
Neurofibromatosis Type 1 Has a Wide Spectrum of Growth Hormone Excess

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Endocrine, Radiographic and Genetic Investigations
2.3. Immunohistochemical Analysis
3. Results
3.1. Clinical Studies
3.1.1. Patient 1
3.1.2. Patient 2
3.1.3. Patient 3
3.1.4. Patient 4
3.1.5. Patient 5
3.1.6. Patient 6
3.1.7. Patient 7
3.1.8. Patient 8
3.1.9. Patient 9
3.1.10. Patient 10
3.2. Biochemical Testing
3.3. Genetic Results
3.4. Immunohistochemical Testing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, J.M. Neurofibromatosis 1. In GeneReviews®; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993–2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1109/ (accessed on 20 February 2022).
- Stewart, D.R.; Korf, B.R.; Nathanson, K.L.; Stevenson, D.A.; Yohay, K. Care of adults with neurofibromatosis type 1: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammert, M.; Friedman, J.M.; Kluwe, L.; Mautner, V.F. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch. Dermatol. 2005, 141, 71–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, S.; Thompson, P.; Frayling, I.; Upadhyaya, M. Molecular diagnosis of neurofibromatosis type 1: 2 years experience. Fam. Cancer 2007, 6, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Costin, G.; Fefferman, R.A.; Kogut, M.D. Hypothalamic gigantism. J. Pediatr. 1973, 83, 419–425. [Google Scholar] [CrossRef]
- Bruzzi, P.; Sani, I.; Albanese, A. Reversible Growth Hormone Excess in Two Girls with Neurofibromatosis Type 1 and Optic Pathway Glioma. Horm. Res. Paediatr. 2015, 84, 414–422. [Google Scholar] [CrossRef]
- Hozumi, K.; Fukuoka, H.; Odake, Y.; Takeuchi, T.; Uehara, T.; Sato, T.; Inoshita, N.; Yoshida, K.; Matsumoto, R.; Bando, H.; et al. Acromegaly caused by a somatotroph adenoma in patient with neurofibromatosis type 1. Endocr. J. 2019, 66, 853–857. [Google Scholar] [CrossRef] [Green Version]
- Adeloye, A. Coexistence of acromegaly and neurofibromatosis in a Nigerian. East Afr. Med. J. 1979, 56, 38–39. [Google Scholar]
- Barberis, M.; Gambacorta, M.; Versari, P.; Filizzolo, F. About a case of Recklinghausen’s disease associated with pituitary adenoma (author’s transl). Pathologica 1979, 71, 265–272. [Google Scholar]
- Checa Garrido, A.; del Pozo Pico, C. Acromegaly and type 1 neurofibromatosis. Is association of both conditions due to chance? Endocrinol. Nutr. 2013, 60, 144–145. [Google Scholar] [CrossRef]
- Fuqua, J.S.; Berkovitz, G.D. Growth hormone excess in a child with neurofibromatosis type 1 and optic pathway tumor: A patient report. Clin. Pediatr. 1998, 37, 749–752. [Google Scholar] [CrossRef]
- Josefson, J.; Listernick, R.; Fangusaro, J.R.; Charrow, J.; Habiby, R. Growth hormone excess in children with neurofibromatosis type 1-associated and sporadic optic pathway tumors. J. Pediatr. 2011, 158, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Hannah-Shmouni, F.; Demidowich, A.P.; Rowell, J.; Lodish, M.; Stratakis, C.A. Large pituitary gland with an expanding lesion in the context of neurofibromatosis 1. BMJ Case Rep. 2017, 2017, bcr-2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambiaso, P.; Galassi, S.; Palmiero, M.; Mastronuzzi, A.; Del Bufalo, F.; Capolino, R.; Cacchione, A.; Buonuomo, P.S.; Gonfiantini, M.V.; Bartuli, A.; et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am. J. Med. Genet. A 2017, 173, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, C.; Bottaro, G. Endocrine implications of neurofibromatosis 1 in childhood. Horm. Res. Paediatr. 2015, 83, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Sani, I.; Albanese, A. Endocrine Long-Term Follow-Up of Children with Neurofibromatosis Type 1 and Optic Pathway Glioma. Horm. Res. Paediatr. 2017, 87, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, C.L.; Drohan, L.A.; Sergott, R.C. Optic-nerve gliomas, chiasmal gliomas and neurofibromatosis type 1. Curr. Opin. Ophthalmol. 2006, 17, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, K.; Tabuchi, A.; Ono, Y.; Tamiya, T.; Ohmoto, T.; Furuta, T.; Hamasaki, S. Pituitary adenoma associated with neurofibromatosis type 1: Case report. No Shinkei Geka 2002, 30, 741–745. [Google Scholar] [PubMed]
- Nakajima, M.; Nakasu, Y.; Nakasu, S.; Matsuda, M.; Handa, J. Pituitary adenoma associated with neurofibromatosis: Case report. Nihon Geka Hokan 1990, 59, 278–282. [Google Scholar]
- Pinnamaneni, K.; Birge, S.J.; Avioli, L.V. Prolactin-secreting pituitary tumor associated with von Recklinghausen’s disease. Arch. Intern. Med. 1980, 140, 397–399. [Google Scholar] [CrossRef]
- Pei, L.; Melmed, S. The neurofibromatosis gene in human pituitary adenomas. Endocr. Pathol. 1994, 5, 229–232. [Google Scholar] [CrossRef]
- Manski, T.J.; Haworth, C.S.; Duval-Arnould, B.J.; Rushing, E.J. Optic pathway glioma infiltrating into somatostatinergic pathways in a young boy with gigantism. Case report. J. Neurosurg. 1994, 81, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Duchowny, M.S.; Katz, R.; Bejar, R.L. Hypothalamic mass and gigantism in neurofibromatosis: Treatment with bromocriptine. Ann. Neurol. 1984, 15, 302–304. [Google Scholar] [CrossRef]
- Josefson, J.L.; Listernick, R.; Charrow, J.; Habiby, R.L. Growth Hormone Excess in Children with Optic Pathway Tumors Is a Transient Phenomenon. Horm. Res. Paediatr. 2016, 86, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Hannah-Shmouni, F.; Stratakis, C.A. Growth hormone excess in neurofibromatosis 1. Genet. Med. 2019, 21, 1254–1255. [Google Scholar] [CrossRef] [PubMed]
- Trivellin, G.; Daly, A.F.; Faucz, F.R.; Yuan, B.; Rostomyan, L.; Larco, D.O.; Schernthaner-Reiter, M.H.; Szarek, E.; Leal, L.F.; Caberg, J.H.; et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N. Engl. J. Med. 2014, 371, 2363–2374. [Google Scholar] [CrossRef]
- Beckers, A.; Lodish, M.B.; Trivellin, G.; Rostomyan, L.; Lee, M.; Faucz, F.R.; Yuan, B.; Choong, C.S.; Caberg, J.H.; Verrua, E.; et al. X-linked acrogigantism syndrome: Clinical profile and therapeutic responses. Endocr. Relat. Cancer 2015, 22, 353–367. [Google Scholar] [CrossRef]
- Trivellin, G.; Bjelobaba, I.; Daly, A.F.; Larco, D.O.; Palmeira, L.; Faucz, F.R.; Thiry, A.; Leal, L.F.; Rostomyan, L.; Quezado, M.; et al. Characterization of GPR101 transcript structure and expression patterns. J. Mol. Endocrinol. 2016, 57, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Drimmie, F.M.; MacLennan, A.C.; Nicoll, J.A.; Simpson, E.; McNeill, E.; Donaldson, M.D. Gigantism due to growth hormone excess in a boy with optic glioma. Clin. Endocrinol. 2000, 53, 535–538. [Google Scholar] [CrossRef]
- National Institutes of Health. National Institutes of Health Consensus Development Conference Statement: Neurofibromatosis. Bethesda, Md., USA, July 13–15, 1987. Neurofibromatosis 1988, 1, 172–178. [Google Scholar]
- Rose, S.R.; Municchi, G.; Barnes, K.M.; Cutler, G.B., Jr. Overnight growth hormone concentrations are usually normal in pubertal children with idiopathic short stature--a Clinical Research Center study. J. Clin. Endocrinol. Metab 1996, 81, 1063–1068. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Kipnis, D.M.; Daughaday, W.H. Growth hormone secretion during sleep. J. Clin. Investig. 1968, 47, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.Y.; Evans, W.S.; Blizzard, R.M.; Veldhuis, J.D.; Merriam, G.R.; Samojlik, E.; Furlanetto, R.; Rogol, A.D.; Kaiser, D.L.; Thorner, M.O. Effects of sex and age on the 24-hour profile of growth hormone secretion in man: Importance of endogenous estradiol concentrations. J. Clin. Endocrinol. Metab. 1987, 64, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.Y.; Weissberger, A.J. Characterization of 24-hour growth hormone secretion in acromegaly: Implications for diagnosis and therapy. Clin. Endocrinol. 1994, 41, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Zadik, Z.; Chalew, S.A.; McCarter, R.J., Jr.; Meistas, M.; Kowarski, A.A. The influence of age on the 24-hour integrated concentration of growth hormone in normal individuals. J. Clin. Endocrinol. Metab. 1985, 60, 513–516. [Google Scholar] [CrossRef]
- Quabbe, H.J.; Schilling, E.; Helge, H. Pattern of growth hormone secretion during a 24-hour fast in normal adults. J. Clin. Endocrinol. Metab. 1966, 26, 1173–1177. [Google Scholar] [CrossRef]
- Riva, P.; Corrado, L.; Natacci, F.; Castorina, P.; Wu, B.L.; Schneider, G.H.; Clementi, M.; Tenconi, R.; Korf, B.R.; Larizza, L. NF1 microdeletion syndrome: Refined FISH characterization of sporadic and familial deletions with locus-specific probes. Am. J. Hum. Genet. 2000, 66, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Fan, Z.; Liu, Q.; Li, J.; Du, J.; Shen, Y.; Wang, S. Two novel mutations of the NF1 gene in Chinese Han families with type 1 neurofibromatosis. J. Dermatol. Sci. 2005, 39, 125–127. [Google Scholar] [CrossRef]
- Hannah-Shmouni, F.; Trivellin, G.; Stratakis, C.A. Genetics of gigantism and acromegaly. Growth Horm. IGF Res. 2016, 30–31, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Thorner, M.O.; Frohman, L.A.; Leong, D.A.; Thominet, J.; Downs, T.; Hellmann, P.; Chitwood, J.; Vaughan, J.M.; Vale, W. Extrahypothalamic growth-hormone-releasing factor (GRF) secretion is a rare cause of acromegaly: Plasma GRF levels in 177 acromegalic patients. J. Clin. Endocrinol. Metab. 1984, 59, 846–849. [Google Scholar] [CrossRef]
- Eigler, T.; Ben-Shlomo, A. Somatostatin system: Molecular mechanisms regulating anterior pituitary hormones. J. Mol. Endocrinol. 2014, 53, R1–R19. [Google Scholar] [CrossRef] [Green Version]
- Boudin, G.; Pepin, B.; Vernant, C.L. Multiple tumours of the nervous system in Recklinghausen’s disease. An anatomo-clinical case with chromophobe adenoma of the pituitary gland. Presse Med. 1970, 78, 1427–1430. [Google Scholar] [PubMed]
- Smith, N.; Santoreneos, S. Non-functioning pituitary macroadenoma in a child with neurofibromatosis type 1. ANZ J. Surg. 2017, 87, E220–E221. [Google Scholar] [CrossRef] [PubMed]
- Filopanti, M.; Verga, U.; Ermetici, F.; Natacci, F.; Lalatta, F.; Avignone, S.; Trespidi, L.; Beck-Peccoz, P.; Mantovani, G.; Lania, A.G.; et al. Double pituitary and conserved function in an adult patient with neurofibromatosis type 1. J. Clin. Endocrinol. Metab. 2011, 96, 1953–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, H.Y.; Wu, T.; Wang, C.J.; Chin, S.C.; Chen, Y.R. Neurological complications involving the central nervous system in neurofibromatosis type 1. Acta Neurol. Taiwan 2007, 16, 68–73. [Google Scholar] [PubMed]
- Ueharu, H.; Yoshida, S.; Kikkawa, T.; Kanno, N.; Higuchi, M.; Kato, T.; Osumi, N.; Kato, Y. Gene tracing analysis reveals the contribution of neural crest-derived cells in pituitary development. J. Anat. 2017, 230, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Asa, S.L.; Kovacs, K. Growth hormone-releasing hormone-producing tumors: Clinical, biochemical, and morphological manifestations. Endocr. Rev. 1988, 9, 357–373. [Google Scholar] [CrossRef]
- Kovacs, K.; Horvath, E.; Thorner, M.O.; Rogol, A.D. Mammosomatotroph hyperplasia associated with acromegaly and hyperprolactinemia in a patient with the McCune-Albright syndrome. A histologic, immunocytologic and ultrastructural study of the surgically-removed adenohypophysis. Virchows Arch. A Pathol. Anat. Histopathol. 1984, 403, 77–86. [Google Scholar] [CrossRef]
- Moran, A.; Asa, S.L.; Kovacs, K.; Horvath, E.; Singer, W.; Sagman, U.; Reubi, J.C.; Wilson, C.B.; Larson, R.; Pescovitz, O.H. Gigantism due to pituitary mammosomatotroph hyperplasia. N. Engl. J. Med. 1990, 323, 322–327. [Google Scholar] [CrossRef]
- Boikos, S.A.; Stratakis, C.A. Pituitary pathology in patients with Carney Complex: Growth-hormone producing hyperplasia or tumors and their association with other abnormalities. Pituitary 2006, 9, 203–209. [Google Scholar] [CrossRef]
- Pack, S.D.; Kirschner, L.S.; Pak, E.; Zhuang, Z.; Carney, J.A.; Stratakis, C.A. Genetic and histologic studies of somatomammotropic pituitary tumors in patients with the “complex of spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas” (Carney complex). J. Clin. Endocrinol. Metab. 2000, 85, 3860–3865. [Google Scholar] [CrossRef] [Green Version]
- Trouillas, J.; Labat-Moleur, F.; Sturm, N.; Kujas, M.; Heymann, M.F.; Figarella-Branger, D.; Patey, M.; Mazucca, M.; Decullier, E.; Verges, B.; et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): A case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am. J. Surg. Pathol. 2008, 32, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Lagonigro, M.S.; Magri, F.; Koziak, M.; Jaffrain-Rea, M.L.; Brauner, R.; Bouligand, J.; Junier, M.P.; Di Rocco, F.; Sainte-Rose, C.; et al. Hyperplasia-adenoma sequence in pituitary tumorigenesis related to aryl hydrocarbon receptor interacting protein gene mutation. Endocr. Relat. Cancer 2011, 18, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirosh, A.; Papadakis, G.Z.; Chittiboina, P.; Lyssikatos, C.; Belyavskaya, E.; Keil, M.; Lodish, M.B.; Stratakis, C.A. 3D Volumetric Measurements of GH Secreting Adenomas Correlate with Baseline Pituitary Function, Initial Surgery Success Rate, and Disease Control. Horm. Metab. Res. 2017, 49, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Segal, L.; Darvish-Zargar, M.; Dilenge, M.E.; Ortenberg, J.; Polomeno, R.C. Optic pathway gliomas in patients with neurofibromatosis type 1: Follow-up of 44 patients. J. AAPOS 2010, 14, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.W.; Phipps, K.; Aquilina, K.; Gaze, M.N.; Hayward, R.; Spoudeas, H.A. Neuroendocrine Morbidity after Pediatric Optic Gliomas: A Longitudinal Analysis of 166 Children over 30 Years. J. Clin. Endocrinol. Metab. 2015, 100, 3787–3799. [Google Scholar] [CrossRef]
- Cnossen, M.H.; Stam, E.N.; Cooiman, L.C.; Simonsz, H.J.; Stroink, H.; Oranje, A.P.; Halley, D.J.; de Goede-Bolder, A.; Niermeijer, M.F.; de Muinck Keizer-Schrama, S.M. Endocrinologic disorders and optic pathway gliomas in children with neurofibromatosis type 1. Pediatrics 1997, 100, 667–670. [Google Scholar] [CrossRef] [Green Version]
- Collet-Solberg, P.F.; Sernyak, H.; Satin-Smith, M.; Katz, L.L.; Sutton, L.; Molloy, P.; Moshang, T., Jr. Endocrine outcome in long-term survivors of low-grade hypothalamic/chiasmatic glioma. Clin. Endocrinol. 1997, 47, 79–85. [Google Scholar] [CrossRef]
- Habiby, R.; Silverman, B.; Listernick, R.; Charrow, J. Precocious puberty in children with neurofibromatosis type 1. J. Pediatr. 1995, 126, 364–367. [Google Scholar] [CrossRef]
- Vassilopoulou-Sellin, R.; Klein, M.J.; Slopis, J.K. Growth hormone deficiency in children with neurofibromatosis type 1 without suprasellar lesions. Pediatr. Neurol. 2000, 22, 355–358. [Google Scholar] [CrossRef]
- Parsa, C.F.; Hoyt, C.S.; Lesser, R.L.; Weinstein, J.M.; Strother, C.M.; Muci-Mendoza, R.; Ramella, M.; Manor, R.S.; Fletcher, W.A.; Repka, M.X.; et al. Spontaneous regression of optic gliomas: Thirteen cases documented by serial neuroimaging. Arch. Ophthalmol. 2001, 119, 516–529. [Google Scholar] [CrossRef] [Green Version]
- Iacovazzo, D.; Caswell, R.; Bunce, B.; Jose, S.; Yuan, B.; Hernandez-Ramirez, L.C.; Kapur, S.; Caimari, F.; Evanson, J.; Ferrau, F.; et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: A clinico-pathological and genetic study. Acta Neuropathol. Commun. 2016, 4, 56. [Google Scholar] [CrossRef]
- Franke, M.; Daly, A.F.; Palmeira, L.; Tirosh, A.; Stigliano, A.; Trifan, E.; Faucz, F.R.; Abboud, D.; Petrossians, P.; Tena, J.J.; et al. Duplications disrupt chromatin architecture and rewire GPR101-enhancer communication in X-linked acrogigantism. Am. J. Hum. Genet. 2022, 109, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Hysinger, J.D.; Barron, T.; Schindler, N.F.; Cobb, O.; Guo, X.; Yalcin, B.; Anastasaki, C.; Mulinyawe, S.B.; Ponnuswami, A.; et al. NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature 2021, 594, 277–282. [Google Scholar] [CrossRef]
- Bates, B.; Zhang, L.; Nawoschik, S.; Kodangattil, S.; Tseng, E.; Kopsco, D.; Kramer, A.; Shan, Q.; Taylor, N.; Johnson, J.; et al. Characterization of Gpr101 expression and G-protein coupling selectivity. Brain Res. 2006, 1087, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nilaweera, K.N.; Wilson, D.; Bell, L.; Mercer, J.G.; Morgan, P.J.; Barrett, P. G protein-coupled receptor 101 mRNA expression in supraoptic and paraventricular nuclei in rat hypothalamus is altered by pregnancy and lactation. Brain Res. 2008, 1193, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Trivellin, G.; Faucz, F.R.; Daly, A.F.; Beckers, A.; Stratakis, C.A. GPR101, an orphan GPCR with roles in growth and pituitary tumorigenesis. Endocr. Relat. Cancer 2020, 27, T87–T97. [Google Scholar] [CrossRef]
- Daly, A.F.; Lysy, P.A.; Desfilles, C.; Rostomyan, L.; Mohamed, A.; Caberg, J.H.; Raverot, V.; Castermans, E.; Marbaix, E.; Maiter, D.; et al. GHRH excess and blockade in X-LAG syndrome. Endocr. Relat. Cancer 2016, 23, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Nunley, K.S.; Gao, F.; Albers, A.C.; Bayliss, S.J.; Gutmann, D.H. Predictive value of cafe au lait macules at initial consultation in the diagnosis of neurofibromatosis type 1. Arch. Dermatol 2009, 145, 883–887. [Google Scholar] [CrossRef] [Green Version]
- DeBella, K.; Poskitt, K.; Szudek, J.; Friedman, J.M. Use of “unidentified bright objects” on MRI for diagnosis of neurofibromatosis 1 in children. Neurology 2000, 54, 1646–1651. [Google Scholar] [CrossRef]
- DeBella, K.; Szudek, J.; Friedman, J.M. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000, 105, 608–614. [Google Scholar] [CrossRef]
- Carmi, D.; Shohat, M.; Metzker, A.; Dickerman, Z. Growth, puberty, and endocrine functions in patients with sporadic or familial neurofibromatosis type 1: A longitudinal study. Pediatrics 1999, 103, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Mautner, V.F.; Kluwe, L.; Friedrich, R.E.; Roehl, A.C.; Bammert, S.; Hogel, J.; Spori, H.; Cooper, D.N.; Kehrer-Sawatzki, H. Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions. J. Med. Genet. 2010, 47, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sex | Age (Years) at NF1 Diagnosis | Age (Years) at GH Excess Diagnosis | Acromegaly | Height SDS | Random GH (ng/mL) | Random IGF-1 a (ng/mL) | GHRH b (pg/mL) |
|---|---|---|---|---|---|---|---|---|
| 1 | F | Birth | 4.8 | No | 1.8 | 2.3 | 540 (Z-score: 9) | 31 |
| 2 | M | 2 | 3 | No | 3 | 13.6 | 659 (Z-score: 15.3) | N/A |
| 3 | M | 1 | 4 | No | 2.7 | 1.55 | 431 (Z-score: 3.8) | 5 |
| 4 | F | 3 | 3 | No | 2.7 | 3.5 | 353 (Z-score: 6.5) | N/A |
| 5 | M | 38 | 52 | Yes | 2.1 | 1.41 | 538 (Z-score: 9.9) | 75 |
| 6 | M | 3 | 3 | No | 2.3 | 9.86 | 683 (Z-score: 15.9) | N/A |
| 7 | F | 3 | 29 | Yes | 0.3 | 4.6 | 451 (Z-score: 4.3) | 11 |
| 8 | M | 25 | 14 | No | 3.2 | 6 | 268 (Z-score: −0.62) | N/A |
| 9 | F | 42 | 42 | Yes | 2.0 | 3.1 | 958 (Z-score: 9.7) | N/A |
| 10 | F | 5 months | 5 | No | 1.1 | 1.14 | 270 (Z-score: 3.5) | 27 |
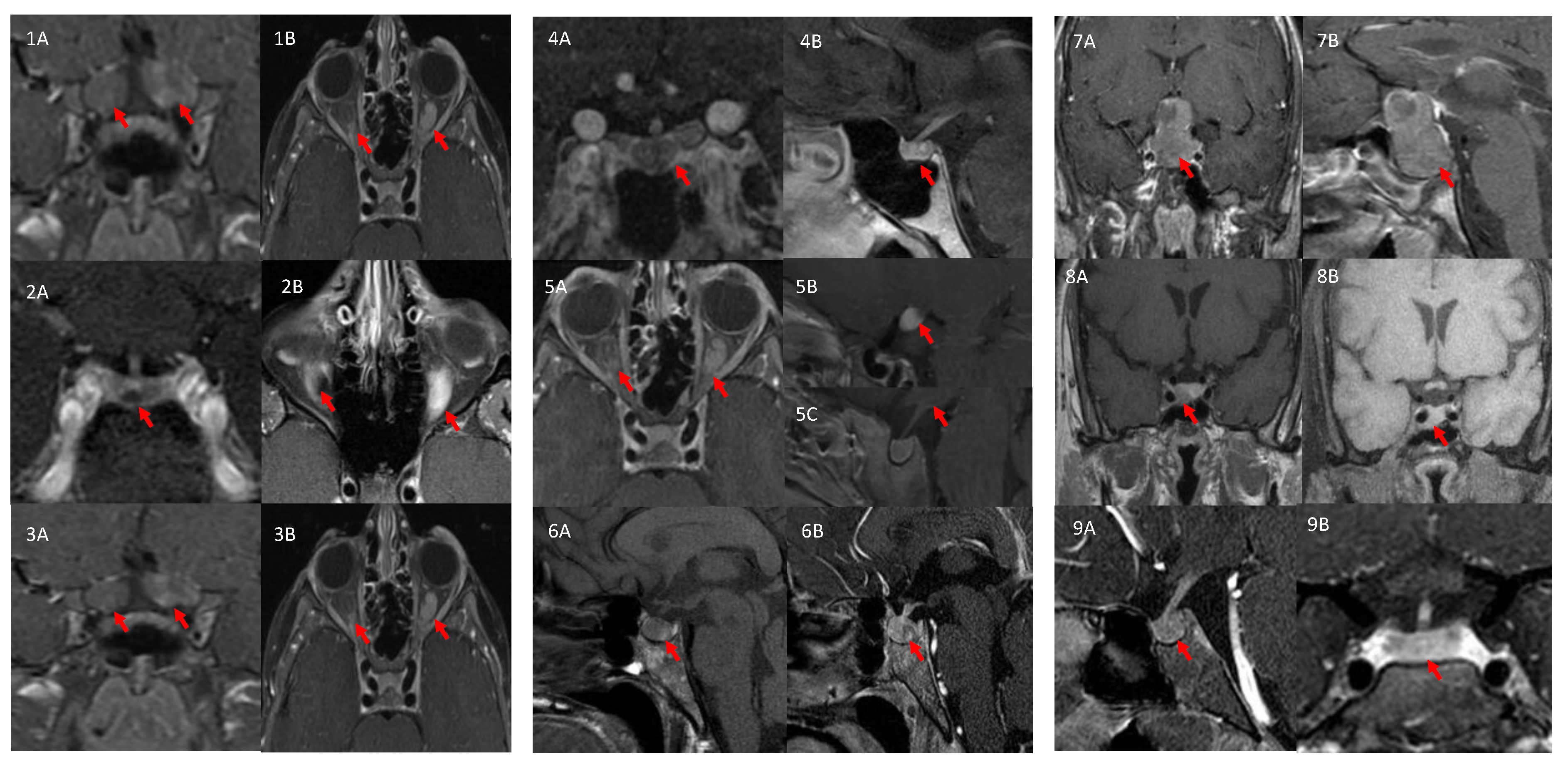
| ID | Pituitary MRI | Pathology | Family History of NF1 |
|---|---|---|---|
| 1 | No tumor | N/A | Positive |
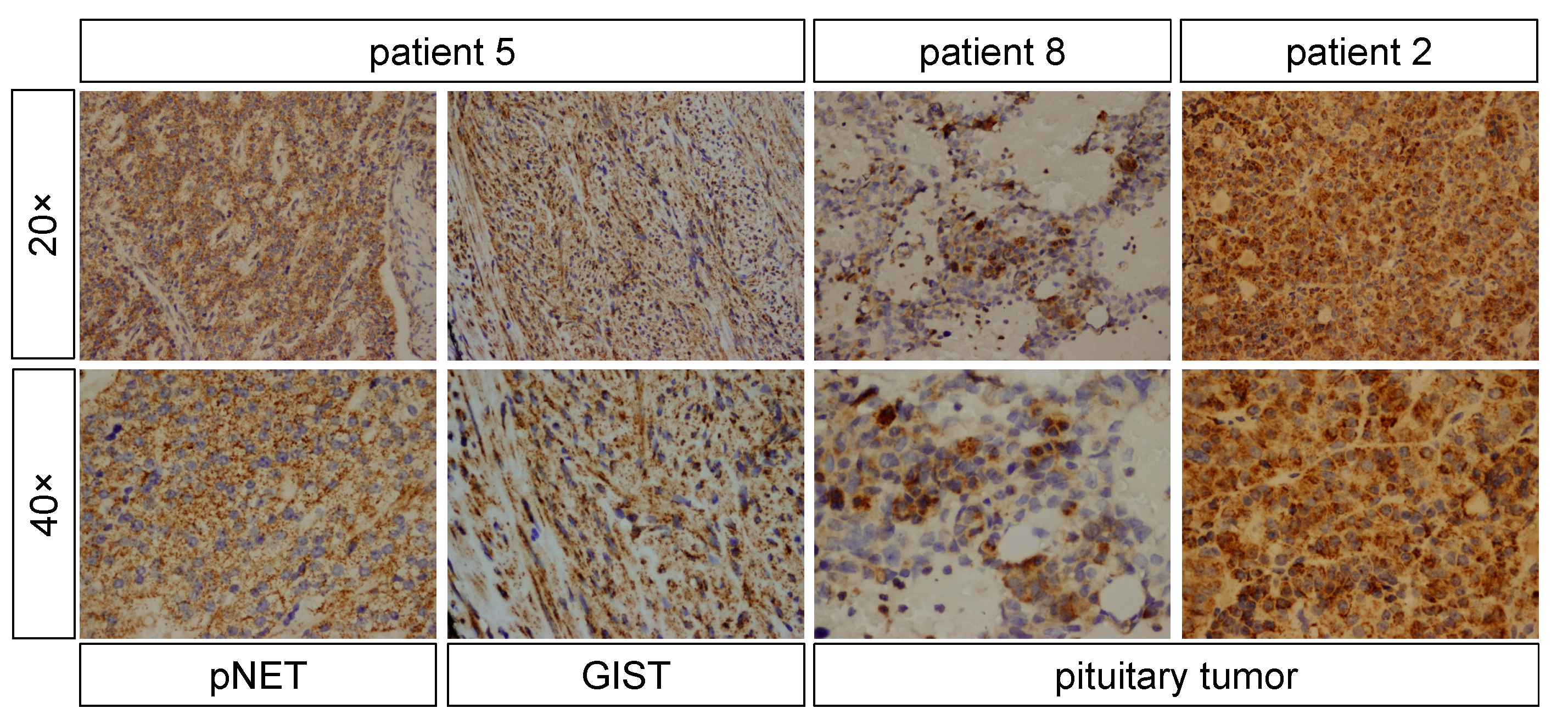
| 2 | Microadenoma (~5 mm) | Pituitary tumor stained negative for GH, patchy positive for GPR101, positive for NF1 | Positive |
| 3 | No tumor | N/A | Negative |
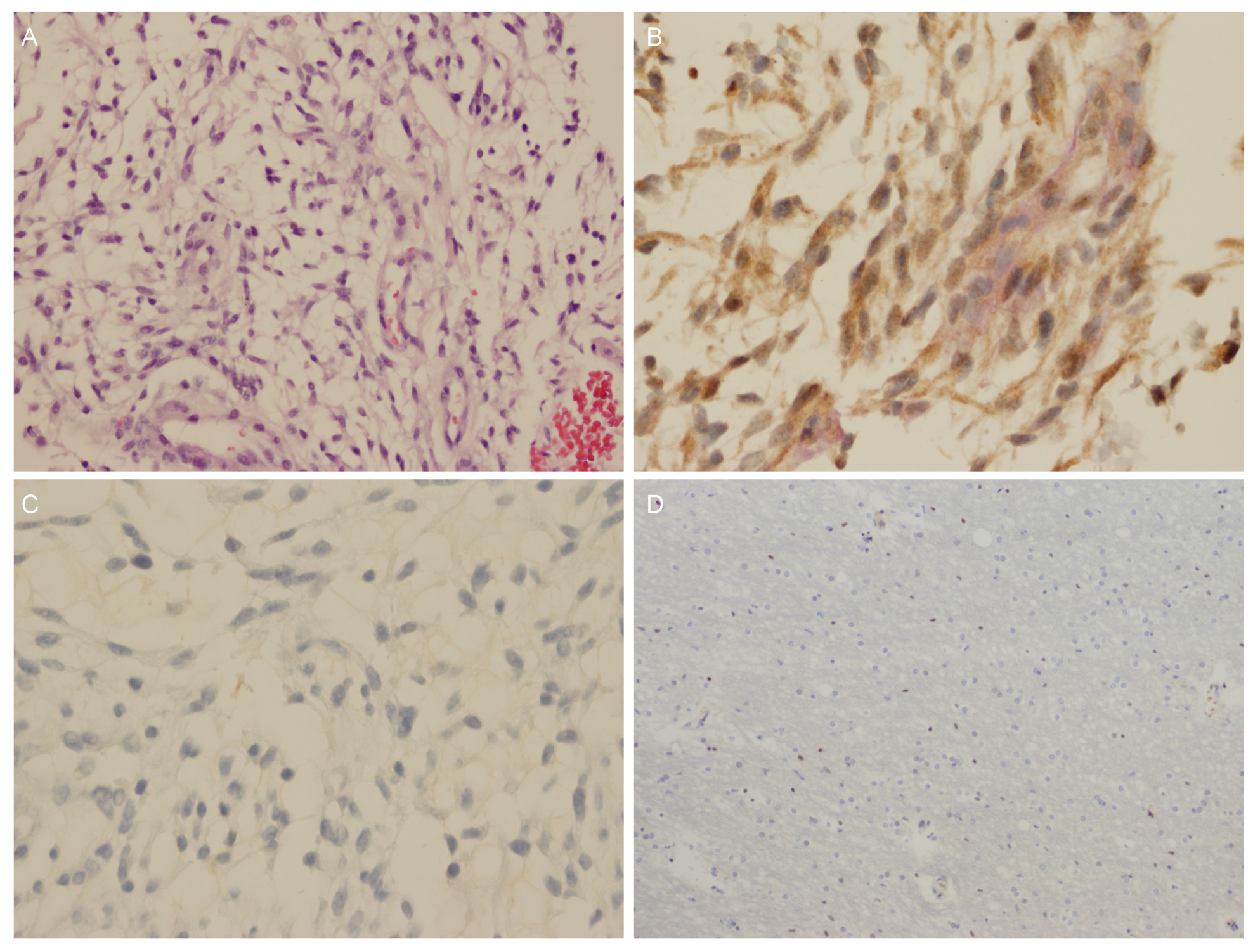
| 4 | No tumor | OPG stained positive for GPR101 (cytoplasmic signal), negative for GHRH, GH, and somatostatin and weakly positive for GH | Positive |
| 5 | No tumor | GIST stained patchy positive for GPR101 and small nuclear staining in pNET | Positive |
| 6 | No tumor; hypothalamic infiltration | N/A | Negative |
| 7 | Voluminous pituitary gland with Rathke’s cyst | N/A | Negative |
| 8 | Macroadenoma (~1 × 0.5 cm) | Pituitary tumor stained positive for GH, PRL and NF1, and patchy positive for GPR101 | Negative |
| 9 | Macroadenoma (~1.4 cm) | N/A | Negative |
| 10 | No tumor; Right inferior hypothalamic enhancement | N/A | Negative |
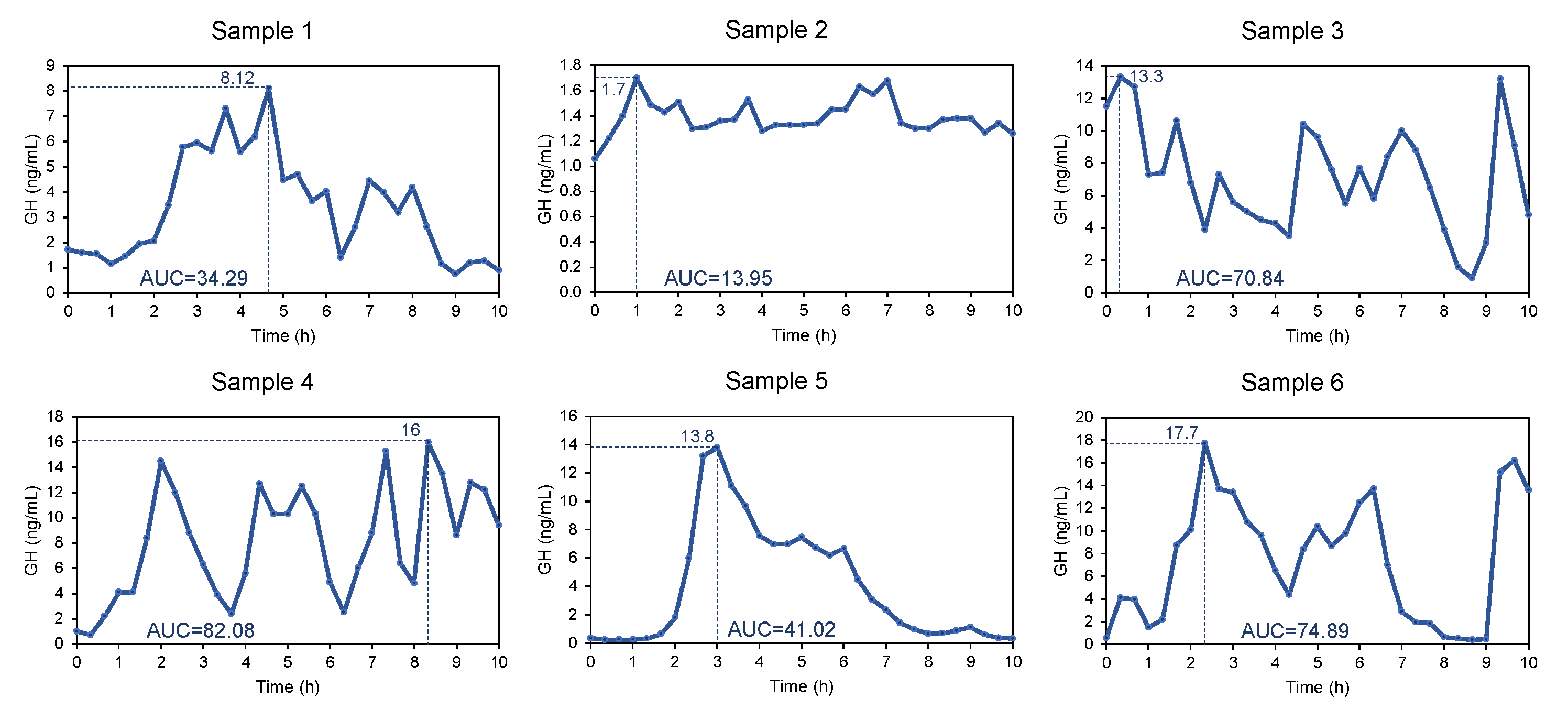
| Sample 1 | Sample 2 | Sample 3 | Sample 4 | Sample 5 | Sample 6 | |
|---|---|---|---|---|---|---|
| Patient | 3 | 5 | 2 | 1 | 7 | 10 |
| N | 31 | 31 | 31 | 31 | 31 | 31 |
| Mean GH (ng/mL) | 3.3 | 1.4 | 7.1 | 8.1 | 3.9 | 7.5 |
| 95% CI | 2.6–4.1 | 1.3–1.4 | 5.913–8.3 | 6.4–9.7 | 2.5–5.5 | 5.5–9.6 |
| Variance | 4.2 | 0.01 | 10.8 | 19.6 | 17.0 | 29.3 |
| SD | 2.0 | 0.1 | 3.3 | 4.4 | 4.1 | 5.4 |
| Minimum GH (ng/mL) | 0.8 | 1.1 | 0.9 | 0.7 | 0.3 | 0.4 |
| Maximum GH (ng/mL) | 8.1 | 1.7 | 13.3 | 16 | 13.8 | 17.7 |
| AUC (baseline = 0; ng/mL·h) | 34.3 | 13.9 | 70.8 | 82.1 | 41.0 | 74.9 |
| ID | NF1 | In Silico Prediction | Other Genetic Findings | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DNA Change | Protein Change | dbSNP ID | ClinVar ID | Location in Gene | LOH | Total MAF (%) | ||||
| 1 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | [17] |
| 2 | c.(576_617)_(785_958)del | N/A | N/A | N/A | exons 6–9 | N/A | N/A | pathogenic | No GPR101 SNVs or CNVs | N/A |
| 3 | c.6406dupT | p.Ser2136Phefs*12 | N/A | N/A | exon 42 | N/A | N/A | pathogenic | N/A | N/A |
| 4 | c.6791dupA | p.Tyr2264* | rs876657715 | RCV000213933.4 RCV000486063.1 | exon 45 | N/A | N/A | pathogenic | No GPR101 SNVs or CNVs, 57 kb deletion at 6q12 | [12] |
| 5 | c.2329T>A | p.Trp777Arg | rs876658853 | RCV000219741.1 | exon 20 | N/A | N/A | likely pathogenic | N/A | [21] |
| 6 | c.2339C>G | p.Thr780Arg | rs199474746 | VCV000457581 | exon 20 | N/A | N/A | pathogenic | N/A | [22] |
| 7 | 17q11.2(29,039,980–30,352,755) × 1 | N/A | N/A | N/A | entire gene | N/A | N/A | pathogenic | N/A | N/A |
| 8 | c.1541A>C | p.Gln514Pro | rs775369084 | VCV000232968.4 | exon 14 | no | 0.01349 | VUS | No GPR101 SNVs, no AIP SNVs or CNVs, no MEN1 SNVs or CNVs | N/A |
| 9 | c.2329T>A | p.Trp777Arg | rs876658853 | RCV000219741.1 | exon 20 | N/A | N/A | likely pathogenic | N/A | N/A |
| 10 | c.7285C>T | p.Arg2429* | rs786202457 | RCV000414562.1 RCV000165277.1 RCV000414830.1 RCV000532076.6 RCV000999932.1 | exon 49 | N/A | N/A | pathogenic | N/A | [23] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannah-Shmouni, F.; Trivellin, G.; Beckers, P.; Karaviti, L.P.; Lodish, M.; Tatsi, C.; Adesina, A.M.; Adamidou, F.; Mintziori, G.; Josefson, J.L.; et al. Neurofibromatosis Type 1 Has a Wide Spectrum of Growth Hormone Excess. J. Clin. Med. 2022, 11, 2168. https://doi.org/10.3390/jcm11082168
Hannah-Shmouni F, Trivellin G, Beckers P, Karaviti LP, Lodish M, Tatsi C, Adesina AM, Adamidou F, Mintziori G, Josefson JL, et al. Neurofibromatosis Type 1 Has a Wide Spectrum of Growth Hormone Excess. Journal of Clinical Medicine. 2022; 11(8):2168. https://doi.org/10.3390/jcm11082168
Chicago/Turabian StyleHannah-Shmouni, Fady, Giampaolo Trivellin, Pablo Beckers, Lefkothea P. Karaviti, Maya Lodish, Christina Tatsi, Adekunle M. Adesina, Fotini Adamidou, Gesthimani Mintziori, Jami L. Josefson, and et al. 2022. "Neurofibromatosis Type 1 Has a Wide Spectrum of Growth Hormone Excess" Journal of Clinical Medicine 11, no. 8: 2168. https://doi.org/10.3390/jcm11082168