
Infections as Novel Risk Factors of Atherosclerotic Cardiovascular Diseases: Pathophysiological Links and Therapeutic Implications

 , , , , , and
, , , , , and
Abstract
:1. Introduction
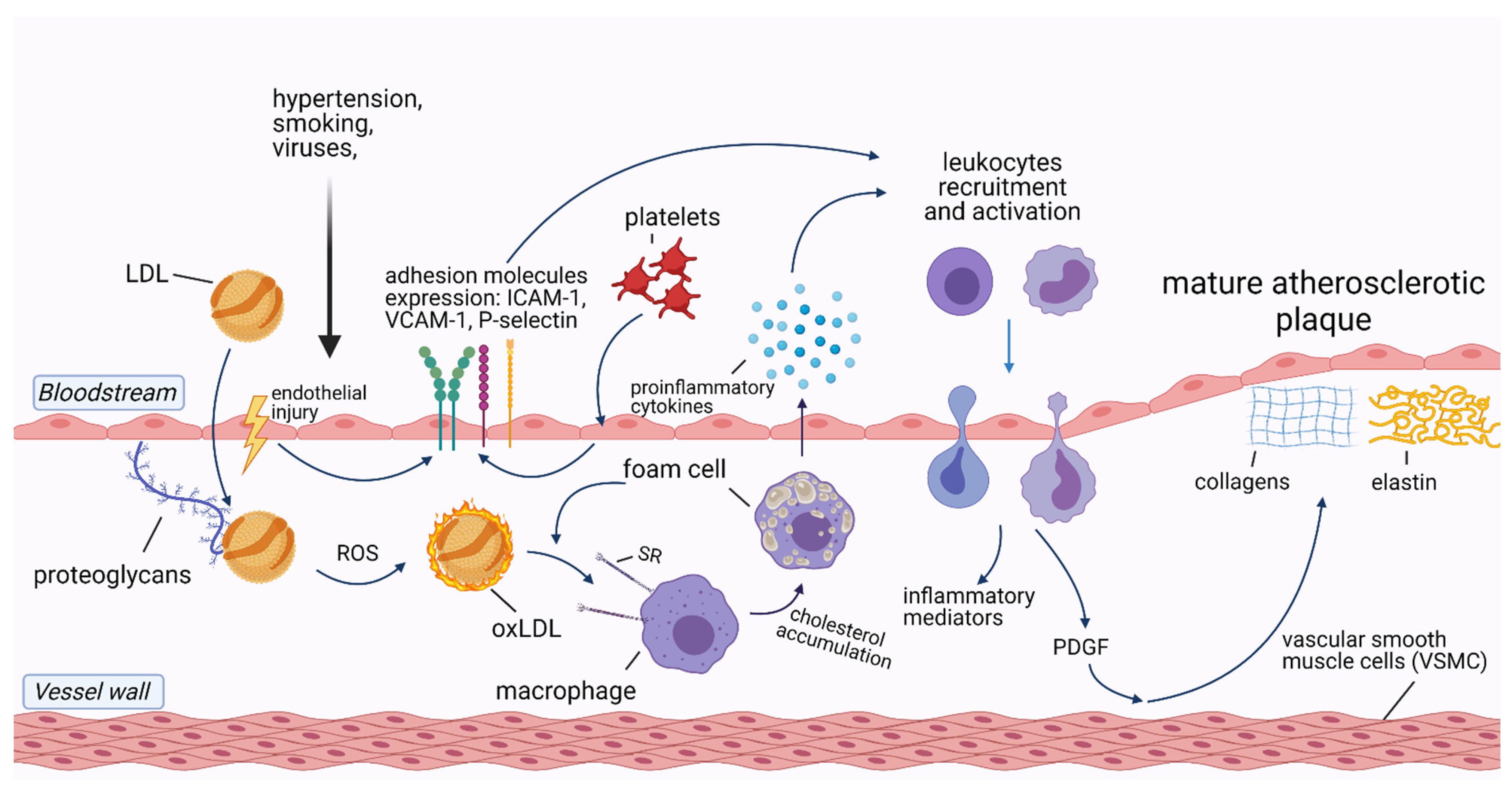
2. The Role of Inflammation in Cardiovascular Diseases
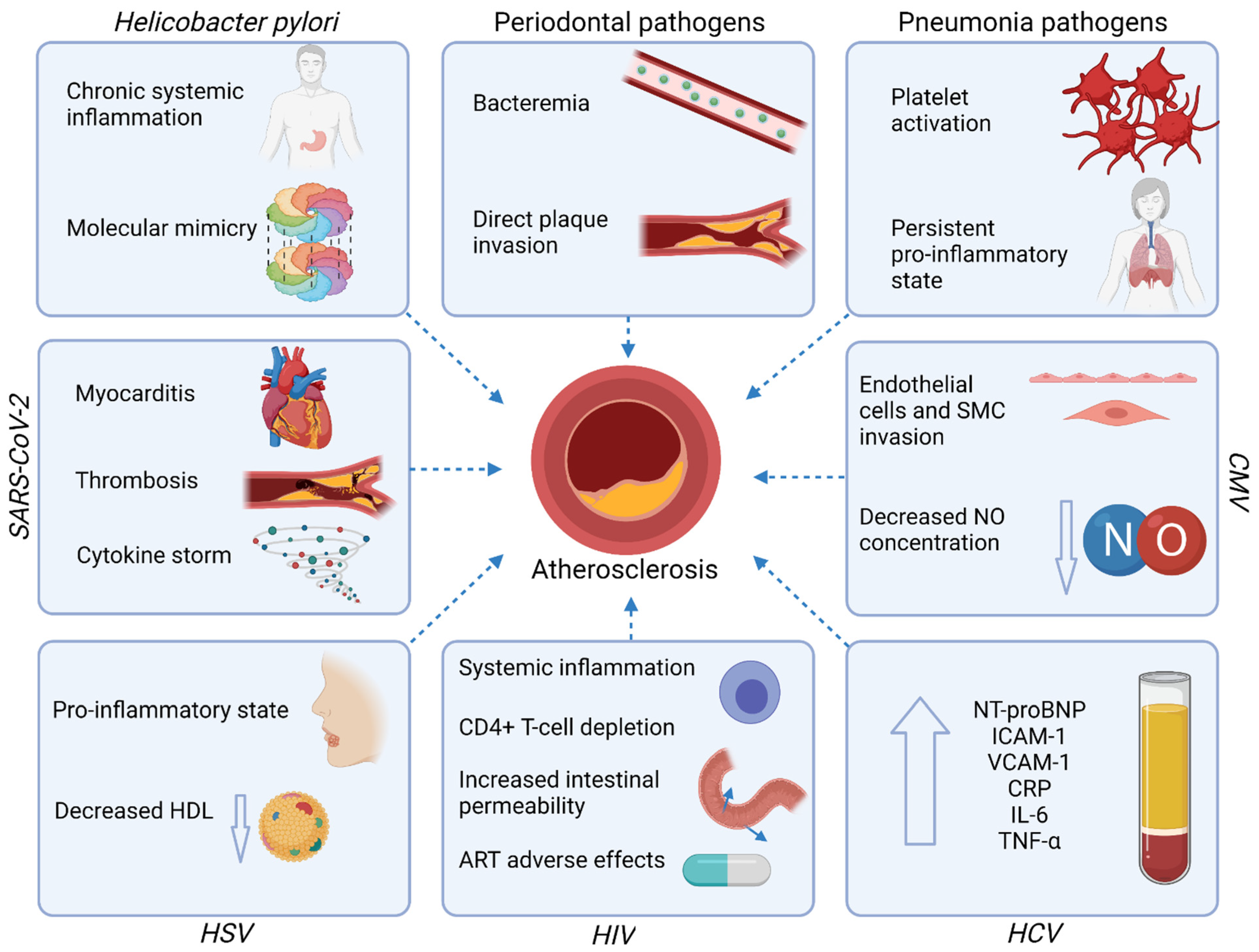
3. Infections and Atherosclerotic Cardiovascular Diseases
3.1. Gastrointestinal Tract Infections
3.1.1. Periodontal Disease
3.1.2. Helicobacter Pylori
3.1.3. Hepatitis C Virus (HCV)
3.2. Respiratory Tract Infections
3.2.1. Pneumonia
3.2.2. Cytomegalovirus (CMV)
3.2.3. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2)
3.3. Immune System Infections
Human Immunodeficiency Virus (HIV)
3.4. Dermatologic Infections
Herpes Simplex Virus (HSV)
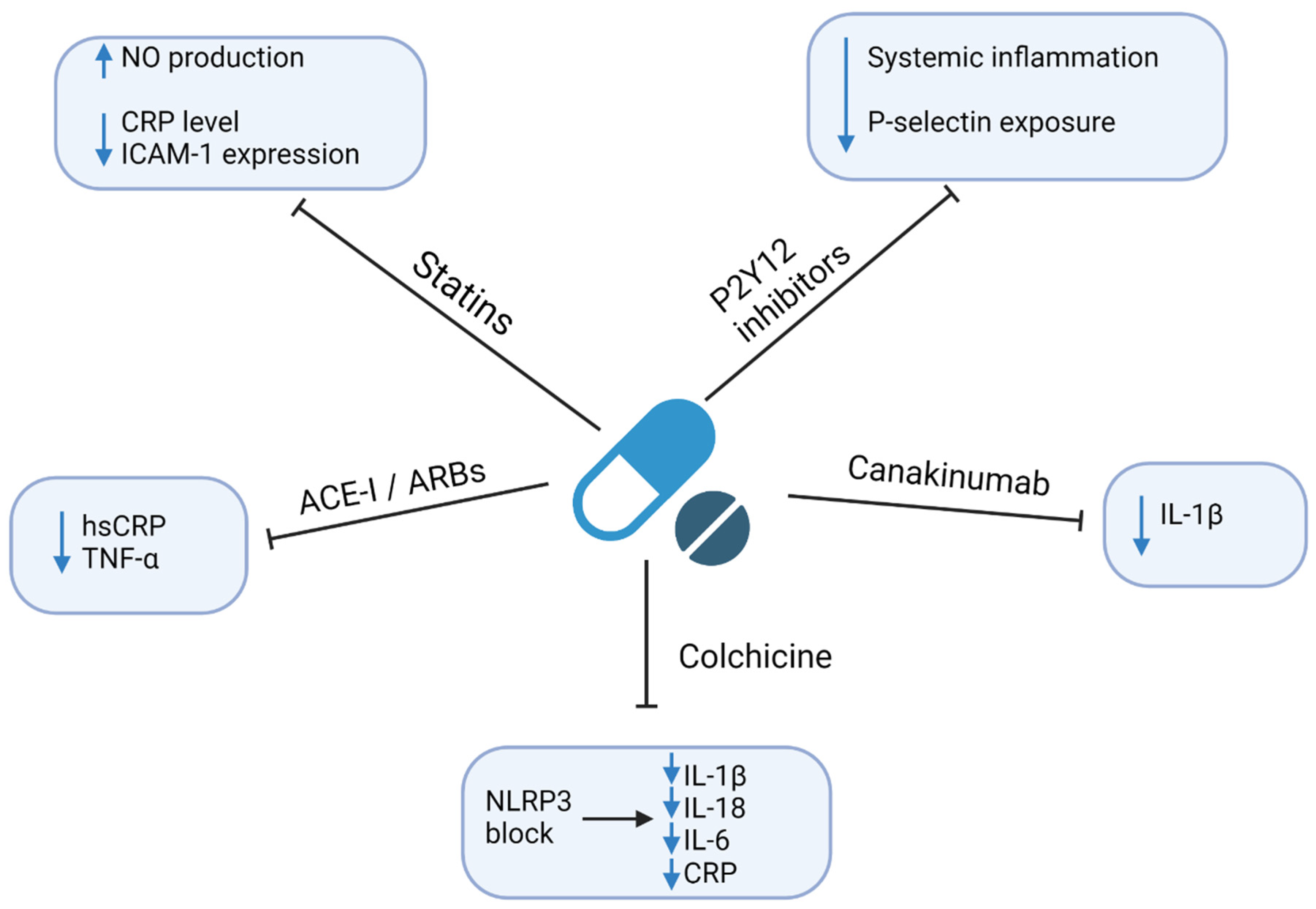
4. Therapeutic Implications
4.1. Statins
4.2. P2Y12 Inhibitors
4.3. Angiotensin-Converting Enzyme Inhibitors (ACE-I) and Angiotensin Receptor Blockers (ARBs)
4.4. Colchicine
4.5. Anti-Cytokine Drugs
4.6. Methotrexate
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Data. Available online: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1 (accessed on 20 March 2021).
- Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; Baigent, C.; et al. The Effects of Lowering LDL Cholesterol with Statin Therapy in People at Low Risk of Vascular Disease: Meta-Analysis of Individual Data from 27 Randomised Trials. Lancet 2012, 380, 581–590. [Google Scholar] [CrossRef]
- Kaasenbrood, L.; Boekholdt, S.M.; Van Der Graaf, Y.; Ray, K.K.; Peters, R.J.G.; Kastelein, J.J.P.; Amarenco, P.; Larosa, J.C.; Cramer, M.J.M.; Westerink, J.; et al. Distribution of Estimated 10-Year Risk of Recurrent Vascular Events and Residual Risk in a Secondary Prevention Population. Circulation 2016, 134, 1419–1429. [Google Scholar] [CrossRef]
- Lechner, K.; von Schacky, C.; McKenzie, A.L.; Worm, N.; Nixdorff, U.; Lechner, B.; Kränkel, N.; Halle, M.; Krauss, R.M.; Scherr, J. Lifestyle Factors and High-Risk Atherosclerosis: Pathways and Mechanisms beyond Traditional Risk Factors. Eur. J. Prev. Cardiol. 2020, 27, 394–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, E.; Novo, S. Role of Inflammation and Infection in Vascular Disease. Acta Chir. Belg. 2005, 105, 567–579. [Google Scholar] [CrossRef]
- Alfarisi, H.A.H.; Mohamed, Z.B.H.; Ibrahim, M. Bin. Basic Pathogenic Mechanisms of Atherosclerosis. Egypt. J. Basic Appl. Sci. 2020, 7, 116–125. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109 (Suppl. 23). [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leishman, S.J.; Do, H.L.; Ford, P.J. Cardiovascular Disease and the Role of Oral Bacteria. J. Oral Microbiol. 2010, 2, 5781. [Google Scholar] [CrossRef]
- Xu, Z.; Li, J.; Wang, H.; Xu, G. Helicobacter Pylori Infection and Atherosclerosis: Is There a Causal Relationship? Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2293–2301. [Google Scholar] [CrossRef]
- Lebedeva, A.M.; Shpektor, A.V.; Vasilieva, E.Y.; Margolis, L.B. Cytomegalovirus Infection in Cardiovascular Diseases. Biochemistry 2018, 83, 1437–1447. [Google Scholar] [CrossRef]
- Restrepo, M.I.; Reyes, L.F. Pneumonia as a Cardiovascular Disease. Respirology 2018, 23, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Berquist, V.; Hoy, J.F.; Trevillyan, J.M. Contribution of Common Infections to Cardiovascular Risk in HIV-Positive Individuals. AIDS Rev. 2017, 19, 72–80. [Google Scholar] [PubMed]
- Wu, Y.P.; Sun, D.D.; Wang, Y.; Liu, W.; Yang, J. Herpes Simplex Virus Type 1 and Type 2 Infection Increases Atherosclerosis Risk: Evidence Based on a Meta-Analysis. BioMed Res. Int. 2016, 2016, 2630865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakal, B.P.; Sweitzer, N.K.; Indik, J.H.; Acharya, D.; William, P. SARS-CoV-2 Infection and Cardiovascular Disease: COVID-19 Heart. Heart Lung Circ. 2020, 29, 973–987. [Google Scholar] [CrossRef]
- WHO Data. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 2 May 2021).
- Williams, K.J.; Tabas, I. The Response-to-Retention Hypothesis of Early Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, G.K.; Hermansson, A. The Immune System in Atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Golia, E.; Limongelli, G.; Natale, F.; Fimiani, F.; Maddaloni, V.; Pariggiano, I.; Bianchi, R.; Crisci, M.; D’Acierno, L.; Giordano, R.; et al. Inflammation and Cardiovascular Disease: From Pathogenesis to Therapeutic Target. Curr. Atheroscler. Rep. 2014, 16, 435. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M. Inflammation and Atherothrombosis. From Population Biology and Bench Research to Clinical Practice. J. Am. Coll. Cardiol. 2006, 48, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Lorenzatti, A.J.; Servato, M.L. New Evidence on the Role of Inflammation in CVD Risk. Curr. Opin. Cardiol. 2019, 34, 418–423. [Google Scholar] [CrossRef]
- Raggi, P.; Genest, J.; Giles, J.T.; Rayner, K.J.; Dwivedi, G.; Beanlands, R.S.; Gupta, M. Role of Inflammation in the Pathogenesis of Atherosclerosis and Therapeutic Interventions. Atherosclerosis 2018, 276, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Gisterå, A.; Robertson, A.K.L.; Andersson, J.; Ketelhuth, D.F.J.; Ovchinnikova, O.; Nilsson, S.K.; Lundberg, A.M.; Li, M.O.; Flavell, R.A.; Hansson, G.K. Transforming Growth Factor-β Signaling in T Cells Promotes Stabilization of Atherosclerotic Plaques through an Interleukin-17-Dependent Pathway. Sci. Transl. Med. 2013, 5, 18–23. [Google Scholar] [CrossRef]
- Libby, P.; Hansson, G.K. Taming Immune and Inflammatory Responses to Treat Atherosclerosis. J. Am. Coll. Cardiol. 2018, 71, 173–176. [Google Scholar] [CrossRef]
- Lordan, R.; Tsoupras, A.; Zabetakis, I. Platelet Activation and Prothrombotic Mediators at the Nexus of Inflammation and Atherosclerosis: Potential Role of Antiplatelet Agents. Blood Rev. 2021, 45, 100694. [Google Scholar] [CrossRef] [PubMed]
- Gąsecka, A.; Rogula, S.; Szarpak, Ł.; Filipiak, K.J. LDL-Cholesterol and Platelets: Insights into Their Interactions in Atherosclerosis. Life 2021, 11, 39. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Heijman, J.; Muna, A.P.; Veleva, T.; Molina, C.E.; Sutanto, H.; Tekook, M.; Wang, Q.; Abu-Taha, I.H.; Gorka, M.; Künzel, S.; et al. Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Postoperative Atrial Fibrillation. Circ. Res. 2020, 127, 1036–1055. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, Functions, and Clinical Relevance of Extracellular Vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [Green Version]
- Hafiane, A.; Daskalopoulou, S.S. Extracellular Vesicles Characteristics and Emerging Roles in Atherosclerotic Cardiovascular Disease. Metabolism 2018, 85, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Gąsecka, A.; Pluta, K.; Solarska, K.; Rydz, B.; Eyileten, C.; Postula, M.; Van Der Pol, E.; Nieuwland, R.; Budnik, M.; Kochanowski, J.; et al. Plasma Concentrations of Extracellular Vesicles Are Decreased in Patients with Post-Infarct Cardiac Remodelling. Biology 2021, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Gąsecka, A.; Van Der Pol, E.; Nieuwland, R.; Stępień, E. Extracellular Vesicles in Post-Infarct Ventricular Remodelling. Kardiol. Pol. 2018, 76, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Gasecka, A.; Nieuwland, R.; Siljander, P.R.M. Platelet-Derived Extracellular Vesicles. In Platelets, 4th ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 401–416. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kamińska, K. MicroRNA in Cardiovascular Biology and Disease. Adv. Clin. Exp. Med. 2017, 26, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Yuan, J.; Zhang, F.; Lei, Q.; Zhang, T.; Li, K.; Guo, J.; Hong, Y.; Bu, G.; Lv, X.; et al. MicroRNA-181a-5p and MicroRNA-181a-3p Cooperatively Restrict Vascular Inflammation and Atherosclerosis. Cell Death Dis. 2019, 10, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soh, J. MicroRNA-30c Reduces Hyperlipidemia and Atherosclerosis by Decreasing Lipid Synthesis and Lipoprotein Secretion. Physiol. Behav. 2016, 176, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Schober, A.; Nazari-Jahantigh, M.; Wei, Y.; Bidzhekov, K.; Gremse, F.; Grommes, J.; Megens, R.T.A.; Heyll, K.; Noels, H.; Hristov, M.; et al. MicroRNA-126-5p Promotes Endothelial Proliferation and Limits Atherosclerosis by Suppressing Dlk1. Nat. Med. 2014, 20, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.C.; Tang, Y.Y.; Peng, J.; Zhao, G.J.; Yang, J.; Yao, F.; Ouyang, X.P.; He, P.P.; Xie, W.; Tan, Y.L.; et al. MicroRNA-19b Promotes Macrophage Cholesterol Accumulation and Aortic Atherosclerosis by Targeting ATP-Binding Cassette Transporter A1. Atherosclerosis 2014, 236, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Ediriweera, H.; Afonso, M.S.; Ramkhelawon, B.; Singaravelu, R.; Liao, X.; Bandler, R.C.; Rahman, K.; Fisher, E.A.; Rayner, K.J.; et al. Microrna-33 regulates macrophage autophagy in atherosclerosis. Arterioscler. Thrombo. Vasc. Biol. 2017, 37, 1058–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyer, X.; Potteaux, S.; Vion, A.C.; Guérin, C.L.; Boulkroun, S.; Rautou, P.E.; Ramkhelawon, B.; Esposito, B.; Dalloz, M.; Paul, J.L.; et al. Inhibition of MicroRNA-92a Prevents Endothelial Dysfunction and Atherosclerosis in Mice. Circ. Res. 2014, 114, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.W.; Hu, Y.R.; Zhao, J.Y.; Li, S.F.; Ma, X.; Wu, S.G.; Lu, J.B.; Qiu, Y.R.; Sha, Y.H.; Wang, Y.C.; et al. An Agomir of MiR-144-3p Accelerates Plaque Formation through Impairing Reverse Cholesterol Transport and Promoting pro-Inflammatory Cytokine Production. PLoS ONE 2014, 9, e94997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 Promotes Atherosclerosis by Repressing Bcl6 in Macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Nazari-Jahantigh, M.; Chan, L.; Zhu, M.; Heyll, K.; Corbalán-Campos, J.; Hartmann, P.; Thiemann, A.; Weber, C.; Schober, A. The MicroRNA-342-5p Fosters Inflammatory Macrophage Activation through an Akt1- and MicroRNA-155-Dependent Pathway during Atherosclerosis. Circulation 2013, 127, 1609–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacante, F.; Denby, L.; Sluimer, J.C.; Baker, A.H. The Function of MiR-143, MiR-145 and the MiR-143 Host Gene in Cardiovascular Development and Disease. Vascul. Pharmacol. 2019, 112, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Gozdowska, R.; Makowska, A.; Gąsecka, A.; Chabior, A.; Marchel, M. Circulating MicroRNA in Heart Failure—Practical Guidebook to Clinical Application. Cardiol Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tomaniak, M.; Sygitowicz, G.; Filipiak, K.J.; Błaszczyk, O.; Kołtowski, L.; Gasecka, A.; Kochanowski, J.; Sitkiewicz, D. Dysregulations of MiRNAs and Galectin-3 May Underlie Left Ventricular Dilatation in Patients with Systolic Heart Failure. Kardiol. Pol. 2018, 76, 1012–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arida, A.; Protogerou, A.D.; Kitas, G.D.; Sfikakis, P.P. Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. Int. J. Mol. Sci. 2018, 19, 1890. [Google Scholar] [CrossRef] [Green Version]
- Adawi, M.; Firas, S.; Blum, A. Rheumatoid Arthritis and Atherosclerosis. Isr. Med. Assoc. J. 2019, 21, 460–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liccardo, D.; Cannavo, A.; Spagnuolo, G.; Ferrara, N.; Cittadini, A.; Rengo, C.; Rengo, G. Periodontal Disease: A Risk Factor for Diabetes and Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 1414. [Google Scholar] [CrossRef] [Green Version]
- Tomita, S.; Komiya-Ito, A.; Imamura, K.; Kita, D.; Ota, K.; Takayama, S.; Makino-Oi, A.; Kinumatsu, T.; Ota, M.; Saito, A. Prevalence of Aggregatibacter Actinomycetemcomitans, Porphyromonas Gingivalis and Tannerella Forsythia in Japanese Patients with Generalized Chronic and Aggressive Periodontitis. Microb. Pathog. 2013, 61, 11–15. [Google Scholar] [CrossRef]
- Larvin, H.; Kang, J.; Aggarwal, V.R.; Pavitt, S.; Wu, J. Risk of Incident Cardiovascular Disease in People with Periodontal Disease: A Systematic Review and Meta-Analysis. Clin. Exp. Dent. Res. 2021, 7, 109–122. [Google Scholar] [CrossRef]
- Carrizales-Sepúlveda, E.F.; Ordaz-Farías, A.; Vera-Pineda, R.; Flores-Ramírez, R. Periodontal Disease, Systemic Inflammation and the Risk of Cardiovascular Disease. Heart Lung Circ. 2018, 27, 1327–1334. [Google Scholar] [CrossRef]
- Sanz, M.; Marco del Castillo, A.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and Cardiovascular Diseases: Consensus Report. J. Clin. Periodontol. 2020, 47, 268–288. [Google Scholar] [CrossRef]
- Schenkein, H.A.; Loos, B.G. Inflammatory Mechanisms Linking Periodontal Diseases to Cardiovascular Diseases. J. Clin. Periodontol. 2013, 40, S51–S69. [Google Scholar] [CrossRef] [PubMed]
- Roca-Millan, E.; González-Navarro, B.; Del Mar Sabater-Recolons, M.; Marí-Roig, A.; Jané-Salas, E.; López-López, J. Periodontal Treatment on Patients with Cardiovascular Disease: Systematic Review and Meta-Analysis. Med. Oral Patol. Oral Cir. Bucal 2018, 23, E681–E690. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.G.; Schmidt, M.M.; Lopes, R.D.; Dipp, T.; Feijó, I.P.; Schmidt, K.E.S.; Gazeta, C.A.; Azeredo, M.L.; Markoski, M.; Pellanda, L.C.; et al. Treating Periodontal Disease in Patients with Myocardial Infarction: A Randomized Clinical Trial. Eur. J. Intern. Med. 2020, 71, 76–80. [Google Scholar] [CrossRef] [Green Version]
- Tonetti, M.S.; D’Aiuto, F.; Nibali, L. Treatment of Periodontitis and Endothelial Function. Jpn. J. Chest Dis. 2008, 67, 353. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Sridhar, S.; Mcintosh, A.; Messow, C.-M.; Aguilera, E.M.; Del Pinto, R.; Pietropaoli, D.; Górska, R.; Siedlinski, M.; Maffia, P.; et al. Periodontal Therapy and Treatment of Hypertensio—Alternative to the Pharmacological Approach. A Systematic Review and Meta-Analysis. Pharmacol. Res. 2021, 19, 105511. [Google Scholar] [CrossRef]
- Czerniuk, M.R.; Górska, R.; Filipiak, K.J.; Opolski, G. C-Reactive Protein in Patients with Coexistent Periodontal Disease and Acute Coronary Syndromes. J. Clin. Periodontol. 2006, 33, 415–420. [Google Scholar] [CrossRef]
- Surma, S.; Romańczyk, M.; Witalińska-Łabuzek, J.; Czerniuk, M.R.; Łabuzek, K.; Filipiak, K.J. Periodontitis, Blood Pressure, and the Risk and Control of Arterial Hypertension: Epidemiological, Clinical, and Pathophysiological Aspects-Review of the Literature and Clinical Trials. Curr. Hypertens. Rep. 2021, 23, 27. [Google Scholar] [CrossRef]
- Cave, D.R.; Go, M.; Cutler, A.; Goldstein, J.; Dunn, B.; Mobley, H.; Barkin, J.; Fennerty, B.; Hunt, R.; Peura, D.; et al. Transmission and Epidemiology of Helicobacter Pylori. Am. J. Med. 1996, 100, S12–S18. [Google Scholar] [CrossRef]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter Pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Fan, C.; Xie, H.; Bil, J. Effect of Helicobacter Pylori Infection on the Risk of Acute Coronary Syndrome: A Systematic Review and Meta-Analysis. Medecine 2019, 98. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yu, M.; Zhang, R.; Chen, S.; Xi, Y.; Duan, G. A Meta-Analysis of the Association between Helicobacter Pylori Infection and Risk of Atherosclerotic Cardiovascular Disease. Helicobacter 2020, 25, 12761. [Google Scholar] [CrossRef]
- Gravina, A.G.; Zagari, R.M.; De Musis, C.; Romano, L.; Loguercio, C.; Romano, M. Helicobacter Pylori and Extragastric Diseases: A Review. World J. Gastroenterol. 2018, 24, 3204–3221. [Google Scholar] [CrossRef] [PubMed]
- Chmiela, M.; Miszczyk, E.; Rudnicka, K. Structural Modifications of Helicobacter Pylori Lipopolysaccharide: An Idea for How to Live in Peace. World J. Gastroenterol. 2014, 20, 9882–9897. [Google Scholar] [CrossRef]
- Sutanto, H.; Lyon, A. Predicting the Neuro-Cardio-Haemodynamic Outcomes of Sepsis and Its Pharmacological Interventions: Get to the Future through Numerical Equations. J. Physiol. 2021, 1–3. [Google Scholar] [CrossRef]
- Matsuura, E.; Kobayashi, K.; Matsunami, Y.; Shen, L.; Quan, N.; Makarova, M.; Suchkov, S.V.; Ayada, K.; Oguma, K.; Lopez, L.R. Autoimmunity, Infectious Immunity, and Atherosclerosis. J. Clin. Immunol. 2009, 29, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Farah, R.; Hamza, H.; Khamisy-farah, R. A Link between Platelet to Lymphocyte Ratio and Helicobacter Pylori Infection. J. Clin. Lab. Anal. 2018, 32, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farah, R.; Khamisy-Farah, R. Association of Neutrophil to Lymphocyte Ratio with Presence and Severity of Gastritis Due to Helicobacter Pylori Infection. J. Clin. Lab. Anal. 2014, 28, 219–223. [Google Scholar] [CrossRef]
- Lee, K.K.; Stelzle, D.; Bing, R.; Anwar, M.; Strachan, F.; Bashir, S.; Newby, D.E.; Shah, J.S.; Chung, M.H.; Bloomfield, G.S.; et al. Global Burden of Atherosclerotic Cardiovascular Disease in People with Hepatitis C Virus Infection: A Systematic Review, Meta-Analysis, and Modelling Study. Lancet Gastroenterol. Hepatol. 2019, 4, 794–804. [Google Scholar] [CrossRef] [Green Version]
- Wen, D.; Du, X.; Dong, J.Z.; Ma, C.S. Hepatitis C Virus Infection and Risk of Coronary Artery Disease: A Meta-Analysis. Eur. J. Intern. Med. 2019, 63, 69–73. [Google Scholar] [CrossRef]
- Babiker, A.; Hassan, M.; Muhammed, S.; Taylor, G.; Poonia, B.; Shah, A.; Bagchi, S. Inflammatory and Cardiovascular Diseases Biomarkers in Chronic Hepatitis C Virus Infection: A Review. Clin. Cardiol. 2020, 43, 222–234. [Google Scholar] [CrossRef]
- Jabeen, S.; Rasheed, A.; Jabeen, N.; Naz, S.A.; Raza, A. Prevalence and Association of HBV and HCV Infection with Cardiovascular Disease Risk Factors in a Peri-Urban Population. J. Pak. Med. Assoc. 2020, 70, 58–63. [Google Scholar] [CrossRef]
- Mehta, D.A.; Cohen, E.; Charafeddine, M.; Cohen, D.E.; Bao, Y.; Sanchez Gonzalez, Y.; Tran, T.T. Effect of Hepatitis C Treatment with Ombitasvir/Paritaprevir/R + Dasabuvir on Renal, Cardiovascular and Metabolic Extrahepatic Manifestations: A Post-Hoc Analysis of Phase 3 Clinical Trials. Infect. Dis. Ther. 2017, 6, 515–529. [Google Scholar] [CrossRef]
- Wernly, B.; Wernly, S.; Niederseer, D.; Datz, C. Hepatitis C Virus (HCV) Infection and Cardiovascular Disease: Hepatologists and Cardiologists Need to Talk! Eur. J. Intern. Med. 2020, 71, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Corrales-Medina, V.F. Association Between Hospitalization for Pneumonia and Subsequent Risk of Cardiovascular Disease. Physiol. Behav. 2014, 63, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gilley, R.P.; González-Juarbe, N.; Shenoy, A.T.; Reyes, L.F.; Dube, P.H.; Restrepo, M.I.; Orihuela, C.J. Infiltrated Macrophages Die of Pneumolysin-Mediated Necroptosis Following Pneumococcal Myocardial Invasion. Infect. Immun. 2016, 84, 1457–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.O.; Millett, E.R.C.; Quint, J.K.; Orihuela, C.J. Cardiotoxicity during Invasive Pneumococcal Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portier, I.; Campbell, R.A. Role of Platelets in Detection and Regulation of Infection. Arterioscler. Thromb. Vasc. Biol. 2020, 41, 70–78. [Google Scholar] [CrossRef]
- Cognasse, F.; Nguyen, K.A.; Damien, P.; McNicol, A.; Pozzetto, B.; Hamzeh-Cognasse, H.; Garraud, O. The Inflammatory Role of Platelets via Their TLRs and Siglec Receptors. Front. Immunol. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cangemi, R.; Casciaro, M.; Rossi, E.; Calvieri, C.; Bucci, T.; Calabrese, C.M.; Taliani, G.; Falcone, M.; Palange, P.; Bertazzoni, G.; et al. Platelet Activation Is Associated with Myocardial Infarction in Patients with Pneumonia. J. Am. Coll. Cardiol. 2014, 64, 1917–1925. [Google Scholar] [CrossRef] [Green Version]
- Rae, N.; Finch, S.; Chalmers, J.D. Cardiovascular Disease as a Complication of Community-Acquired Pneumonia. Curr. Opin. Pulm. Med. 2016, 22, 212–218. [Google Scholar] [CrossRef]
- Polgreen, L.A.; Riedle, B.N.; Cavanaugh, J.E.; Girotra, S.; London, B.; Schroeder, M.C.; Polgreen, P.M. Estimated Cardiac Risk Associated with Macrolides and Fluoroquinolones Decreases Substantially When Adjusting for Patient Characteristics and Comorbidities. J. Am. Heart Assoc. 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Marra, F.; Zhang, A.; Gillman, E.; Bessai, K.; Parhar, K.; Vadlamudi, N.K. The Protective Effect of Pneumococcal Vaccination on Cardiovascular Disease in Adults: A Systematic Review and Meta-Analysis. Int. J. Infect. Dis. 2020, 99, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, G.; Bai, J.; He, B.; Huang, K.; Hu, X.; Liu, D. Cytomegalovirus Infection and Relative Risk of Cardiovascular Disease (Ischemic Heart Disease, Stroke, and Cardiovascular Death): A Meta-Analysis of Prospective Studies up to 2016. J. Am. Heart Assoc. 2017, 6, 1–10. [Google Scholar] [CrossRef]
- Lv, Y.L.; Han, F.F.; Gong, L.L.; Liu, H.; Ma, J.; Yu, W.Y.; Wan, Z.R.; Jia, Y.J.; Zhang, W.; Shi, M.; et al. Human Cytomegalovirus Infection and Vascular Disease Risk: A Meta-Analysis. Virus Res. 2017, 227, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.L.; Lederman, M.M.; Gianella, S. Partners in Crime: The Role of CMV in Immune Dysregulation and Clinical Outcome During HIV Infection. Curr. HIV/AIDS Rep. 2016, 13, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Nikitskaya, E.; Lebedeva, A.; Ivanova, O.; Maryukhnich, E.; Shpektor, A.; Grivel, J.C.; Margolis, L.; Vasilieva, E. Cytomegalovirus-Productive Infection Is Associated with Acute Coronary Syndrome. J. Am. Heart Assoc. 2016, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, B.; Sinzger, C. Endothelial Cells in Human Cytomegalovirus Infection: One Host Cell out of Many or a Crucial Target for Virus Spread? Thromb. Haemost. 2009, 102, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Weis, M.; Kledal, T.N.; Lin, K.Y.; Panchal, S.N.; Gao, S.Z.; Valantine, H.A.; Mocarski, E.S.; Cooke, J.P. Cytomegalovirus Infection Impairs the Nitric Oxide Synthase Pathway: Role of Asymmetric Dimethylarginine in Transplant Arteriosclerosis. Circulation 2004, 109, 500–505. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, R.B.; Botelho, B.G.; de Hollanda, J.V.G.; Ferreira, L.V.L.; Junqueira de Andrade, L.Z.; Oei, S.S.M.L.; de Souza Mello, T.; Muxfeldt, E.S. Covid-19 and the Cardiovascular System: A Comprehensive Review. J. Hum. Hypertens. 2021, 35, 4–11. [Google Scholar] [CrossRef]
- Szarpak, L.; Filipiak, K.J.; Gasecka, A.; Pruc, M.; Drozd, A.; Jaguszewski, M.J. Correlation between Takotsubo Cardiomyopathy and SARS-CoV-2 Infection. Med. Hypotheses 2021, 146, 110454. [Google Scholar] [CrossRef]
- Gąsecka, A.; Borovac, J.A.; Guerreiro, R.A.; Giustozzi, M.; Parker, W.; Caldeira, D.; Chiva-Blanch, G. Thrombotic Complications in Patients with COVID-19: Pathophysiological Mechanisms, Diagnosis, and Treatment. Cardiovasc. Drugs Ther. 2021, 35, 215–229. [Google Scholar] [CrossRef]
- Zhao, Y.-H.; Zhao, L.; Yang, X.-C.; Wang, P. Cardiovascular Complications of SARS-CoV-2 Infection (COVID-19): A Systematic Review and Meta-Analysis. Rev. Cardiovasc. Med. 2021, 22, 159. [Google Scholar] [CrossRef]
- Gąsecka, A.; Filipiak, K.J.; Jaguszewski, M.J. Impaired Microcirculation Function in Covid-19 and Implications for Potential Therapies. Cardiol. J. 2020, 27, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Szarpak, Ł.; Nowak, B.; Kosior, D.; Zaczynski, A.; Filipiak, K.J.; Jaguszewski, M.J. Cytokines as Predictors of COVID-19 Severity: Evidence from a Meta-Analysis. Polish Arch. Intern. Med. 2021, 131, 98–99. [Google Scholar] [CrossRef]
- Szarpak, L.; Zaczynski, A.; Kosior, D.; Bialka, S.; Ladny, J.R.; Gilis-Malinowska, N.; Smereka, J.; Kanczuga-Koda, L.; Gasecka, A.; Filipiak, K.J.; et al. Evidence of Diagnostic Value of Ferritin in Patients with COVID-19. Cardiol. J. 2020, 27, 886–887. [Google Scholar] [CrossRef]
- Hertanto, D.M.; Sutanto, H.; Wungu, C.D.K. Immunomodulation as a Potent COVID-19 Pharmacotherapy: Past, Present and Future. Preprints 2021. [Google Scholar] [CrossRef]
- Evans, P.C.; Ed Rainger, G.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial Dysfunction in COVID-19: A Position Paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020, 116, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Ruetzler, K.; Szarpak, L.; Ladny, J.R.; Gasecka, A.; Malinowska, N.G.; Pruc, M.; Smereka, J.; Nowak, B.; Filipiak, K.J.; Jaguszewski, M.J. D-Dimer Levels Predict COVID-19 Severity and Mortality. Kardiol. Pol. 2021, 79, 217–219. [Google Scholar] [CrossRef]
- Long, B.; Brady, W.J.; Koyfman, A.; Gottlieb, M. Cardiovascular complications in COVID-19. Am. J. Emerg. Med. 2020, 38, 1504–1507. [Google Scholar] [CrossRef]
- Gasecka, A.; Pruc, M.; Kukula, K.; Gilis-Malinowska, N.; Filipiak, K.J.; Jaguszewski, M.J.; Szarpak, L. Post-Covid-19 Heart Syndrome. Cardiol. J. 2021, 28, 353–354. [Google Scholar] [CrossRef]
- Freiberg, M.S.; Chang, C.H.; Kuller, L.H.; Goetz, M.B.; Leaf, D.; Oursler, K.A.; Mcginnis, K.; Crothers, K.; Sico, J.; Crane, H.; et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern. Med. 2013, 173, 614–622. [Google Scholar] [CrossRef] [PubMed]
- So-Armah, K.; Benjamin, L.A.; Bloomfield, G.S.; Feinstein, M.J.; Hsue, P.; Njuguna, B.; Freiberg, M.S. HIV and Cardiovascular Disease. Lancet HIV 2020, 7, E279–E293. [Google Scholar] [CrossRef]
- Visser, M.R.; Vercellotti, G.M. Herpes Simplex Virus and Atherosclerosis. Eur. Heart J. 1993, 14 (SuppL K), 39–42. [Google Scholar]
- Vilkuna-Rautiainen, T.; Pussinen, P.J.; Roivainen, M.; Petäys, T.; Jousilahti, P.; Hovi, T.; Vartiainen, E.; Asikainen, S. Serum Antibody Response to Periodontal Pathogens and Herpes Simplex Virus in Relation to Classic Risk Factors of Cardiovascular Disease. Int. J. Epidemiol. 2006, 35, 1486–1494. [Google Scholar] [CrossRef]
- Mendy, A.; Vieira, E.R.; Gasana, J. Seropositivity to Herpes Simplex Virus Type 2, but Not Type 1 Is Associated with Premature Cardiovascular Diseases: A Population-Based Cross-Sectional Study. Atherosclerosis 2013, 231, 18–21. [Google Scholar] [CrossRef]
- Rosenfeld, M.E.; Campbell, L.A. Pathogens and Atherosclerosis: Update on the Potential Contribution of Multiple Infectious Organisms to the Pathogenesis of Atherosclerosis. Thromb. Haemost. 2011, 106, 858–867. [Google Scholar] [CrossRef]
- Pothineni, N.V.K.; Subramany, S.; Kuriakose, K.; Shirazi, L.F.; Romeo, F.; Shah, P.K.; Mehta, J.L. Infections, Atherosclerosis, and Coronary Heart Disease. Eur. Heart J. 2017, 38, 3195–3201. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Reduction in C-Reactive Protein and LDL Cholesterol and Cardiovascular Event Rates after Initiation of Rosuvastatin: A Prospective Study of the JUPITER Trial. Lancet 2009, 373, 1175–1182. [Google Scholar] [CrossRef]
- Thomas, M.R.; Outteridge, S.N.; Ajjan, R.A.; Phoenix, F.; Sangha, G.K.; Faulkner, R.E.; Ecob, R.; Judge, H.M.; Khan, H.; West, L.E.; et al. Platelet P2Y12 Inhibitors Reduce Systemic Inflammation and Its Prothrombotic Effects in an Experimental Human Model. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2562–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.; Solomon, S.; Pieper, K.; Reed, S.; Rouleau, J.; Velazquez, E.; White, H.; Howlett, J.; Swedberg, K.; Maggioni, A.; et al. The Effect of Valsartan, Captopril, or Both on Atherosclerotic Events after Acute Myocardial Infarction: An Analysis of the Valsartan in Acute Myocardial Infarction Trial (VALIANT). J. Am. Coll. Cardiol. 2006, 47, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Greenberg, J.D.; Kremer, J.M.; Curtis, J.R.; Hochberg, M.C.; Reed, G.; Tsao, P.; Farkouh, M.E.; Nasir, A.; Setoguchi, S.; Solomon, D.H. Tumour Necrosis Factor Antagonist Use and Associated Risk Reduction of Cardiovascular Events among Patients with Rheumatoid Arthritis. Ann. Rheum. Dis. 2011, 70, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; Macfadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Heart, N.; Rosenberg, Y.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Welsh, P.; Grassia, G.; Botha, S.; Sattar, N.; Maffia, P. Targeting Inflammation to Reduce Cardiovascular Disease Risk: A Realistic Clinical Prospect? Br. J. Pharmacol. 2017, 174, 3898–3913. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, A.D.; Aday, A.W.; Rose, L.M.; Ridker, P.M. Residual Inflammatory Risk on Treatment with PCSK9 Inhibition and Statin Therapy. Circulation 2018, 138, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Tokgözoğlu, L.; Corsini, A.; Sirtori, C.R. PCSK9 Inhibition and Inflammation: A Narrative Review. Atherosclerosis 2019, 288, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Pertzov, B.; Eliakim-Raz, N.; Atamna, H.; Trestioreanu, A.Z.; Yahav, D.; Leibovici, L. Hydroxymethylglutaryl-CoA Reductase Inhibitors (Statins) for the Treatment of Sepsis in Adults—A Systematic Review and Meta-Analysis. Clin. Microbiol. Infect. 2019, 25, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Lowenstern, A.; Storey, R.F.; Neely, M.; Sun, J.L.; Angiolillo, D.J.; Cannon, C.P.; Himmelmann, A.; Huber, K.; James, S.K.; Katus, H.A.; et al. Platelet-Related Biomarkers and Their Response to Inhibition with Aspirin and P2y12-Receptor Antagonists in Patients with Acute Coronary Syndrome. J. Thromb. Thrombolysis 2017, 44, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Husted, S.; Storey, R.F.; Harrington, R.A.; Emanuelsson, H.; Cannon, C.P. Changes in Inflammatory Biomarkers in Patients Treated with Ticagrelor or Clopidogrel. Clin. Cardiol. 2010, 33, 206–212. [Google Scholar] [CrossRef]
- Kiers, D.; van der Heijden, W.A.; van Ede, L.; Gerretsen, J.; de Mast, Q.; van der Ven, A.J.; El Messaoudi, S.; Rongen, G.A.; Gomes, M.; Kox, M.; et al. A Randomized Trial on the Effect of Anti-Platelet Therapy on the Systemic Inflammatory Response in Human Endotoxemia. Thrombo. Haemost. 2017, 117, 1798–1807. [Google Scholar] [CrossRef]
- Gasecka, A.; Nieuwland, R.; Budnik, M.; Dignat-George, F.; Eyileten, C.; Harrison, P.; Lacroix, R.; Leroyer, A.; Opolski, G.; Pluta, K.; et al. Ticagrelor Attenuates the Increase of Extracellular Vesicle Concentrations in Plasma after Acute Myocardial Infarction Compared to Clopidogrel. J. Thromb. Haemost. 2020, 18, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Gąsecka, A.; Rogula, S.; Eyileten, C.; Postuła, M.; Jaguszewski, M.J.; Kochman, J.; Mazurek, T.; Nieuwland, R.; Filipiak, K.J. Role of P2y Receptors in Platelet Extracellular Vesicle Release. Int. J. Mol. Sci. 2020, 21, 6065. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tikellis, C.; Thomas, M.C.; Golledge, J. Angiotensin Converting Enzyme 2 and Atherosclerosis. Atherosclerosis 2013, 226, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar, R.; Shafiee, M.; Hesari, A.R.; Ferns, G.A.; Ghasemi, F.; Avan, A. The Potential Therapeutic Use of Renin–Angiotensin System Inhibitors in the Treatment of Inflammatory Diseases. J. Cell. Physiol. 2019, 234, 2277–2295. [Google Scholar] [CrossRef]
- Mitrovic, V.; Klein, H.H.; Krekel, N.; Kreuzer, J.; Fichtlscherer, S.; Schirmer, A.; Paar, W.D.; Hamm, C.W. Influence of the Angiotensin Converting Enzyme Inhibitor Ramipril on High-Sensitivity C-Reactive Protein (Hs-CRP) in Patients with Documented Atherosclerosis. Z. Kardiol. 2005, 94, 336–342. [Google Scholar] [CrossRef]
- Ceconi, C.; Fox, K.M.; Remme, W.J.; Simoons, M.L.; Deckers, J.W.; Bertrand, M.; Parrinello, G.; Kluft, C.; Blann, A.; Cokkinos, D.; et al. ACE Inhibition with Perindopril and Biomarkers of Atherosclerosis and Thrombosis: Results from the PERTINENT Study. Atherosclerosis 2009, 204, 273–275. [Google Scholar] [CrossRef]
- Han, S.H.; Chung, W.J.; Kang, W.C.; Lee, K.; Park, Y.M.; Shin, M.S.; Ahn, T.H.; Choi, I.S.; Shin, E.K. Rosuvastatin Combined with Ramipril Significantly Reduced Atheroma Volume by Anti-Inflammatory Mechanism: Comparative Analysis with Rosuvastatin Alone by Intravascular Ultrasound. Int. J. Cardiol. 2012, 158, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.J.; Robertson, S.; Barraclough, J.; Xia, Q.; Mallat, Z.; Bursill, C.; Celermajer, D.S.; Patel, S. Colchicine Acutely Suppresses Local Cardiac Production of Inflammatory Cytokines in Patients with an Acute Coronary Syndrome. J. Am. Heart Assoc. 2015, 4, e002128. [Google Scholar] [CrossRef] [Green Version]
- Kajikawa, M.; Higashi, Y.; Tomiyama, H.; Maruhashi, T.; Kurisu, S.; Kihara, Y.; Mutoh, A.; Ueda, S. ichiro. Effect of Short-Term Colchicine Treatment on Endothelial Function in Patients with Coronary Artery Disease. Int. J. Cardiol. 2019, 281, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Kurup, R.; Barraclough, J.; Henriquez, R.; Cartland, S.; Arnott, C.; Misra, A.; Martínez, G.; Kavurma, M.; Patel, S. Colchicine as a Novel Therapy for Suppressing Chemokine Production in Patients with an Acute Coronary Syndrome: A Pilot Study. Clin. Ther. 2019, 41, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Fiolet, A.T.L.; Nidorf, S.M.; Mosterd, A.; Cornel, J.H. Colchicine in Stable Coronary Artery Disease. Clin. Ther. 2019, 41, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, A.H.; Katzenberger, D.R.; Britnell, S.R. Colchicine in Acute Coronary Syndrome: A Systematic Review. Ann. Pharmacother. 2021, 55, 187–197. [Google Scholar] [CrossRef]
- Harrington, R.A. Targeting Inflammation in Coronary Artery Disease. N. Engl. J. Med. 2017, 377, 1197–1198. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Authors | Therapy | Mechanism of Action | Information about Study | Outcomes | Effect |
|---|---|---|---|---|---|
| Ridker et al., 2009 [111] | Rosuvastatin vs. placebo | HMG-CoA inhibitor, pleiotropic effects | A randomized, double-blind, placebo-controlled trial including 15,548 initially healthy men and women | Cardiovascular death, non-fatal stroke, non-fatal AMI, hospitalization due to unstable angina, revascularization | ↓ risk of adverse outcomes (HR = 0.35; 95% CI: 0.23–0.54; p < 0.0001) |
| Thomas et al., 2015 [112] | Ticagrelor vs. clopidogrel vs. placebo | Inhibition of P2Y12 receptor | Randomized injection of E. coli endotoxins to 30 healthy volunteers (10-ticagrelor, 10-clopidogrel, 10-placeboes) | Concentrations of inflammatory biomarkers | Ticagrelor and clopidogrel: ↓ IL6, TNF-α, CCL2 Only ticagrelor: ↓ G-CSF, IL-8; ↑ IL-10; ↔ hsCRP |
| McMurray et al., 2006 [113] | Valsartan vs. captopril | ARB or ACE inhibition | Randomized 14,703 high-risk patients with acute MI to receive captopril or valsartan or the combination of the two | All-cause mortality, cardiovascular mortality, non-fatal cardiovascular events | ↓ risk of adverse outcomes; similar effect of ARBs and ACE-I (HR = 0.97; 95% CI:0.91–1.03; p = 0.286) |
| Tardif et al., 2020 [114] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A randomized, double-blind, placebo-controlled trial including 4745 patients with recent AMI (~2 weeks before) | Cardiovascular death, resuscitated cardiac arrest, AMI, stroke, coronary revascularization | ↓ risk of adverse outcomes (HR = 0.77; 95% CI: 0.61–0.96; p = 0.02) |
| Nidorf et al., 2019 [115] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A randomized, placebo-controlled, double-blind trial including 5522 patients with chronic coronary syndrome | Cardiovascular death, MI, ischemic stroke, coronary revascularization | ↓ risk of adverse outcomes (HR = 0.69; 95% CI: 0.57–0.83; p < 0.001) |
| Nidorf et al., 2013 [116] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A prospective, randomized, observer-blinded, placebo-controlled clinical trial including 532 patients with stable coronary disease | Acute coronary syndrome, out-of-hospital cardiac arrest, ischemic stroke | ↓ risk of adverse outcomes (HR = 0.33; 95% CI: 0.18–0.59; p < 0.001) |
| Ridker et al., 2017 [117] | Canakinumab 150 mg every 3 months vs. placebo | Monoclonal anti-IL-1β antibody | A randomized, double-blind, placebo-controlled trial including 10,061 patients with previous AMI and hsCRP ≥ 2 mg/L | Non-fatal myocardial infarction, nonfatal stroke, cardiovascular death | ↓ risk of adverse outcomes HR = 0.85 (95% CI: 0.74–0.98; p = 0.021) |
| Greenberg et al., 2010 [118] | TNF-α antagonists vs. DMARDs | TNF-α inhibition | A longitudinal cohort study of 10,156 rheumatoid arthritis patients enrolled in the US-based CORRONA database | Non-fatal MI, transient ischemic attack, stroke, cardiovascular death | ↓ risk of adverse outcomes by TNF-α (HR = 0.39; 95% CI 0.19–0.82) |
| Ridker et al., 2019 [119] | Methotrexate 15–20 mg/week vs. placebo | Antimetabolite, immune-system suppressant | A randomized, double-blind, placebo-controlled trial including 4786 patients with previous MI or multivessel coronary disease, additionally with type 2 diabetes or metabolic syndrome | Nonfatal MI, nonfatal stroke, cardiovascular death, unstable angina | ↔ adverse outcomes (HR = 0.96; 95% CI: 0.79–1.16; p = 0.67) ↔ hsCRP, IL-1β, IL-6 ↑ ALT, AST |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szwed, P.; Gąsecka, A.; Zawadka, M.; Eyileten, C.; Postuła, M.; Mazurek, T.; Szarpak, Ł.; Filipiak, K.J. Infections as Novel Risk Factors of Atherosclerotic Cardiovascular Diseases: Pathophysiological Links and Therapeutic Implications. J. Clin. Med. 2021, 10, 2539. https://doi.org/10.3390/jcm10122539
Szwed P, Gąsecka A, Zawadka M, Eyileten C, Postuła M, Mazurek T, Szarpak Ł, Filipiak KJ. Infections as Novel Risk Factors of Atherosclerotic Cardiovascular Diseases: Pathophysiological Links and Therapeutic Implications. Journal of Clinical Medicine. 2021; 10(12):2539. https://doi.org/10.3390/jcm10122539
Chicago/Turabian StyleSzwed, Piotr, Aleksandra Gąsecka, Mateusz Zawadka, Ceren Eyileten, Marek Postuła, Tomasz Mazurek, Łukasz Szarpak, and Krzysztof J. Filipiak. 2021. "Infections as Novel Risk Factors of Atherosclerotic Cardiovascular Diseases: Pathophysiological Links and Therapeutic Implications" Journal of Clinical Medicine 10, no. 12: 2539. https://doi.org/10.3390/jcm10122539