. Extracellular vesicles from Helicobacter pylori-infected cells and Helicobacter pylori outer membrane vesicles in atherosclerosis. Helicobacter 2022, 27, e12877. [Google Scholar] [CrossRef]
- Franceschi, F.; Zuccalà, G.; Roccarina, D.; Gasbarrini, A. Clinical effects of Helicobacter pylori outside the stomach. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 234–242. [Google Scholar] [CrossRef]
- Lee, M.; Baek, H.; Park, J.S.; Kim, S.; Kyung, C.; Baik, S.J.; Lee, B.K.; Kim, J.-H.; Ahn, C.W.; Kim, K.R.; et al. Current Helicobacter pylori infection is significantly associated with subclinical coronary atherosclerosis in healthy subjects: A cross-sectional study. PLoS ONE 2018, 13, e0193646. [Google Scholar] [CrossRef]
- Yokota, K.; Osaki, T.; Hayashi, S.; Yokota, S.; Takeuchi, H.; Rimbara, E.; Ojima, H.; Sato, T.; Yonezawa, H.; Shibayama, K.; et al. Establishment of a reference panel of Helicobacter pylori strains for antimicrobial susceptibility testing. Helicobacter 2022, 27, e12874. [Google Scholar] [CrossRef]
- Neunert, C.E. Current management of immune thrombocytopenia. Hematology 2013, 2013, 276–282. [Google Scholar] [CrossRef]
- Semple, J.W.; Rebetz, J.; Maouia, A.; Kapur, R. An update on the pathophysiology of immune thrombocytopenia. Curr. Opin. Hematol. 2020, 27, 423–429. [Google Scholar] [CrossRef]
- Gasbarrini, A.; Franceschi, F.; Tartaglione, R.; Landolfi, R.; Pola, P.; Gasbarrini, G. Regression of autoimmune thrombocytopenia after eradication of Helicobacter pylori. Lancet 1998, 352, 878. [Google Scholar] [CrossRef]
- O’Neill, C.M.; Weitz, I.C.; O’Connell, C.; Liebman, H.A. Ethnic and racial difference in Helicobacter pylori infection in patients with immune thrombocytopenia treated at a major urban medical center. Platelets 2019, 30, 413–417. [Google Scholar] [CrossRef]
- Frydman, G.H.; Davis, N.; Beck, P.L.; Fox, J.G. Helicobacter pylori Eradication in Patients with Immune Thrombocytopenic Purpura: A Review and the Role of Biogeography. Helicobacter 2015, 20, 239–251. [Google Scholar] [CrossRef]
- Vishnu, P.; Duncan, J.; Connell, N.; Cooper, N.; Lim, W.; Rodeghiero, F.; Tomiyama, Y.; Grace, R.F.; Bakchoul, T.; Arnold, D.M.; et al. International survey on Helicobacter pylori testing in patients with immune thrombocytopenia: Communication of the platelet immunology scientific and standardization committee. J. Thromb. Haemost. 2021, 19, 287–296. [Google Scholar] [CrossRef]
- Pezeshki, S.M.S.; Saki, N.; Ghandali, M.V.; Ekrami, A.; Avarvand, A.Y. Effect of Helicobacter pylori eradication on patients with ITP: A meta-analysis of studies conducted in the Middle East. Blood Res. 2021, 56, 38–43. [Google Scholar] [CrossRef]
- Amedei, A.; Bergman, M.P.; Appelmelk, B.J.; Azzurri, A.; Benagiano, M.; Tamburini, C.; van der Zee, R.; Telford, J.L.; Vandenbroucke-Grauls, C.M.J.E.; D’Elios, M.M.; et al. Molecular Mimicry between Helicobacter pylori Antigens and H+,K+–Adenosine Triphosphatase in Human Gastric Autoimmunity. J. Exp. Med. 2003, 198, 1147–1156. [Google Scholar] [CrossRef]
- Faller, G.; Winter, M.; Steininger, H.; Lehn, N.; Meining, A.; Bayerdörffer, E.; Kirchner, T. Decrease of Antigastric Autoantibodies in Helicobacter pylori Gastritis after Cure of Infection. Pathol. Res. Pract. 1999, 195, 243–246. [Google Scholar] [CrossRef]
- Wang, L.; Cao, Z.-M.; Zhang, L.-L.; Dai, X.-C.; Liu, Z.-J.; Zeng, Y.-X.; Li, X.-Y.; Wu, Q.-J.; Lv, W.-L. Helicobacter pylori and Autoimmune Diseases: Involving Multiple Systems. Front. Immunol. 2022, 13, 833424. [Google Scholar] [CrossRef] [PubMed]
- Ihtesham, A.; Maqbool, S.; Nadeem, M.; Janjua, M.B.A.; Sundus, O.; Naqqash, A.B.; Mohamed, W.I.; Haider, S.T.; Ahmad, M.; Mustafa, M.A.T.; et al. Helicobacter pylori induced Immune Thrombocytopenic Purpura and perspective role of Helicobacter pylori eradication therapy for treating Immune Thrombocytopenic Purpura. AIMS Microbiol. 2021, 7, 284–303. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Khellaf, M.; Desforges, L.; Lee, K.; Schaeffer, A.; Godeau, B.; Bierling, P. Autoimmune Thrombocytopenic Purpura and Helicobacter pylori Infection. Arch. Intern. Med. 2002, 162, 1033–1036. [Google Scholar] [CrossRef]
- Kim, B.J.; Kim, H.S.; Jang, H.J.; Kim, J.H. Helicobacter pylori Eradication in Idiopathic Thrombocytopenic Purpura: A Meta-Analysis of Randomized Trials. Gastroenterol. Res. Pract. 2018, 2018, 6090878. [Google Scholar] [CrossRef]
- Takahashi, T.; Yujiri, T.; Shinohara, K.; Inoue, Y.; Sato, Y.; Fujii, Y.; Okubo, M.; Zaitsu, Y.; Ariyoshi, K.; Nakamura, Y.; et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori -associated chronic idiopathic thrombocytopenic purpura. Br. J. Haematol. 2004, 124, 91–96. [Google Scholar] [CrossRef]
- Kodama, M.; Kitadai, Y.; Ito, M.; Kai, H.; Masuda, H.; Tanaka, S.; Yoshihara, M.; Fujimura, K.; Chayama, K. Immune Response to CagA Protein is Associated with Improved Platelet Count after Helicobacter pylori Eradication in Patients with Idiopathic Thrombocytopenic Purpura. Helicobacter 2007, 12, 36–42. [Google Scholar] [CrossRef]
- Satoh, K.; Hirayama, T.; Takano, K.; Suzuki-Inoue, K.; Sato, T.; Ohta, M.; Nakagomi, J.; Ozaki, Y. VacA, the vacuolating cytotoxin of Helicobacter pylori, binds to multimerin 1 on human platelets. Thromb. J. 2013, 11, 23. [Google Scholar] [CrossRef]
- Franceschi, F.; Christodoulides, N.; Kroll, M.H.; Genta, R.M. Helicobacter pylori and Idiopathic Thrombocytopenic Purpura. Ann. Intern. Med. 2004, 140, 766–767. [Google Scholar] [CrossRef]
- Shimoda, A.; Ueda, K.; Nishiumi, S.; Murata-Kamiya, N.; Mukai, S.-A.; Sawada, S.-I.; Azuma, T.; Hatakeyama, M.; Akiyoshi, K. Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci. Rep. 2016, 6, 18346. [Google Scholar] [CrossRef]
- Jackson, S.; Beck, P.L.; Pineo, G.F.; Poon, M.-C. Helicobacter pylori eradication: Novel therapy for immune thrombocytopenic purpura? A review of the literature. Am. J. Hematol. 2005, 78, 142–150. [Google Scholar] [CrossRef]
- Morimoto, N.; Takeuchi, H.; Takahashi, T.; Ueta, T.; Tanizawa, Y.; Kumon, Y.; Kobayashi, M.; Sugiura, T. Helicobacter pylori-associated chronic idiopathic thrombocytopenic purpura and low molecular weight H. pylori proteins. Scand. J. Infect. Dis. 2007, 39, 409–416. [Google Scholar] [CrossRef]
- Takeuchi, H.; Islam, J.; Kaneko, A.; Kimura, A.; Shida, T.; Oboshi, W.; Katayama, H.; Oishi, T.; Fujieda, M.; Morimoto, N. Helicobacter pylori protein that binds to and activates platelet specifically reacts with sera of H. pylori-associated chronic immune thrombocytopenia. Platelets 2021, 32, 1120–1123. [Google Scholar] [CrossRef]
- Cao, P.; McClain, M.S.; Forsyth, M.H.; Cover, T.L. Extracellular Release of Antigenic Proteins by Helicobacter pylori. Infect. Immun. 1998, 66, 2984–2986. [Google Scholar] [CrossRef]
- Vallese, F.; Mishra, N.M.; Pagliari, M.; Berto, P.; Codolo, G.; de Bernard, M.; Zanotti, G. Helicobacter pylori antigenic Lpp20 is a structural homologue of Tipα and promotes epithelial-mesenchymal transition. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 3263–3271. [Google Scholar] [CrossRef]
- Kostrzynska, M.; O’Toole, P.W.; Taylor, D.E.; Trust, T.J. Molecular characterization of a conserved 20-kilodalton membrane-associated lipoprotein antigen of Helicobacter pylori. J. Bacteriol. 1994, 176, 5938–5948. [Google Scholar] [CrossRef]
- Byrne, M.F.; Kerrigan, S.W.; Corcoran, P.A.; Atherton, J.C.; Murray, F.E.; Fitzgerald, D.J.; Cox, D.M. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology 2003, 124, 1846–1854. [Google Scholar] [CrossRef]
- Riad, M. Association of Helicobacter pylori infection with coronary artery disease: Is it an independent risk factor? Egypt. Heart J. 2021, 73, 61. [Google Scholar] [CrossRef]
- Scopel-Guerra, A.; Olivera-Severo, D.; Staniscuaski, F.; Uberti, A.F.; Callai-Silva, N.; Jaeger, N.; Porto, B.N.; Carlini, C.R. The Impact of Helicobacter pylori Urease upon Platelets and Consequent Contributions to Inflammation. Front. Microbiol. 2017, 8, 2447. [Google Scholar] [CrossRef]
- Kowalski, M.; Rees, W.; Konturek, P.; Grove, R.; Scheffold, T.; Meixner, H.; Brunec, M.; Franz, N.; Konturek, J.; Pieniazek, P.; et al. Detection of Helicobacter pylori specific DNA in human atheromatous coronary arteries and its association to prior myocardial infarction and unstable angina. Dig. Liver Dis. 2002, 34, 398–402. [Google Scholar] [CrossRef]
- Li, N.; Liu, S.-F.; Dong, K.; Zhang, G.-C.; Huang, J.; Wang, Z.-H.; Wang, T.-J. Exosome-Transmitted miR-25 Induced by H. pylori Promotes Vascular Endothelial Cell Injury by Targeting KLF2. Front. Cell. Infect. Microbiol. 2019, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, P.A.; Atherton, J.C.; Kerrigan, S.W.; Wadstrom, T.; Murray, F.E.; Peek, R.M.; Fitzgerald, D.J.; Cox, D.M.; Byrne, M.F. The effect of different strains of Helicobacter pylori on platelet aggregation. Can. J. Gastroenterol. 2007, 21, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Asahi, A.; Nishimoto, T.; Okazaki, Y.; Suzuki, H.; Masaoka, T.; Kawakami, Y.; Ikeda, Y.; Kuwana, M. Helicobacter pylori eradication shifts monocyte Fcγ receptor balance toward inhibitory FcγRIIB in immune thrombocytopenic purpura patients. J. Clin. Investig. 2008, 118, 2939–2949. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, S.; Iizumi, T.; Watanabe, E.; Shimizu, M.; Kamiya, S.; Nagata, K.; Kumagai, Y.; Fukunaga, Y.; Takahashi, H. Implications for Induction of Autoimmunity via Activation of B-1 Cells by Helicobacter pylori Urease. Infect. Immun. 2006, 74, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Yokohama, A.; Osaki, Y.; Ogawa, Y.; Nakahashi, H.; Toyama, K.; Mitsui, T.; Hashimoto, Y.; Koiso, H.; Uchiumi, H.; et al. Circulating plasmacytoid dendritic cells in patients with primary and Helicobacter pylori-associated immune thrombocytopenia. Eur. J. Haematol. 2012, 88, 340–349. [Google Scholar] [CrossRef]
- Appelmelk, B.J.; van Die, I.; van Vliet, S.J.; Vandenbroucke-Grauls, C.M.J.E.; Geijtenbeek, T.B.H.; van Kooyk, Y. Cutting Edge: Carbohydrate Profiling Identifies New Pathogens That Interact with Dendritic Cell-Specific ICAM-3-Grabbing Nonintegrin on Dendritic Cells. J. Immunol. 2003, 170, 1635–1639. [Google Scholar] [CrossRef]
- Voland, P.; Hafsi, N.; Zeitner, M.; Laforsch, S.; Wagner, H.; Prinz, C. Antigenic Properties of HpaA and Omp18, Two Outer Membrane Proteins of Helicobacter pylori. Infect. Immun. 2003, 71, 3837–3843. [Google Scholar] [CrossRef]
- Tummuru, M.K.; Sharma, S.A.; Blaser, M.J. Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Mol. Microbiol. 1995, 18, 867–876. [Google Scholar] [CrossRef]
- Asahi, M.; Azuma, T.; Ito, S.; Ito, Y.; Suto, H.; Nagai, Y.; Tsubokawa, M.; Tohyama, Y.; Maeda, S.; Omata, M.; et al. Helicobacter pylori Caga Protein Can Be Tyrosine Phosphorylated in Gastric Epithelial Cells. J. Exp. Med. 2000, 191, 593–602. [Google Scholar] [CrossRef]
- Holland, R.L.; Bosi, K.D.; Harpring, G.H.; Luo, J.; Wallig, M.; Phillips, H.; Blanke, S.R. Chronic in vivo exposure to Helicobacter pylori VacA: Assessing the efficacy of automated and long-term intragastric toxin infusion. Sci. Rep. 2020, 10, 9307. [Google Scholar] [CrossRef]
- Neunert, C.; Terrell, D.R.; Arnold, D.M.; Buchanan, G.; Cines, D.B.; Cooper, N.; Cuker, A.; Despotovic, J.M.; George, J.N.; Grace, R.F.; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3, 3829–3866. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner, P.; Megraud, F.; O’Morain, C.A.; Gisbert, J.P.; Kuipers, E.J.; Axon, A.T.; Bazzoli, F.; Gasbarrini, A.; Atherton, J.; Graham, D.Y.; et al. Management of Helicobacter pylori infection—The Maastricht V/Florence Consensus Report. Gut 2017, 66, 6–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Jung, H.-K.; Lee, H.L.; Jang, J.Y.; Lee, H.; Kim, C.G.; Shin, W.G.; Shin, E.S.; Lee, Y.C. Guidelines for the diagnosis and treatment of Helicobacter pylori infection in Korea, 2013 revised edition. Korean J. Gastroenterol. 2013, 62, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, H.; Kuwana, M.; Hato, T.; Takafuta, T.; Fujimura, K.; Kurata, Y.; Murata, M.; Tomiyama, Y. Reference guide for management of adult immune thrombocytopenia in Japan: 2019 Revision. Int. J. Hematol. 2020, 111, 329–351. [Google Scholar] [CrossRef]
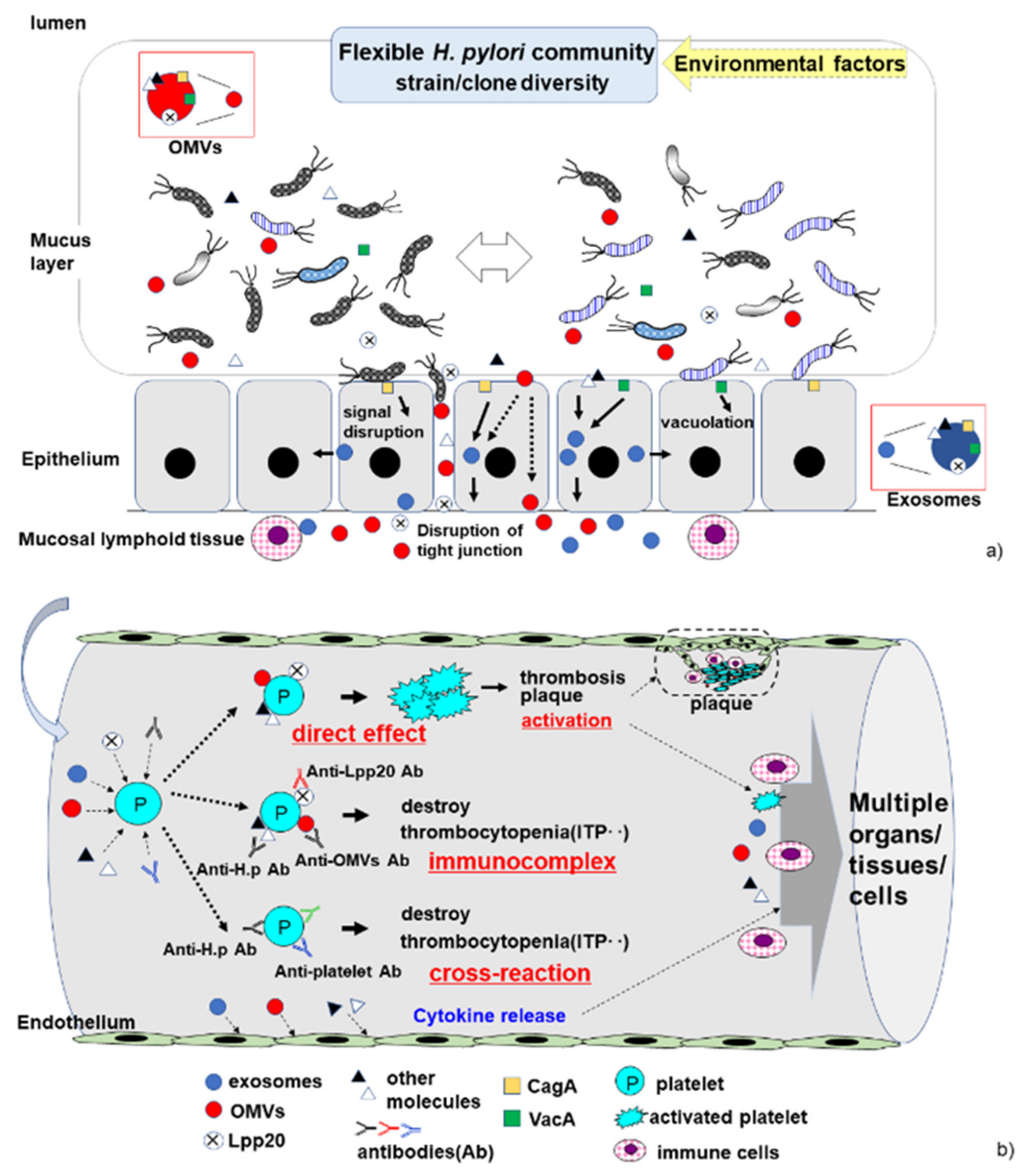
Figure 1.
The continuously changing H. pylori community in the stomach, and proposed pathophysiological pathways by which H. pylori induces cITP. (a) In the stomach, the strain/clone composition of the H. pylori community is highly variable. The bacteria release molecules such as outer membrane vesicles (OMVs) containing pathogenic antigens (i.e., CagA and VacA). Bacterium–epithelial cell interactions can induce host cell damage, disrupt host signal transduction, and lead to the production of exosomes. The bacterial molecules and host cytokines trigger an immune response in mucosal lymphoid tissue. (b) In the vessel, the proposed pathophysiological pathways by which H. pylori induces cITP, including cross-reaction, immunocomplex formation, and direct interaction of H. pylori molecules with platelets in the host blood vessels. Exosomes, OMVs, and molecules released by H. pylori affect platelets and the endothelium in the presence or absence of anti-H. pylori antibodies. These bacterial factors (exosomes, OMVs, Lpp20, and other molecules) may bind directly to platelets, leading to platelet activation. The production of a variety of anti-H. pylori antibodies contributes to platelet destruction via cross-reaction and immunocomplex formation in H. pylori-associated cITP.
Figure 1.
The continuously changing H. pylori community in the stomach, and proposed pathophysiological pathways by which H. pylori induces cITP. (a) In the stomach, the strain/clone composition of the H. pylori community is highly variable. The bacteria release molecules such as outer membrane vesicles (OMVs) containing pathogenic antigens (i.e., CagA and VacA). Bacterium–epithelial cell interactions can induce host cell damage, disrupt host signal transduction, and lead to the production of exosomes. The bacterial molecules and host cytokines trigger an immune response in mucosal lymphoid tissue. (b) In the vessel, the proposed pathophysiological pathways by which H. pylori induces cITP, including cross-reaction, immunocomplex formation, and direct interaction of H. pylori molecules with platelets in the host blood vessels. Exosomes, OMVs, and molecules released by H. pylori affect platelets and the endothelium in the presence or absence of anti-H. pylori antibodies. These bacterial factors (exosomes, OMVs, Lpp20, and other molecules) may bind directly to platelets, leading to platelet activation. The production of a variety of anti-H. pylori antibodies contributes to platelet destruction via cross-reaction and immunocomplex formation in H. pylori-associated cITP.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Takeuchi, H.; Okamoto, A. Helicobacter pylori Infection and Chronic Immune Thrombocytopenia. J. Clin. Med. 2022, 11, 4822. https://doi.org/10.3390/jcm11164822
AMA Style
Takeuchi H, Okamoto A. Helicobacter pylori Infection and Chronic Immune Thrombocytopenia. Journal of Clinical Medicine. 2022; 11(16):4822. https://doi.org/10.3390/jcm11164822
Chicago/Turabian StyleTakeuchi, Hiroaki, and Aoi Okamoto. 2022. "Helicobacter pylori Infection and Chronic Immune Thrombocytopenia" Journal of Clinical Medicine 11, no. 16: 4822. https://doi.org/10.3390/jcm11164822
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.