
Pediatric Rhabdomyosarcoma: Epidemiology and Genetic Susceptibility
by
 , , and
, , and
Bailey A. Martin-Giacalone
1,2,† ,
,
P. Adam Weinstein
1,3,†,
Sharon E. Plon
1,4 and
Philip J. Lupo
1,* 1
Department of Pediatrics, Section of Hematology-Oncology, Baylor College of Medicine, Houston, TX 77030, USA
2
Program in Translational Biology and Molecular Medicine, Graduate School of Biomedical Sciences, Baylor College of Medicine, Houston, TX 77030, USA
3
Genetics and Genomics Graduate Program, Graduate School of Biomedical Sciences, Baylor College of Medicine, Houston, TX 77030, USA
4
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
J. Clin. Med. 2021, 10(9), 2028; https://doi.org/10.3390/jcm10092028
Submission received: 29 March 2021
/
Revised: 30 April 2021
/
Accepted: 5 May 2021
/
Published: 9 May 2021
(This article belongs to the Special Issue Current Advances in Pediatric Rhabdomyosarcoma)
Abstract
:Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma in children, yet little is known about its etiology. Studies that examine either environmental exposures or germline genetic predisposition in RMS have begun to identify factors that contribute to this malignancy. Here, we summarize epidemiological reports of RMS incidence in terms of several factors, including age at diagnosis, biological sex, and geographic location. We then describe findings from association studies, which explore the role of parental exposures, birth and perinatal characteristics, and childhood exposures in RMS. Further, we discuss RMS predisposition syndromes and large-scale sequencing studies that have further identified RMS-associated genes. Finally, we propose future directions of study, which aim to advance our understanding of the origin of RMS and can provide knowledge for novel RMS therapies.
1. Introduction
Soft-tissue sarcomas account for 7% of all pediatric cancer diagnoses in the United States (U.S.) [1], and approximately 50% of all pediatric soft-tissue sarcoma diagnoses are rhabdomyosarcoma (RMS) [2]. RMS has an incidence of approximately 4.71 per million children and adolescents less than 20 years of age in the United States. This is similar to other sarcomas, including osteosarcoma (5.09 per million) and Ewing sarcoma (2.95 per million), but lower than the most frequent pediatric cancer—acute lymphoblastic leukemia (ALL, 32.59 per million) [3]. Importantly, survival for RMS is poor. In the most recently completed Children’s Oncology Group (COG) intermediate-risk study, the four-year event-free survival was 63% [4]. For individuals with metastatic disease, the three-year event-free survival is less than 20%, despite multi-agent therapies [5].
RMS can occur anywhere in the body, including the head and neck, genitourinary organs, extremities, and abdomen. Although still debated, evidence suggests that RMS arises from the mesenchymal cell lineage, which is typically fated to become skeletal muscle tissue [6]. The expressions of myogenic factors are the primary support for this hypothesis [7,8,9]. However, recent reports have shown that RMS can also arise from endothelial progenitors, which suggests a mechanism for tumors that originate in areas that are devoid of skeletal muscle tissue [10].
RMS is traditionally classified into two major histological subtypes, embryonal RMS (ERMS) and alveolar RMS (ARMS), of which 60–70% of cases are ERMS and 20–30% are ARMS [11]. Currently, the World Health Organization (WHO) also recognizes two other less common subtypes, namely, pleomorphic and spindle cell/sclerosing RMS [12]. Notably, 80% of ARMS cases are defined by a chromosomal translocation between PAX3 or PAX7 and FOXO1 genes; these translocations result in fusion genes that largely drive oncogenic activity. The other 20% of ARMS cases are similar to ERMS in terms of clinical outcomes and the pattern of somatic mutations [13]. There is an overwhelming consensus among experts that fusion status is a stronger prognostic factor for risk stratification compared to tumor histology. Therefore, fusion status is now integrated into risk stratification for treatment protocols [13,14,15].
Despite the clinical significance of RMS, the underlying etiologies remain unclear. Evidence supports that environmental and genetic factors individually contribute to this malignancy. Therefore, in this review, we present the current knowledge of the factors that play a role in RMS.
2. Epidemiology of RMS
A primary objective of epidemiology is to study the distribution and determinants of disease in human populations. Assessments evaluating the distribution of disease (e.g., incidence, overall survival) are considered descriptive epidemiology, whereas studies evaluating the determinants of disease (e.g., environmental exposures, nutritional status) are considered analytic epidemiology. One of the most comprehensive assessments of the distribution and survival of RMS using a large population-based sample was published in 2009 using data from the Surveillance, Epidemiology, and End Results (SEER) program [16]. The analysis spanned from 1975 to 2005 and included nine U.S. cancer registries, which together represent approximately 35% of the U.S. population; these registries ascertain >98% of cases in their area. However, it is important to note that studies evaluating the determinants of RMS are much more limited, which is likely due to the rarity of this tumor (there are approximately 350 newly diagnosed RMS cases per year in the United States). Additionally, these studies often present equivocal or relatively modest findings. In this section, we summarize studies that have evaluated the distribution and/or determinants of RMS.
2.1. Trends in Incidence
Between 1975 and 2005, there was no significant change in the incidence of RMS overall or ERMS in the United States [11]. This is in contrast with the steady increases in the incidences observed for other pediatric cancers [3,17,18]. While there has been an increase in the incidence of ARMS (annual percent change (APC) = 4.20, 95% confidence interval (CI) = 2.60–5.82), this is likely due to changes in diagnostic criteria. While there is no conclusive evidence that the incidence of RMS has changed over time, there are data suggesting that the incidence of RMS varies significantly by demographic factors, including age, biological sex, and geographic location.
2.2. Incidence by Age and Sex
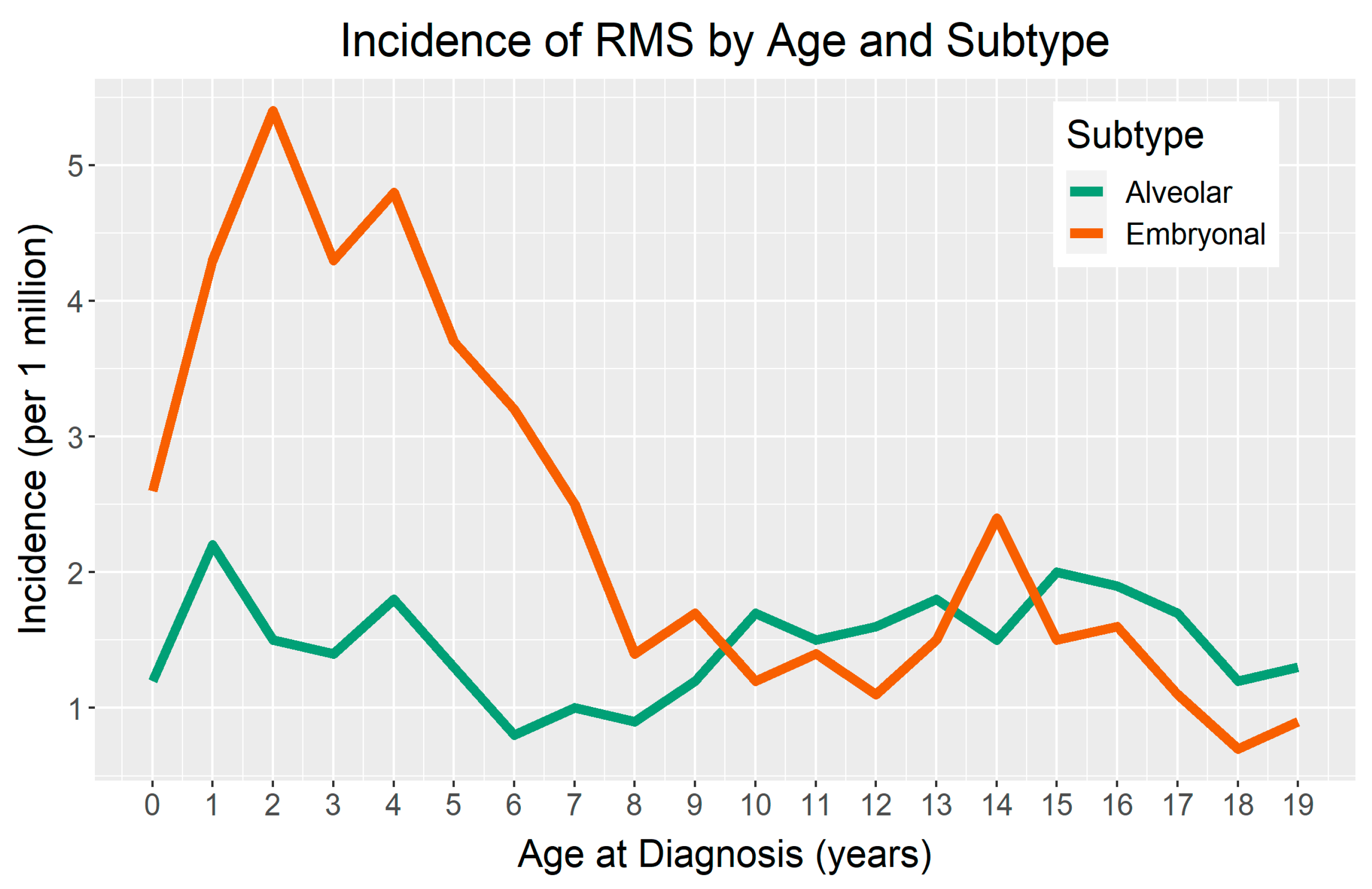
There is a demonstrated difference in the incidence of RMS by age, with peaks occurring during early childhood. For children four years of age or younger, RMS incidence is 6.5 per million in the United States [11]. Notably, there appears to be a second, less pronounced peak in incidence for ERMS during adolescence (Figure 1) [2,11,19]. Osteosarcoma and Ewing sarcoma also illustrate this age-related, second peak in incidence. Valberg et al. suggested that an accelerated pubertal growth period may play a role in susceptibility to sarcomas during adolescence [20].
The incidence of RMS also varies by biological sex. Specifically, RMS incidence is higher in males compared to females (rate ratio (RR) = 1.37, 95% CI = 1.21–1.56); this phenomenon is driven by the predominance of male ERMS diagnoses (RR = 1.51 male to female, 95% CI = 1.27–1.80) [11]. Notably, this disparity (male predominance in risk) is consistent with most other pediatric cancers [21,22], which could yield insights into the etiologies of these malignancies.
2.3. Global Incidence
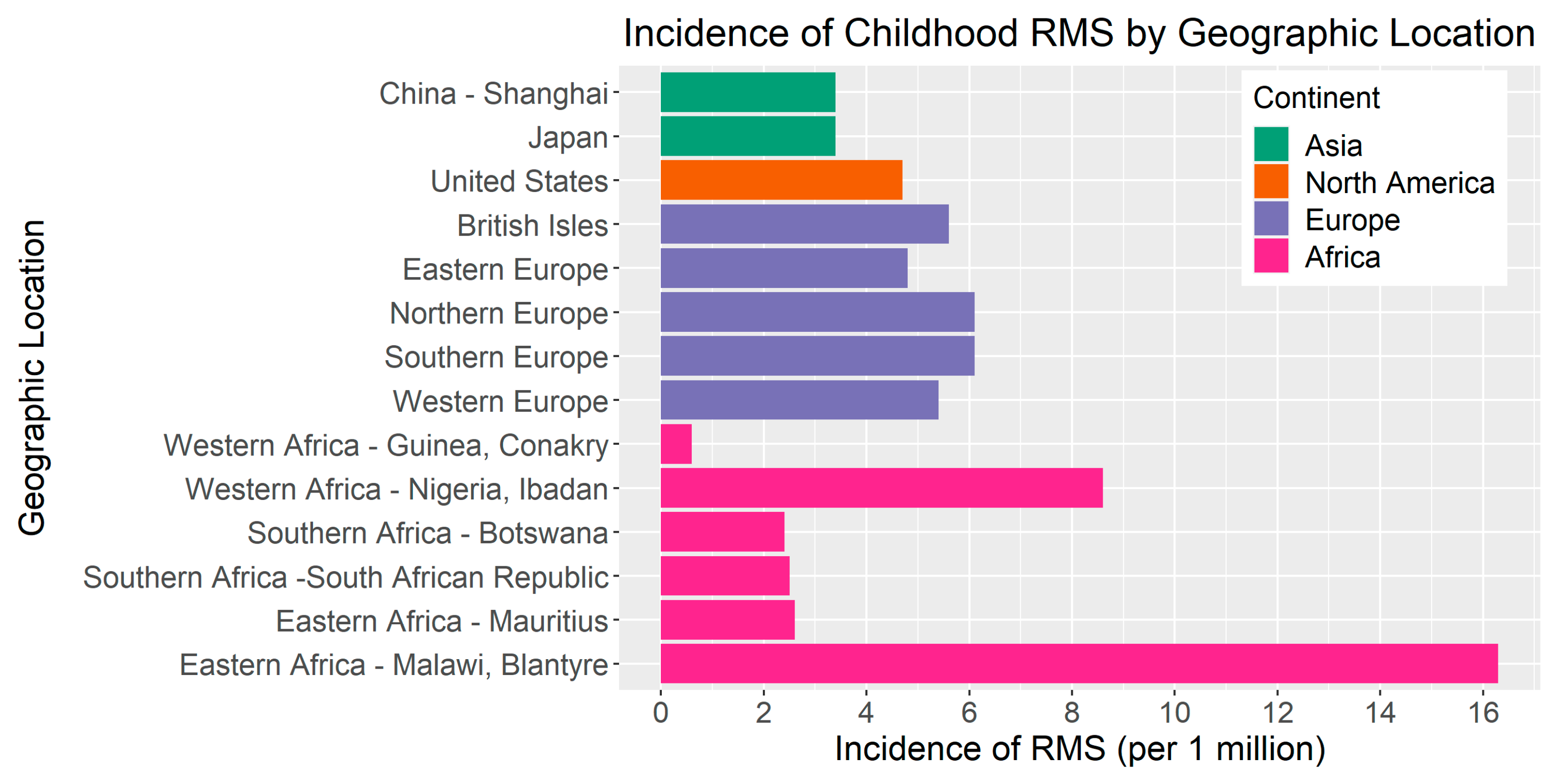
The RMS incidence is modestly elevated in European countries compared to the United States (Figure 2). Based on a report including 59 cancer registries from 19 European countries [23], the overall incidence of RMS in Europe was 5.4 cases per million in children <15 years of age from 1978–1997. The incidence ranged from 4.8 per million in Eastern Europe to 6.1 per million in both Northern Europe and Southern Europe. In a separate study from 1984 to 2010, the incidence of RMS in Sweden was 4.9 cases per million [24]. In contrast, parts of Asia appear to have a lower incidence of RMS compared to the United States and Europe. For instance, in a study of the Shanghai Cancer Registry, the incidence of RMS was 3.4 per million between 2002 and 2005 [25]. Further, a comparative study of the incidence of pediatric cancers in England and Japan estimated that the RMS incidence from 1993–2010 was 3.4 per million (95% CI = 2.8–3.9) in Japan compared to 5.0 per million in England (95% CI = 4.6–5.3) [26].
In sub-Saharan Africa, the RMS incidence is highly variable. For example, a study of 16 population-based registries within the African Cancer Registry Network estimated that the incidence rate of childhood RMS in Western Africa ranged from 0.6 to 8.6 per million compared to 2.4 to 2.5 per million in Southern Africa and 2.6 to 16.3 per million in Eastern Africa. The differences in incidence between and within regions may be due to the variability in environmental factors, cancer surveillance, or diagnostic practices [27]. There are ongoing efforts to improve cancer registration efforts and access to diagnostics in low- and middle-income countries [28].
2.4. Incidence by Race and Ethnicity
Despite the literature showing racial disparities in other pediatric cancers, there is little evidence to suggest that the incidence of RMS varies by race/ethnicity [17]. For instance, Ognjanovic et al. estimated that the overall incidence of RMS for White children was 4.6 per one million person-years compared to 4.9 in Black children [11]. This is notably different than Ewing sarcoma, where the incidence is low among Black children compared to White children [29].
3. Non-Genetic Risk Factors
More than half of all RMS cases occur before 10 years of age [11], which indicates that in utero and early-life environmental exposures may play a large role in RMS etiology. Here, we discuss prenatal exposures, birth characteristics, and childhood exposures that may be associated with RMS risk (Table 1).
3.1. Parental Exposures
3.1.1. Parental Age
Young and advanced parental age is a suggested risk factor for several pediatric cancers. However, RMS study findings have been equivocal in relation to this factor. In a large assessment including linked data from birth certificates and cancer registries from five U.S. states, each five-year increase in maternal age was modestly associated with RMS risk, even after adjusting for paternal age (adjusted odds ratio (aOR) = 1.19, 95% CI = 1.05–1.34). The association of a five-year increase in paternal age was not significant after adjusting for maternal age (aOR = 1.00, 95% CI = 0.90–1.11) [30]. Most recently, in a study of 198 RMS cases, Lupo et al. reported no significant associations of RMS with maternal age or paternal age. For ERMS, risk was slightly reduced for paternal age ≥ 35 years at delivery (incidence rate ratio (IRR) = 0.51, 95% CI = 0.27–0.97) [31].
3.1.2. Additional Parental Exposures
Some reports have evaluated the role of parental exposures on the risk of RMS in the offspring. These exposures include recreational drug use [32,33], prenatal diagnostic radiation [34], and various occupational exposures [35,36]. For example, a case–control study involving 319 RMS cases found that any maternal X-ray examination during pregnancy was associated with an increased risk of RMS in their children (OR = 1.9, 95% CI = 1.1–3.4). The risk of RMS was greatest for X-ray exposure in the first trimester, with a 5.7-fold increase (95% CI = 1.2–27.8), and specifically, for the association of X-ray exposure in the first trimester and ERMS (OR = 10.5, 95% CI = 1.5–458.4) [34]. Additionally, a recent study that examined 1923 RMS cases from the U.K. National Registry of Childhood Tumors found that paternal occupational exposure to electromagnetic fields was associated with RMS risk in the fathers’ children (OR = 1.67, 95% CI = 1.22–2.28) [37]. However, these associations have yet to be replicated.
3.2. Perinatal and Birth Characteristics
3.2.1. Birth Weight and Birth Term
Ognjanovic et al. analyzed data from 583 RMS cases and reported that high birth weight, measured for each 500 g increase, was associated with an increase in the odds of RMS overall (OR = 1.18, 95% CI = 1.09–1.29) and ERMS (OR = 1.27, 95% CI = 1.14–1.42) [38]. In contrast, a study of 722 pediatric RMS cases in California showed that high birth weight, measured as >4000 g, was not associated with RMS overall nor with either histological subtype [39]. Similarly, Lupo et al. did not find a significant association between high birth weight, measured as ≥4000 g, and the odds of overall RMS (OR = 1.35, 95% CI = 0.77–2.37) [40]. However, high birth weight and low birth weight (<2500 g) were significant for ARMS (high: OR = 2.41, 95% CI = 1.09–5.35; low: OR = 4.46, 95% CI = 1.41–14.1).
Neither Ognjanovic et al. nor Shrestha et al. found any significant associations of preterm birth (≤36 weeks of age) and the risk of RMS [38,41]. Conversely, a recent study reported that preterm birth (<37 weeks) was significantly associated with RMS incidence (IRR = 1.74, 95% CI = 1.08–2.79, p = 0.02) [31]. Based on these assessments, there is no clear association between birth weight or term and RMS, as there is with some other pediatric cancers, including hepatoblastoma and ALL [42,43].
3.2.2. Birth Defects
Both chromosomal and non-chromosomal birth defects are relatively well-established risk factors for pediatric cancer [44,45]. In a 1995 study of 249 RMS cases, the cases had greater odds of birth defects compared to the controls (OR = 2.36, 95% CI = 0.92–6.52, p = 0.05), and this association was significant, specifically in males (OR = 3.16, 95% CI = 1.02–10.41, p = 0.02) [46]. However, in a recent study of the risk of cancer in children with birth defects, Lupo et al. assessed 418 cases of RMS and did not find any significant associations with any major chromosomal or non-chromosomal birth defects [44]. The evidence for birth defects and RMS is less clear than for other pediatric cancers, suggesting more work is needed to elucidate these relationships.
3.3. Exposures during Childhood
3.3.1. Immune-Related Factors
Immune-related factors are emerging as significant exposures that may contribute to RMS. A report that evaluated allergies and atopy found significant inverse associations with other atopic conditions, such as allergies (OR = 0.60, 95% CI = 0.41–0.87) and hives (OR = 0.61, 95% CI = 0.38–0.97) [40]. Further, the protective relationship of vaccinations against pediatric cancer is also gaining interest. A study of the association of immunizations and the risk of RMS showed that having any incomplete immunizations significantly increased the risk of RMS (OR = 5.30, 95% CI = 2.47–11.33) [47]. Interestingly, this report did not find any significant associations between infections and childhood RMS.
Breast milk is a known source of immune-related components, such as antibodies and prebiotics. Breastfeeding for ≥12 months was significantly associated with a decreased risk of childhood RMS (OR = 0.36, 95% CI = 0.18–0.70). Further, the analysis showed that there was a statistically significant association between increasing breastfeeding duration and a decreased risk of RMS (p = 0.01) [40].
3.3.2. Additional Childhood Exposures
There is a lack of knowledge of the associations between childhood environmental exposures and RMS. One case–control study from 1982 found that children with diets that included organ meats were at a significantly greater risk of RMS (RR = 3.7, 95% CI = 1.5–8.3, p = 0.04). They also reported that children exposed to chemicals other than pesticides and rodenticides were at a 3.2-fold greater risk of RMS (95% CI = 1.1–9.2). However, this study only included 33 RMS cases [48]. Compared to other pediatric cancers, there is a dearth of studies that evaluated the impact of exposures during early life and the risk of RMS.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of suggested risk factors for pediatric rhabdomyosarcoma.
| Risk Factor | Odds Ratio (95% CI) | No. of Cases | References |
|---|---|---|---|
| Parental exposures | |||
| Parental age | |||
| Each 5-year increase in maternal age | 1.19 (1.05–1.34) * | 556 | [30] |
| Recreational drug use | |||
| Maternal | 3.1 (1.4–6.7) * | 322 | [32] |
| Paternal | 2.0 (1.3–3.3) * | 322 | [32] |
| Prenatal diagnostic radiation | 1.9 (1.1–3.4) * | 319 | [34] |
| Occupational exposures | |||
| Agent Orange exposure | 1.72 (0.55–5.41) | 319 | [36] |
| Electromagnetic fields | 1.67 (1.22–2.28) * | 1923 | [37] |
| Perinatal/birth characteristics | |||
| Birth weight | |||
| Overall RMS a, each 500 g increase | 1.18 (1.09–1.29) * | 583 | [38] |
| ERMS b, each 500 g increase | 1.27 (1.14–1.42) * | 363 | [38] |
| ARMS c, ≥4000 g | 2.41 (1.09–5.35) * | 66 | [49] |
| ARMS, <2500 g | 4.46 (1.41–14.1) * | 66 | [49] |
| Preterm birth | |||
| Overall RMS, GA d <37 weeks | 1.74 (1.08–2.79) *,e | 198 | [31] |
| ERMS, GA <37 weeks | 1.97 (0.98–3.94) e | 198 | [31] |
| Childhood exposures | |||
| Allergies | 0.60 (0.41–0.87) * | 322 | [40] |
| Hives | 0.61(0.38–0.97) * | 322 | [40] |
| Incomplete immunizations | 5.30 (2.47–11.33) * | 322 | [47] |
| Breastfeeding, ≥12 months | 0.36 (0.18–0.70) * | 322 | [40] |
* Statistically significant, a rhabdomyosarcoma (RMS), b embryonal RMS (ERMS), c alveolar RMS (ARMS), d gestational age (GA), e incidence rate ratio (IRR).
4. Future Directions
Elucidating the role of prenatal/birth characteristics and environmental exposures in RMS is essential to providing a comprehensive understanding of RMS etiology. However, compared to other pediatric cancers, these associations are highly understudied and replications of previous exposure assessments are scarce. Therefore, novel assessments are needed to understand the effects of non-genetic exposures on RMS risk. Future studies should also consider stratifying their analyses by histology and fusion status. Finally, there is a need for comparative studies that evaluate the associations of exposures and RMS risk using data from multiple countries and diverse populations. Obtaining more representative global estimates of these associations would significantly enhance the current understanding of the factors that contribute to RMS etiology.
5. Genetic Risk
While somatic alterations associated with RMS, such as the PAX/FOXO1 fusion, have been well characterized, less is known about germline susceptibility to RMS. A 2015 study from our group compared 322 individuals with RMS to 322 population-based controls to evaluate whether there was an association between family history of cancer and RMS [50]. We found that individuals with a first-degree relative who had any history of cancer had an increased risk of developing ERMS (OR = 2.44, 95% CI = 1.54–3.86). Additionally, individuals with a first-degree relative that had a cancer diagnosis when they were younger than 30 years old were also at an increased risk of developing RMS (OR = 2.37, 95% CI = 1.34–4.18). These findings suggest that inherited variants in cancer predisposition genes may play a role in increasing pediatric RMS risk. To date, there have been no genome-wide association studies (GWASs) of individuals with RMS to further explore potential common genetic variations that are specific to this malignancy.
Unlike cancers such as breast cancer or retinoblastoma, there is no Mendelian (single-gene) disorder where RMS is the primary manifestation. Thus, we review a diverse set of Mendelian disorders that have each been linked to an increased risk of RMS. Further, we discuss recently completed pediatric cancer and RMS-specific sequencing studies that have identified germline variants of interest amongst RMS patients.
5.1. Autosomal Dominant Mendelian Disorders
5.1.1. Li–Fraumeni Syndrome (LFS)
LFS is a cancer predisposition syndrome with an autosomal dominant inheritance pattern that is associated with deleterious germline mutations (pathogenic variants) in the TP53 gene. These variants are reported to either inactivate the tumor suppression activity of TP53 or have a dominant negative impact [51]. Multiple different pediatric and adult-onset cancers are associated with LFS, many of which have an earlier age of onset compared to the general population.
The first report of LFS by Li and Fraumeni in 1969 included a cohort of four RMS patients [52]. Diller et al. reported three children with germline TP53 missense mutations among 33 sporadic RMS patients [53]. Further, these three cases were all noted to be under the age of three years, indicating that germline TP53 mutations may play a role in developing RMS earlier in life. Ognjanovic et al. also determined that children under the age of three years with a germline TP53 mutation are at the greatest risk of developing RMS compared to other sarcomas (OR = 16.7, 95% CI = 9.4–29.7) [54]. Hettmer et al. further explored whether germline TP53 mutations were associated with a specific RMS histological subtype [55]. They evaluated eight consecutive children with TP53 mutations and RMS and found they all displayed the anaplastic RMS subtype. Furthermore, they evaluated seven additional patients with anaplastic RMS and unknown TP53 status and discovered that three of these individuals harbored germline TP53 pathogenic variants. While these findings suggest an association between anaplastic RMS and TP53 mutations, the authors note that these conclusions should be confirmed in larger cohorts.
5.1.2. Costello Syndrome (CS)
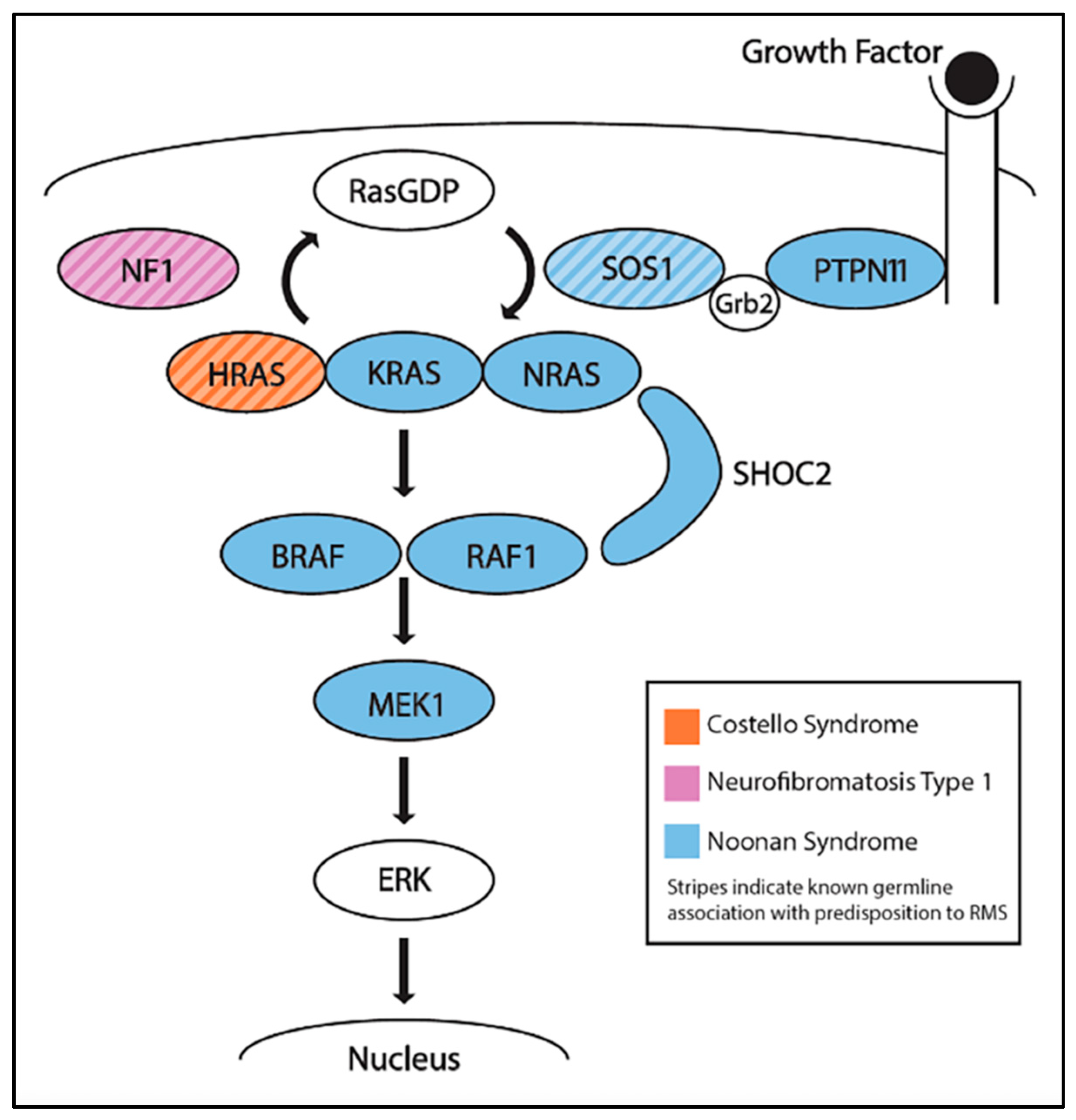
CS is a rare, developmental disorder and cancer predisposition syndrome that is caused by heterozygous activating mutations in HRAS. CS is one of many conditions, including Noonan syndrome and neurofibromatosis type 1, that result in the overactivation of the Ras pathway and are often referred to as RASopathies (Figure 3) [56]. Individuals with CS can have a variety of phenotypes, including significant intellectual disability, a broad neck, short stature, heart defects, and pubertal delay [57]. In a retrospective study of 29 patients with CS and a cancer diagnosis, RMS was the most commonly seen, occurring in 19 patients: 9 EMRS, 1 ARMS, 1 pleomorphic, 1 spindle-cell, 1 mixed histology, and 6 unclassified [58]. Other tumors that were detected included neuroblastoma (n = 5), bladder cancer (n = 4), and fibrosarcoma (n = 1). Kratz et al. later performed a retrospective analysis of cancer incidence in a larger cohort of 784 individuals with known mutations in the Ras pathway [59]. Two of twelve individuals with cancer had germline mutations in HRAS and developed ERMS.
5.1.3. Neurofibromatosis Type 1 (NF1)
NF1 is an autosomal dominant syndrome associated with inactivating mutations in NF1, a gene whose product is involved in the Ras pathway (Figure 3). NF1 includes multiple different features, including café au lait spots, benign tumors in nervous tissues (neurofibromas), optic gliomas, and learning disabilities [60]. The protein neurofibromin, encoded by NF1, typically negatively regulates the Ras pathway [61], thus inactivating mutations in NF1 also in hyperactivation of the Ras pathway. Urogenital, and to a lesser extent, orbital, ERMS have been associated with NF1 [62]. Although studies have shown that the prevalence of RMS amongst NF1 patients is less than 1%, it is still greater than in individuals without NF1 [63]. In a retrospective study of 16 RMS patients with NF1, all 16 patients were identified as having ERMS, providing further evidence that patients with NF1 have a small but significant ERMS predisposition [64].
5.1.4. Noonan Syndrome (NS)
NS is another autosomal dominant syndrome that is caused by defects in several genes in the Ras pathway, including KRAS, NRAS, RAF1, BRAF, PTPN11, SOS1, SHOC2, or MEK1, which result in Ras pathway activation (Figure 3). Individuals with NS can display developmental delay, intellectual disabilities, distinctive facial features, and congenital heart defects [65]. Despite the multiple genes that are associated with NS, the only gene with reported germline mutations in two patients with NS and ERMS is SOS1 [66,67]. In a study of 45 individuals with NS and a history of cancer, the top three malignancies were ALL (n = 8), neuroblastoma (n = 8), and RMS (n = 6, 1 botryoid and 5 ERMS) [58].
5.1.5. DICER1 Tumor Syndrome
DICER1 tumor syndrome, also known as pleuropulmonary blastoma familial tumor susceptibility syndrome, is an autosomal dominant disorder resulting from germline inactivating mutations of DICER1. DICER1 is a miRNA processing enzyme, and as such, broadly influences gene expression [68]. Individuals with mutations in DICER1 are at an increased risk of developing RMS and many other cancer types, including pleuropulmonary blastoma (the highest cancer risk), cystic nephroma, thyroid cancer, and ovarian Sertoli–Leydig cell tumors [69]. RMS tumors that are associated with DICER1 mutation often arise in the cervix [70]. In these individuals, the somatic variant (second hit) in the tumor is frequently a missense variant in the RNAse III domain [71]. Due to the association of RMS with germline loss-of-function DICER1 mutations, Doros et al. investigated the prevalence of somatic DICER1 mutations in a cohort of 52 sporadic, unselected ERMS patients that did not have DICER1 tumor syndrome [72]. They found that two of the 52 sporadic cases displayed DICER1 mutations in the tumors, one of which was a missense mutation that was predicted to interrupt a splice site and the other was an in-frame deletion of a glutamic acid residue near the RNAse III domains. There was no functional assessment of these mutations, but both were predicted to negatively impact DICER1 function. These findings strengthened the putative association between DICER1 syndrome and increased RMS risk. Stewart et al. surveyed three cohorts of individuals with germline mutations in DICER1 to determine the incidence of neoplasms [73]. Their study revealed six RMS patients among the 148 probands, and RMS was the fourth most commonly detected neoplasm amongst the DICER1 mutation carriers, behind pleuropulmonary blastoma (n = 47), Sertoli–Leydig cell tumor (n = 24), and thyroid cancer (n = 10). The mean age of diagnosis of the six RMS patients was 10 years old.
5.1.6. Rubinstein–Taybi Syndrome (RTS)
RTS is an autosomal dominant disorder that is caused by inactivating mutations, primarily in CREBBP, and to a lesser degree, EP300. Individuals with RTS have multiple congenital anomalies, including broad thumbs and dysmorphic facial features [65]. An early case report from 1981 described a 4-year-old nasopharyngeal RMS patient with RTS [74]. In a more recent study of more than 700 patients with RTS, two further individuals were identified that had nasopharyngeal RMS [75].
5.1.7. Retinoblastoma (RB)
Hereditary RB is an autosomal dominant syndrome that is caused by inactivating mutations in RB1. Approximately 90% of individuals with a germline mutation in RB1 will develop retinoblastoma [76]. After the treatment and eradication of the initial retinoblastoma, these individuals can also develop a number of secondary cancers later in life. RMS, especially ERMS, is occasionally detected as one such secondary neoplasm in individuals with RB1 mutations that have completed retinoblastoma treatment [77]. Temming et al. analyzed 648 retinoblastoma patients from 1940–2008 to determine whether their treatment course (radiotherapy or chemotherapy) influenced the incidence of secondary cancers [78]. They found that soft-tissue sarcomas (including RMS) were the most commonly seen secondary cancers seen in inherited retinoblastoma (RB1) survivors, and that radiotherapy significantly increased the risk of secondary cancer in retinoblastoma patients (HR = 2.97, 95% CI = 1.64–5.36). Kleinermann et al. evaluated 963 survivors (one-year post-diagnosis) of hereditary retinoblastoma to calculate the risk of developing a second soft-tissue sarcoma amongst these individuals [79]. They reported a total of 69 sarcoma diagnoses amongst 68 patients, 8 of which were RMS. They calculated that retinoblastoma survivors were at a significantly increased risk of developing RMS (SIR = 279, 95% CI = 120–551). An example of an RMS transition after radiotherapy treatment was described by Cebulla et al. in a patient who presented with retinoblastoma, received radiotherapy, and subsequently developed RMS in the field of radiation within 2 years of treatment [80].
5.2. Autosomal Recessive Mendelian Disorders
5.2.1. Constitutional Mismatch Repair Deficiency (CMMRD)
CMMRD is an autosomal recessive disorder that is caused by biallelic mutations in one of the DNA mismatch repair genes: MLH1, MSH2, MSH6, and PMS2. Individuals with CMMRD may display café au lait spots, similar to individuals with NF1. They also have a high risk of developing a wide variety of malignancies in childhood [81]. In 2015, Lavoine et al. performed a retrospective study of 31 CMMRD patients with a history of cancer [82]. Although CNS tumors, leukemias, and gastrointestinal tumors predominated, three individuals were diagnosed with sarcomas (one dermatofibrosarcoma and two osteosarcoma). In another study, two patients from consanguineous families with CMMRD developed RMS [83]. Overall, it has been reported that 10–15% of CMMRD patients present “non-canonical” tumors, including soft-tissue sarcomas, such as RMS [81].
5.2.2. Fanconi Anemia (FA)
FA is an autosomal recessive disorder that is characterized by biallelic mutations in the FANC family of genes. Individuals with FA have a variety of hematological problems, including frequent development of acute myelogenous leukemia [84]. A retrospective study of 10 pediatric patients with FA and a history of solid tumors identified two patients with RMS, one of which had ERMS [85]. Human xenograft assays have shown that in fusion-positive ARMS, inhibition of the FANCD2 gene may represent a potentially viable treatment avenue [86].
5.2.3. Mosaic Variegated Aneuploidy Syndrome (MVA)
MVA is a rare autosomal recessive disorder that is characterized by consistent gain or loss of chromosomes (often monosomy or trisomy) in cells throughout a variety of tissues. Those affected with MVA typically display growth deficiencies, microcephaly, and an increased risk of developing pediatric cancer [87]. Hanks et al. surveyed five families with a history of MVA, two of which had instances of RMS: a 7-year-old with ERMS of the soft palate and a 5-month-old with ERMS of the vagina. Both of these patients had biallelic deleterious or loss-of-function mutations in the BUB1B gene, the first of several genes associated with MVA [88]. BUB1B protein plays a critical role in the mitotic spindle checkpoint.
5.3. Epigenetic Mechanisms
Beckwith–Wiedemann Syndrome (BWS)
BWS is caused by a complex mechanism of epigenetic silencing of regions of chromosome 11. Individuals with BWS display overgrowth phenotypes and have an increased risk of developing multiple cancers, including Wilms tumor, hepatoblastoma, neuroblastoma, and RMS [89]. Smith et al. reported three patients with BWS and RMS, all of whom had fusion-negative ARMS [90]. They note these findings were a departure from earlier case reports [91,92,93], which suggested that RMS in BWS may only be associated with ERMS. More recently, Mussa et al. performed a pooled analysis of 1370 BWS patients to determine cancer incidence and whether certain BWS epigenetic changes had different cancer phenotypes [94]. While Wilms tumor (n = 48) and hepatoblastoma (n = 23) were the two most common cancers detected, they also described seven patients with RMS amongst the pooled cohorts, six of whom shared the same epigenetic change, a loss of methylation of ICR2. They documented that patients with BWS have an increased risk of RMS compared to the general population. A follow-up pooled analysis from Maas et al. added a further 229 BWS patients and detected an additional RMS case [95].
6. Large-scale Pediatric Cancer Germline Genetics Studies
Recent studies of larger cohorts of unselected pediatric cancer patients have both identified germline findings relevant to RMS predisposition, as well as somatic mutations that are associated with the previously discussed cancer predisposition syndromes.
Our group completed the Baylor Advancing Sequencing in Childhood Cancer Care (BASIC3) study, which performed paired clinical germline (normal) and somatic (tumor) whole-exome sequencing (WES) on newly diagnosed pediatric cancer patients with solid tumors [96]. Of the first 150 patients, there were 15 patients diagnosed with RMS. There was one germline finding, namely, a patient with a TP53 germline mutation. Three further patients had somatic mutations in genes related to cancer predisposition syndromes, namely, DICER1, HRAS (Costello), and PTPN11 (Noonan).
In a study from St. Jude Research Hospital, Zhang et al. reviewed whole-genome and whole-exome sequencing on 1120 pediatric cancer patients to determine the prevalence of germline mutations in known cancer predisposition genes [97]. Among the 43 RMS patients assessed, they reported germline mutations in three individuals: two in TP53 (Li–Fraumeni Syndrome) and one in BRCA2. BRCA2 belongs to the Fanconi anemia family of genes and is well-known for its association with adult-onset cancer, including breast and ovarian cancer. Although this was the first report of heterozygous BRCA2 mutations and RMS, more recent studies (see below) have strengthened the evidence of this relationship.
In the Peds-MiOncoSeq effort at the University of Michigan, Mody et al. performed germline and tumor WES and transcriptomic analysis on 102 children and young adults with relapsed or rare cancers in an effort to identify mutations in these individuals that may be clinically actionable [98]. They identified two RMS patients with germline findings: one with a frameshift FANCD2 mutation (FA) and another with a stopgain TP53 (LFS) mutation.
Large-Scale RMS-Specific Studies
Although RMS is associated with a variety of cancer predisposition syndromes (as described above), it is not the primary tumor type observed in any disorder. RMS-specific sequencing studies can address this gap in knowledge and identify whether there are genes of interest that are specific to RMS. Two such RMS-specific cohort sequencing studies have recently explored both the frequency and identity of germline mutations in cancer predisposition genes in RMS patients. Our group completed the Genetics of Embryonal and Alveolar Rhabdomyosarcoma Study (GEARS), which performed germline WES analysis of 615 RMS patients from the Children’s Oncology Group in order to determine the prevalence of germline mutations in 70 known cancer predisposition genes [99]. We found that 45 of the 615 patients (7.3%) possessed cancer predisposition germline mutations. ERMS was more common than ARMS in these individuals, and all of this group had fusion-negative RMS. Those with a germline mutation were more likely to be younger at diagnosis. The most common germline findings in CPGs that are known to be associated with RMS were in TP53 (LFS; n = 11), NF1 (n = 9), and HRAS (CS; n = 5). There were 12 patients with germline mutations in cancer predisposition genes that are not currently associated with RMS, including two genes with heterozygous mutations in more than one patient: BRCA2 (n = 6) and SDHA (n = 2). The enrichment of germline variants in BRCA2 among RMS patients compared to a control population was significant (OR = 3.55, 95% CI = 1.43–9.40). There were no patients with biallelic mutations consistent with a recessive syndrome diagnosis.
A second study from Kim et al. used whole-genome sequencing (WGS) to determine the rate and identity of germline mutations in a cohort of 273 intermediate-risk RMS patients from the COG [100]. They also performed WES on a secondary cohort, unselected for risk, that comprised 121 RMS patients from the COG, the Cooperative Human Tissue Network, the Children’s Hospital at Westmead, and NCI for further comparison. Among both cohorts, there were individuals with mutations in RMS-associated cancer predisposition genes, including TP53, DICER1, NF1, MSH2, and MSH6. This study reported one individual with a heterozygous germline mutation in BRCA2 and another with biallelic germline mutations in BRCA2 consistent with Fanconi anemia D2. In this study, germline mutations were also detected in genes that are rarely reported with RMS, including single individuals with an ERCC2, CBL, SMARCA4, RET, and RH mutation. Whether these five newly reported genes play a role in RMS is unknown and replication studies are needed.
7. Future Directions
RMS-specific studies are needed to address the lack of knowledge of RMS susceptibility genes and pathogenic variants. A GWAS of RMS may further identify common variants and novel genes that are associated with an increased risk of developing this tumor. Additionally, exome and/or genome sequencing is emerging as a clinically relevant tool for identifying novel genes in pediatric cancer. To build upon the recent RMS sequencing studies, analysis of larger sets of sarcoma-related genes and the integration of techniques to increase the power of detection (e.g., burden testing) is needed. As more germline susceptibility variants are discovered using these methods, the identification of regulatory variants that cooperate with somatic driver mutations would greatly enhance the approach to multidisciplinary, integrated studies of RMS. This has been a notable area of study in Ewing sarcoma [101,102].
Furthermore, an analysis of the different subtypes of RMS may identify whether certain predisposition syndromes are specifically associated with one specific, or a few, RMS subtypes. For example, in medulloblastoma (MB), children with certain inherited cancer syndromes will present with specific MB subtypes: individuals with Gorlin syndrome have an increased risk to develop Sonic Hedgehog (SHH)-MB; conversely, individuals with familial adenomatous polyposis syndrome are at risk of developing the WNT-MB subtype [103]. Discovering new associations and further studying purported associations will further our understanding of subtype-specific RMS predisposition including lesser-known subtypes, such as pleomorphic and spindle-cell/sclerosing RMS.
To date, RMS has been modeled in human cell lines, mice, and zebrafish. Fusion-positive RMS has been modeled in mouse mesenchymal stem cells [104], as well as human fetal skeletal muscle cells [105]. Fusion-negative RMS has been modeled in zebrafish through the over-activation of the Ras pathway [106]. These cellular and non-human models can similarly be used in the future to further explore both known CPGs for disease mechanisms, as well as novel CPGs derived from GWASs or sequencing studies.
Additionally, studies of the role of genetic variants in cancer outcomes could generate important clinical knowledge. Although the identification of pathogenic germline variants in rare diseases, such as RMS, is challenging, studies of other childhood cancers have shown that pathogenic germline variants may contribute to adverse outcomes. Studies have shown that germline variants in GATA3 and TP53 may predispose children with ALL to outcomes such as secondary malignancies and relapse [107,108,109]. Further, a pan-cancer analysis of childhood cancer revealed that of the 6% of patients who harbored a pathogenic germline variant in a CPG, 3% also carried germline alterations that were considered potentially druggable events (defined as variants in genes with direct or indirect targeted therapy that is currently available or in development). As awareness of the importance of germline genetics for the therapeutic development of cancer increases [110], there are opportunities to integrate the knowledge of germline variants with known somatic targets for RMS patients with particularly poor or severe outcomes.
8. Conclusions
As the breadth of knowledge of the individual effects of environmental exposures and genetic variants on RMS predisposition increases, a significant step for understanding the disease etiology would be to examine the gene–environment interactions that may also contribute to this complex disease. To our knowledge, there have been no published studies of gene–environment interactions in RMS, though there have been some reports in other pediatric cancers [111]. Ultimately, there is a need for increased efforts for both environmental and genetic studies in order to translate findings into clinical practice. Confirming the environmental exposures that play a role in RMS could inform preventative strategies, primarily regarding maternal health and in utero exposures. Additionally, identifying novel RMS-associated genes and germline variants will aid in enhancing risk stratification, developing therapeutic targets, and addressing poor outcomes.
Author Contributions
Conceptualization, B.A.M.-G., P.A.W., S.E.P. and P.J.L.; methodology, B.A.M.-G., P.A.W., S.E.P. and P.J.L.; formal analysis, B.A.M.-G., P.A.W., S.E.P. and P.J.L.; investigation, B.A.M.-G., P.A.W., S.E.P. and P.J.L.; resources, B.A.M.-G., P.A.W., S.E.P. and P.J.L.; data curation, B.A.M.-G., P.A.W., S.E.P. and P.J.L.; writing—original draft preparation, B.A.M.-G., P.A.W., S.E.P. and P.J.L.; writing—review and editing, B.A.M.-G., P.A.W., S.E.P. and P.J.L.; visualization, B.A.M.-G., P.A.W. and P.J.L.; supervision, S.E.P. and P.J.L.; project administration, B.A.M.-G., P.A.W., S.E.P. and P.J.L.; funding acquisition, S.E.P. and P.J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Cancer Prevention and Research Institute of Texas (CPRIT) grant number RP170071, the Children’s Oncology Group Foundation, and the Isabella Santos Foundation.
Conflicts of Interest
S.E.P. is a member of the Scientific Advisory Board of Baylor Genetics Laboratories.
References
- Li, J.; Thompson, T.D.; Miller, J.W.; Pollack, L.A.; Stewart, S.L. Cancer Incidence Among Children and Adolescents in the United States, 2001–2003. Pediatrics 2008, 121, e1470–e1477. [Google Scholar] [CrossRef]
- Gurney, J.; Young, J.; Roffers, S.; Smith, M.; Bunin, G. Soft Tissue Sarcomas. In Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995; Ries, L., Smith, M., Gurney, J., Linet, M., Tamra, T., Young, J., Bunin, G., Eds.; National Cancer Institute, SEER Program: Bethesda, MD, USA, 1999; pp. 111–124. [Google Scholar]
- Siegel, D.A.; King, J.; Tai, E.; Buchanan, N.; Ajani, U.A.; Li, J. Cancer Incidence Rates and Trends Among Children and Adolescents in the United States, 2001–2009. Pediatrics 2014, 134, e945–e955. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, D.S.; Spunt, S.L.; Skapek, S.X. Children’s Oncology Group’s 2013 Blueprint for Research: Soft Tissue Sarcomas. Pediatr. Blood Cancer 2013, 60, 1001–1008. [Google Scholar] [CrossRef] [Green Version]
- Malempati, S.; Hawkins, D.S. Rhabdomyosarcoma: Review of the Children’s Oncology Group (COG) soft-tissue Sarcoma committee experience and rationale for current COG studies. Pediatr. Blood Cancer 2012, 59, 5–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashi, V.P.; Hatley, M.E.; Galindo, R.L. Probing for a deeper understanding of rhabdomyosarcoma: Insights from complementary model systems. Nat. Rev. Cancer 2015, 15, 426–439. [Google Scholar] [CrossRef] [Green Version]
- Charytonowicz, E.; Cordon-Cardo, C.; Matushansky, I.; Ziman, M. Alveolar rhabdomyosarcoma: Is the cell of origin a mesenchymal stem cell? Cancer Lett. 2009, 279, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Linardic, C.M.; Naini, S.; Herndon, J.E.; Kesserwan, C.; Qualman, S.J.; Counter, C.M. The PAX3-FKHR Fusion Gene of Rhabdomyosarcoma Cooperates with Loss of p16INK4A to Promote Bypass of Cellular Senescence. Cancer Res. 2007, 67, 6691–6699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, C.; Arenkiel, B.R.; Coffin, C.M.; El-Bardeesy, N.; Depinho, R.A.; Capecchi, M.R. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: Cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004, 18, 2614–2626. [Google Scholar] [CrossRef] [Green Version]
- Drummond, C.J.; Hanna, J.A.; Garcia, M.R.; Devine, D.J.; Heyrana, A.J.; Finkelstein, D.; Rehg, J.E.; Hatley, M.E. Hedgehog Pathway Drives Fusion-Negative Rhabdomyosarcoma Initiated from Non-myogenic Endothelial Progenitors. Cancer Cell 2018, 33, 108–124.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ognjanovic, S.; Linabery, A.M.; Charbonneau, B.; Ross, J.A. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer 2009, 115, 4218–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudzinski, E.R.; Anderson, J.R.; Hawkins, D.S.; Skapek, S.X.; Parham, D.M.; Teot, L.A. The World Health Organization Classification of Skeletal Muscle Tumors in Pediatric Rhabdomyosarcoma: A Report from the Children’s Oncology Group. Arch. Pathol. Lab. Med. 2015, 139, 1281–1287. [Google Scholar] [CrossRef] [Green Version]
- Rhee, D.S.; Rodeberg, D.A.; Baertschiger, R.M.; Aldrink, J.H.; Lautz, T.B.; Grant, C.; Meyers, R.L.; Tracy, E.T.; Christison-Lagay, E.R.; Glick, R.D.; et al. Update on pediatric rhabdomyosarcoma: A report from the APSA Cancer Committee. J. Pediatr. Surg. 2020, 55, 1987–1995. [Google Scholar] [CrossRef]
- Hibbitts, E.; Chi, Y.; Hawkins, D.S.; Barr, F.G.; Bradley, J.A.; Dasgupta, R.; Meyer, W.H.; Rodeberg, D.A.; Rudzinski, E.R.; Spunt, S.L.; et al. Refinement of risk stratification for childhood rhabdomyosarcoma using FOXO1 fusion status in addition to established clinical outcome predictors: A report from the Children’s Oncology Group. Cancer Med. 2019, 8, 6437–6448. [Google Scholar] [CrossRef] [Green Version]
- Rudzinski, E.R.; Kelsey, A.; Vokuhl, C.; Linardic, C.M.; Shipley, J.; Hettmer, S.; Koscielniak, E.; Hawkins, D.S.; Bisogno, G. Pathology of childhood rhabdomyosarcoma: A consensus opinion document from the Children’s Oncology Group, European Paediatric Soft Tissue Sarcoma Study Group, and the Cooperative Weichteilsarkom Studiengruppe. Pediatr. Blood Cancer 2021, 68, e28798. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute: Surveillance, Epidemiology, and End Results Program. Available online: https://seer.cancer.gov/ (accessed on 2 March 2021).
- Linabery, A.M.; Ross, J.A. Trends in childhood cancer incidence in the U.S. (1992–2004). Cancer 2007, 112, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Ward, Z.J.; Yeh, J.M.; Bhakta, N.; Frazier, A.L.; Atun, R. Estimating the total incidence of global childhood cancer: A simulation-based analysis. Lancet Oncol. 2019, 20, 483–493. [Google Scholar] [CrossRef]
- Altekruse, S.; Kosary, C.; Krapcho, M.; Neyman, N.; Aminou, R.; Waldron, W.; Ruhl, J.; Howlader, N.; Tatalovich, Z.; Cho, H.; et al. Age-Adjusted and Age-Specific SEER Cancer Incidence Rates. In SEER Cancer Statistics Review, 1975–2018; National Cancer Institute: Bethesda, MD, USA, 2020. [Google Scholar]
- Valberg, M.; Grotmol, T.; Tretli, S.; Veierød, M.B.; Devesa, S.S.; Aalen, O.O. Frailty modeling of age-incidence curves of osteosarcoma and Ewing sarcoma among individuals younger than 40 years. Stat. Med. 2012, 31, 3731–3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, L.A.; Richardson, M.; Kehm, R.D.; McLaughlin, C.C.; Mueller, B.A.; Chow, E.J.; Spector, L.G. The association between sex and most childhood cancers is not mediated by birthweight. Cancer Epidemiol. 2018, 57, 7–12. [Google Scholar] [CrossRef]
- Dorak, M.T.; Ekarpuzoglu, E. Gender Differences in Cancer Susceptibility: An Inadequately Addressed Issue. Front. Genet. 2012, 3, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, G.; Peris-Bonet, R.; Carli, M.; Martínez-García, C.; de Toledo, J.S.; Steliarova-Foucher, E. Childhood soft tissue sarcomas incidence and survival in European children (1978–1997): Report from the Automated Childhood Cancer Information System project. Eur. J. Cancer 2006, 42, 2136–2149. [Google Scholar] [CrossRef]
- Lychou, S.E.; Gustafsson, G.G.; Ljungman, G.E. Higher rates of metastatic disease may explain the declining trend in Swedish paediatric rhabdomyosarcoma survival rates. Acta Paediatr. 2015, 105, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Bao, P.-P.; Zheng, Y.; Wang, C.-F.; Gu, K.; Jin, F.; Lu, W. Time trends and characteristics of childhood cancer among children age 0-14 in Shanghai. Pediatr. Blood Cancer 2009, 53, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Ito, Y.; Magadi, W.; Bonaventure, A.; Stiller, C.A.; Katanoda, K.; Matsuda, T.; Miyashiro, I.; Pritchard-Jones, K.; Rachet, B. Childhood cancer incidence and survival in Japan and England: A population-based study (1993–2010). Cancer Sci. 2018, 109, 422–434. [Google Scholar] [CrossRef] [Green Version]
- Stefan, C.; Bray, F.; Ferlay, J.; Liu, B.; Parkin, D.M. Cancer of childhood in sub-Saharan Africa. Ecancermedicalscience 2017, 11, 755. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Stefan, C. Editorial: Childhood Cancer in sub-Saharan Africa. Ecancermedicalscience 2017, 11, ed69. [Google Scholar] [CrossRef] [Green Version]
- Jawad, M.U.; Cheung, M.C.; Min, E.S.; Schneiderbauer, M.M.; Koniaris, L.G.; Scully, S.P. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome. Cancer 2009, 115, 3526–3536. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.J.; Carozza, S.E.; Chow, E.J.; Fox, E.E.; Horel, S.; McLaughlin, C.C.; Mueller, B.A.; Puumala, S.E.; Reynolds, P.; Von Behren, J.; et al. Parental Age and Risk of Childhood Cancer. Epidemiology 2009, 20, 475–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupo, P.J.; Luna-Gierke, R.E.; Chambers, T.M.; Tavelin, B.; Scheurer, M.E.; Melin, B.; Papworth, K. Perinatal and familial risk factors for soft tissue sarcomas in childhood through young adulthood: A population-based assessment in 4 million live births. Int. J. Cancer 2020, 146, 791–802. [Google Scholar] [CrossRef]
- Grufferman, S.; Schwartz, A.G.; Ruymann, F.B.; Maurer, H.M. Parents’ use of cocaine and marijuana and increased risk of rhabdomyosarcoma in their children. Cancer Causes Control. 1993, 4, 217–224. [Google Scholar]
- Rumrich, I.K.; Viluksela, M.; Vähäkangas, K.; Gissler, M.; Surcel, H.-M.; Hänninen, O. Maternal Smoking and the Risk of Cancer in Early Life–A Meta-Analysis. PLoS ONE 2016, 11, e0165040. [Google Scholar] [CrossRef] [Green Version]
- Grufferman, S.; Ruymann, F.; Ognjanovic, S.; Erhardt, E.B.; Maurer, H.M. Prenatal X-ray Exposure and Rhabdomyosarcoma in Children: A Report from the Children’s Oncology Group. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Hicks, N.; Zack, M.; Caldwell, G.G.; Fernbach, D.J.; Falletta, J.M. Childhood cancer and occupational radiation exposure in parents. Cancer 1984, 53, 1637–1643. [Google Scholar] [CrossRef]
- Grufferman, S.; Lupo, P.J.; Vogel, R.I.; Danysh, H.E.; Erhardt, E.B.; Ognjanovic, S. Parental Military Service, Agent Orange Exposure, and the Risk of Rhabdomyosarcoma in Offspring. J. Pediatr. 2014, 165, 1216–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, G.M.; Bunch, K.J.; Stiller, C.A.; Vincent, T.J.; Murphy, M.F.G. Case–control study of paternal occupational exposures and childhood bone tumours and soft-tissue sarcomas in Great Britain, 1962–2010. Br. J. Cancer 2020, 122, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Ognjanovic, S.; Carozza, E.S.; Chow, E.J.; Fox, E.E.; Horel, S.; McLaughlin, C.C.; Mueller, A.B.; Puumala, S.; Reynolds, P.; Von Behren, J.; et al. Birth characteristics and the risk of childhood rhabdomyosarcoma based on histological subtype. Br. J. Cancer 2009, 102, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, L.M.; McCauley, K.; Ma, X.; Wiemels, J.L.; Chokkalingam, A.P.; Metayer, C. Birth weight, fetal growth, and risk of pediatric rhabdomyosarcoma: An updated record linkage study in California. Ann. Epidemiol. 2016, 26, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Lupo, P.J.; Zhou, R.; Skapek, S.X.; Hawkins, D.S.; Spector, L.G.; Scheurer, M.E.; Okcu, M.F.; Melin, B.; Papworth, K.; Erhardt, E.B.; et al. Allergies, atopy, immune-related factors and childhood rhabdomyosarcoma: A report from the children’s oncology group. Int. J. Cancer 2014, 134, 431–436. [Google Scholar] [CrossRef]
- Shrestha, A.; Ritz, B.; Ognjanovic, S.; Lombardi, C.A.; Wilhelm, M.; Heck, J.E. Early Life Factors and Risk of Childhood Rhabdomyosarcoma. Front. Public Health 2013, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Spector, L.G.; Birch, J. The epidemiology of hepatoblastoma. Pediatr. Blood Cancer 2012, 59, 776–779. [Google Scholar] [CrossRef]
- Groves, F.D.; Watkins, B.T.; Roberts, D.J.; Tucker, T.C.; Shen, T.; Flood, T.J. Birth Weight and Risk of Childhood Acute Lymphoblastic Leukemia in Arizona, Illinois, and Kentucky. South. Med. J. 2018, 111, 579–584. [Google Scholar] [CrossRef]
- Lupo, P.J.; Schraw, J.M.; Desrosiers, T.A.; Nembhard, W.N.; Langlois, P.H.; Canfield, M.A.; Copeland, G.; Meyer, R.E.; Brown, A.L.; Chambers, T.M.; et al. Association Between Birth Defects and Cancer Risk Among Children and Adolescents in a Population-Based Assessment of 10 Million Live Births. JAMA Oncol. 2019, 5, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Daltveit, D.S.; Klungsøyr, K.; Engeland, A.; Ekbom, A.; Gissler, M.; Glimelius, I.; Grotmol, T.; Madanat-Harjuoja, L.; Ording, A.G.; Sæther, S.M.M.; et al. Cancer risk in individuals with major birth defects: Large Nordic population based case-control study among children, adolescents, and adults. BMJ 2020, 371, m4060. [Google Scholar] [CrossRef]
- Yang, P.; Grufferman, S.; Khoury, M.J.; Schwartz, A.G.; Kowalski, J.; Ruymann, F.B.; Maurer, H.M. Association of childhood rhabdomyosarcoma with neurofibromatosis type i and birth defects. Genet. Epidemiol. 1995, 12, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Sankaran, H.; Danysh, H.E.; Scheurer, M.E.; Okcu, M.F.; Skapek, S.X.; Hawkins, D.S.; Spector, L.G.; Erhardt, E.B.; Grufferman, S.; Lupo, P.J. The Role of Childhood Infections and Immunizations on Childhood Rhabdomyosarcoma: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer 2016, 63, 1557–1562. [Google Scholar] [CrossRef] [Green Version]
- Oddsberg, J. Environmental Factors in the Etiology of Esophageal Atresia. J. Pediatr. Gastroenterol. Nutr. 2011, 52, S4–S5. [Google Scholar] [CrossRef]
- Lupo, P.J.; Danysh, H.E.; Skapek, S.X.; Hawkins, D.S.; Spector, L.G.; Zhou, R.; Okcu, M.F.; Papworth, K.; Erhardt, E.B.; Grufferman, S. Maternal and birth characteristics and childhood rhabdomyosarcoma: A report from the Children’s Oncology Group. Cancer Causes Control. 2014, 25, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Lupo, P.J.; Danysh, H.E.; Plon, S.E.; Curtin, K.; Malkin, D.; Hettmer, S.; Hawkins, D.S.; Skapek, S.X.; Spector, L.G.; Papworth, K.; et al. Family history of cancer and childhood rhabdomyosarcoma: A report from the Children’s Oncology Group and the Utah Population Database. Cancer Med. 2015, 4, 781–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratz, C.P.; Achatz, M.I.; Brugières, L.; Frebourg, T.; Garber, J.E.; Greer, M.-L.C.; Hansford, J.R.; Janeway, K.A.; Kohlmann, W.K.; McGee, R.; et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin. Cancer Res. 2017, 23, e38–e45. [Google Scholar] [CrossRef] [Green Version]
- Li, F.P. Soft-Tissue Sarcomas, Breast Cancer, and Other Neoplasms. Ann. Intern. Med. 1969, 71, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Diller, L.; Sexsmith, E.; Gottlieb, A.; Li, F.P.; Malkin, D. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J. Clin. Investig. 1995, 95, 1606–1611. [Google Scholar] [CrossRef]
- Ognjanovic, S.; Olivier, M.; Bergemann, T.L.; Hainaut, P. Sarcomas in TP53 germline mutation carriers. Cancer 2012, 118, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Hettmer, S.; Archer, N.M.; Somers, G.R.; Novokmet, A.; Wagers, A.J.; Diller, L.; Rodriguez-Galindo, C.; Teot, L.A.; Malkin, D. Anaplastic rhabdomyosarcoma inTP53germline mutation carriers. Cancer 2014, 120, 1068–1075. [Google Scholar] [CrossRef] [Green Version]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef] [Green Version]
- Gripp, K.W.; Rauen, K.A. Costello Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Kratz, C.P.; Rapisuwon, S.; Reed, H.; Hasle, H.; Rosenberg, P.S. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Kratz, C.P.; Franke, L.; Peters, H.M.; Kohlschmidt, N.; Kazmierczak, B.; Finckh, U.; Bier, A.; Eichhorn, B.W.; De Blank, C.M.; Kraus, C.; et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br. J. Cancer 2015, 112, 1392–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J. Neurofibromatosis 1. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Bollag, G.; Clapp, D.W.; Shih, S.; Adler, F.; Zhang, Y.Y.; Thompson, P.; Lange, B.J.; Freedman, M.H.; McCormick, F.; Jacks, T.; et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat. Genet. 1996, 12, 144–148. [Google Scholar] [CrossRef]
- Schneider, K.W.; Cost, N.G.; Schultz, K.A.P.; Svihovec, S.; Suttman, A. Germline predisposition to genitourinary rhabdomyosarcoma. Transl. Androl. Urol. 2020, 9, 2430–2440. [Google Scholar] [CrossRef]
- Evans, D.G.R.; Salvador, H.; Chang, V.Y.; Erez, A.; Voss, S.D.; Schneider, K.W.; Scott, H.S.; Plon, S.E.; Tabori, U. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis 1. Clin. Cancer Res. 2017, 23, e46–e53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crucis, A.; Richer, W.; Brugières, L.; Bergeron, C.; Marie-Cardine, A.; Stephan, J.-L.; Girard, P.; Corradini, N.; Munzer, M.; Lacour, B.; et al. Rhabdomyosarcomas in children with neurofibromatosis type I: A national historical cohort. Pediatr. Blood Cancer 2015, 62, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C. Rubinstein-Taybi Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Jongmans, M.C.J.; Hoogerbrugge, P.M.; Hilkens, L.; Flucke, U.; Van Der Burgt, I.; Noordam, K.; Ruiterkamp-Versteeg, M.; Yntema, H.G.; Nillesen, W.M.; Ligtenberg, M.J.L.; et al. Noonan syndrome, the SOS1 gene and embryonal rhabdomyosarcoma. Genes Chromosom. Cancer 2010, 49, 635–641. [Google Scholar] [CrossRef]
- Denayer, E.E.; Devriendt, K.K.; De Ravel, T.; Van Buggenhout, G.G.; Smeets, E.E.; Francois, I.; Sznajer, Y.; Craen, M.M.; Leventopoulos, G.G.; Mutesa, L.L.; et al. Tumor spectrum in children with Noonan syndrome andSOS1orRAF1mutations. Genes Chromosom. Cancer 2009, 49, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.A.P.; Stewart, D.R.; Kamihara, J.; Bauer, A.J.; Merideth, M.A.; Stratton, P.; Huryn, L.A.; Harris, A.K.; Doros, L.; Field, A. DICER1 Tumor Predisposition Summary Genetic Counseling. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Robertson, J.C.; Jorcyk, C.L.; Oxford, J.T. DICER1 Syndrome: DICER1 Mutations in Rare Cancers. Cancers 2018, 10, 143. [Google Scholar] [CrossRef] [Green Version]
- Schultz, K.A.P.; Stewart, D.R.; Kamihara, J.; Bauer, A.J.; Merideth, M.A.; Stratton, P.; Huryn, L.A.; Harris, A.K.; Doros, L.; Field, A.; et al. DICER1 Tumor Predisposition. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2014; pp. 1–34. [Google Scholar]
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. [Google Scholar] [CrossRef]
- Henke, L.E.; Perkins, S.M.; Pfeifer, J.D.; Ma, C.; Chen, Y.; Dewees, T.; Grigsby, P.W. BRAF V600E mutational status in pediatric thyroid cancer. Pediatr. Blood Cancer 2014, 61, 1168–1172. [Google Scholar] [CrossRef]
- Stewart, D.R.; Best, A.F.; Williams, G.M.; Harney, L.A.; Carr, A.G.; Harris, A.K.; Kratz, C.P.; Dehner, L.P.; Messinger, Y.H.; Rosenberg, P.S.; et al. Neoplasm Risk Among Individuals with a Pathogenic Germline Variant in DICER1. J. Clin. Oncol. 2019, 37, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Sobel, R.A.; Woerner, S. Rubinstein-Taybi syndrome andnasopharyngeal rhabdomyosarcoma. J. Pediatr. 1981, 99, 1000–1001. [Google Scholar] [CrossRef]
- Miller, R.W.; Rubinstein, J.H. Tumors in Rubinstein-Taybi syndrome. Am. J. Med. Genet. 1995, 56, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, D.R.; Gallie, B.L. Retinoblastoma. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Kohashi, K.; Oda, Y.; Yamamoto, H.; Tamiya, S.; Takahira, T.; Takahashi, Y.; Tajiri, T.; Taguchi, T.; Suita, S.; Tsuneyoshi, M. Alterations of RB1 gene in embryonal and alveolar rhabdomyosarcoma: Special reference to utility of pRB immunoreactivity in differential diagnosis of rhabdomyosarcoma subtype. J. Cancer Res. Clin. Oncol. 2008, 134, 1097–1103. [Google Scholar] [CrossRef]
- Temming, P.; Arendt, M.; Viehmann, A.; Eisele, L.; Le Guin, C.H.D.; Schündeln, M.M.; Biewald, E.; Astrahantseff, K.; Wieland, R.; Bornfeld, N.; et al. Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr. Blood Cancer 2017, 64, 71–80. [Google Scholar] [CrossRef]
- Kleinerman, R.A.; Tucker, M.A.; Abramson, D.H.; Seddon, J.M.; Tarone, R.E.; Fraumeni, J.F. Risk of Soft Tissue Sarcomas by Individual Subtype in Survivors of Hereditary Retinoblastoma. J. Natl. Cancer Inst. 2007, 99, 24–31. [Google Scholar] [CrossRef]
- Cebulla, C.M.; Kleinerman, R.A.; Alegret, A.; Kulak, A.; Dubovy, S.R.; Hess, D.J.; Murray, T.G. Rapid appearance of rhabdomyosarcoma after radiation and chemotherapy for retinoblastoma: A clinicopathologic correlation. Retin. Cases Brief. Rep. 2009, 3, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Tabori, U.; Hansford, J.R.; Achatz, M.I.; Kratz, C.P.; Plon, S.E.; Frebourg, T.; Brugières, L. Clinical Management and Tumor Surveillance Recommendations of Inherited Mismatch Repair Deficiency in Childhood. Clin. Cancer Res. 2017, 23, e32–e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoine, N.; Colas, C.; Muleris, M.; Bodo, S.; Duval, A.; Entz-Werle, N.; Coulet, F.; Cabaret, O.; Andreiuolo, F.; Charpy, C.; et al. Constitutional mismatch repair deficiency syndrome: Clinical description in a French cohort. J. Med. Genet. 2015, 52, 770–778. [Google Scholar] [CrossRef]
- Kratz, C.P.; Holter, S.; Etzler, J.; Lauten, M.; Pollett, A.; Niemeyer, C.M.; Gallinger, S.; Wimmer, K. Rhabdomyosarcoma in patients with constitutional mismatch-repair-deficiency syndrome. J. Med. Genet. 2009, 46, 418–420. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.A.; Tolar, J. Fanconi Anemia. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Malric, A.; Defachelles, A.-S.; Leblanc, T.; Lescoeur, B.; Lacour, B.; Peuchmaur, M.; Maurage, C.-A.; Pierron, G.; Guillemot, D.; D’Enghien, C.D.; et al. Fanconi anemia and solid malignancies in childhood: A national retrospective study. Pediatr. Blood Cancer 2015, 62, 463–470. [Google Scholar] [CrossRef]
- Singh, M.; Leasure, J.M.; Chronowski, C.; Geier, B.; Bondra, K.; Duan, W.; Hensley, L.A.; Villalona-Calero, M.; Li, N.; Vergis, A.M.; et al. FANCD2 Is a Potential Therapeutic Target and Biomarker in Alveolar Rhabdomyosarcoma Harboring the PAX3–FOXO1 Fusion Gene. Clin. Cancer Res. 2014, 20, 3884–3895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemont, S.; Bocéno, M.; Rival, J.M.; Méchinaud, F.; David, A. High risk of malignancy in mosaic variegated aneuploidy syndrome. Am. J. Med. Genet. 2002, 109, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.; Coleman, K.; Reid, S.; Plaja, A.; Firth, H.; Fitzpatrick, D.; Kidd, A.; Méhes, K.; Nash, R.; Robin, N.; et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004, 36, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Shuman, C.; Beckwith, J.B.; Weksberg, R. Beckwith-Wiedemann Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Smith, A.C.; Squire, J.A.; Thorner, P.; Zielenska, M.; Shuman, C.; Grant, R.; Chitayat, D.; Nishikawa, J.L.; Weksberg, R. Association of Alveolar Rhabdomyosarcoma with the Beckwith-Wiedemann Syndrome. Pediatr. Dev. Pathol. 2001, 4, 550–558. [Google Scholar] [CrossRef]
- Sotelo-Avila, C.; Gooch, W.M. Neoplasms associated with the Beckwith-Wiedemann syndrome. Perspect. Pediatr. Pathol. 1976, 3, 255–272. [Google Scholar]
- Vaughan, W.; Sanders, D.W.; Grosfeld, J.L.; Plumley, A.D.; Rescorla, F.J.; Scherer, L.; West, K.W.; Breitfeld, P.P. Favorable outcome in children with Beckwith-Wiedeman syndrome and intraabdominal malignant tumors. J. Pediatr. Surg. 1995, 30, 1042–1045. [Google Scholar] [CrossRef]
- Aideyan, U.; Kao, S. Case report: Urinary bladder rhabdomyosarcoma associated with Beckwith-Wiedemann syndrome. Clin. Radiol. 1998, 53, 457–459. [Google Scholar] [CrossRef]
- Mussa, A.; Molinatto, C.; Baldassarre, G.; Riberi, E.; Russo, S.; Larizza, L.; Riccio, A.; Ferrero, G.B. Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J. Pediatr. 2016, 176, 142–149.e1. [Google Scholar] [CrossRef]
- Maas, S.M.; Vansenne, F.; Kadouch, D.J.M.; Ibrahim, A.; Bliek, J.; Hopman, S.; Mannens, M.M.; Merks, J.H.M.; Maher, E.R.; Hennekam, R.C. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am. J. Med. Genet. Part. A 2016, 170, 2248–2260. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.A.; Gutierrez, S.; Petersen, A.K.; Bavle, A.; et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children with Solid Tumors. JAMA Oncol. 2016, 2, 616–624. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Aman, P.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mody, R.J.; Wu, Y.-M.; Lonigro, R.J.; Cao, X.; Roychowdhury, S.; Vats, P.; Frank, K.M.; Prensner, J.R.; Asangani, A.I.; Palanisamy, N.; et al. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA 2015, 314, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sisoudiya, S.D.; Martin-Giacalone, A.B.; Khayat, M.M.; Dugan-Perez, S.; Marquez-Do, A.D.; Scheurer, E.M.; Muzny, D.; Boerwinkle, E.; Gibbs, A.R.; et al. Germline Cancer Predisposition Variants in Pediatric Rhabdomyosarcoma: A Report from the Children’s Oncology Group. J. Natl. Cancer Inst. 2020. [Google Scholar] [CrossRef]
- Kim, J.; Light, N.; Subasri, V.; Young, E.L.; Wegman-Ostrosky, T.; Barkauskas, D.A.; Hall, D.; Lupo, P.J.; Patidar, R.; Maese, L.D.; et al. Pathogenic Germline Variants in Cancer Susceptibility Genes in Children and Young Adults with Rhabdomyosarcoma. JCO Precis. Oncol. 2021, 75–87. [Google Scholar] [CrossRef]
- Grünewald, T.G.P.; Bernard, V.; Gilardi-Hebenstreit, P.; Raynal, V.; Surdez, D.; Aynaud, M.-M.; Mirabeau, O.; Cidre-Aranaz, F.; Tirode, F.; Zaidi, S.; et al. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat. Genet. 2015, 47, 1073–1078. [Google Scholar] [CrossRef] [Green Version]
- Musa, J.; Cidre-Aranaz, F.; Aynaud, M.-M.; Orth, M.F.; Knott, M.M.L.; Mirabeau, O.; Mazor, G.; Varon, M.; Hölting, T.L.B.; Grossetête, S.; et al. Cooperation of cancer drivers with regulatory germline variants shapes clinical outcomes. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Prim. 2019, 5, 11. [Google Scholar] [CrossRef]
- Ren, Y.-X.; Finckenstein, F.G.; Abdueva, D.A.; Shahbazian, V.; Chung, B.; Weinberg, K.I.; Triche, T.J.; Shimada, H.; Anderson, M.J. Mouse Mesenchymal Stem Cells Expressing PAX-FKHR form Alveolar Rhabdomyosarcomas by Cooperating with Secondary Mutations. Cancer Res. 2008, 68, 6587–6597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linardic, C.M.; Downie, D.L.; Qualman, S.; Bentley, R.C.; Counter, C.M. Genetic Modeling of Human Rhabdomyosarcoma. Cancer Res. 2005, 65, 4490–4495. [Google Scholar] [CrossRef] [Green Version]
- Langenau, D.M.; Keefe, M.D.; Storer, N.Y.; Guyon, J.R.; Kutok, J.L.; Le, X.; Goessling, W.; Neuberg, D.S.; Kunkel, L.M.; Zon, L.I. Effects of RAS on the genesis of embryonal rhabdomyosarcoma. Genes Dev. 2007, 21, 1382–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, A.P.-Y.; Devidas, M.; Lee, S.H.R.; Cao, X.; Pei, D.; Borowitz, M.; Wood, B.; Gastier-Foster, J.M.; Dai, Y.; et al. Association of GATA3 Polymorphisms with Minimal Residual Disease and Relapse Risk in Childhood Acute Lymphoblastic Leukemia. J. Natl. Cancer Inst. 2020, 113, 408–417. [Google Scholar] [CrossRef]
- Qian, M.; Cao, X.; Devidas, M.; Yang, W.; Cheng, C.; Dai, Y.; Carroll, A.; Heerema, N.A.; Zhang, H.; Moriyama, T.; et al. TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J. Clin. Oncol. 2018, 36, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Andreu, V.; Roberts, K.G.; Harvey, R.C.; Yang, W.; Cheng, C.; Pei, D.; Xu, H.; Gastier-Foster, J.; Shuyu, E.; Yew-Suang Lim, J.; et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat. Genet. 2013, 45, 1494–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thavaneswaran, S.; Rath, E.; Tucker, K.; Joshua, A.M.; Hess, D.; Pinese, M.; Ballinger, M.L.; Thomas, D.M. Therapeutic implications of germline genetic findings in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 386–396. [Google Scholar] [CrossRef]
- Lupo, P.J.; Spector, L.G. Cancer Progress and Priorities: Childhood Cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Age-incidence curve of rhabdomyosarcoma (RMS) in terms of the major histological subtypes: embryonal RMS (ERMS) and alveolar RMS (ARMS). The data represent the incidences from 2000–2017 and was published by the Surveillance, Epidemiology, and End Results (SEER) program.
Figure 1.
Age-incidence curve of rhabdomyosarcoma (RMS) in terms of the major histological subtypes: embryonal RMS (ERMS) and alveolar RMS (ARMS). The data represent the incidences from 2000–2017 and was published by the Surveillance, Epidemiology, and End Results (SEER) program.

Figure 2.
Incidence of RMS per 1,000,000 children by geographic location. Bar color indicates geographic location by continent. RMS incidence in Africa is presented as two countries per region, which illustrate the minimum and maximum values of the range by region, as reported by Stefan et al. [27].
Figure 2.
Incidence of RMS per 1,000,000 children by geographic location. Bar color indicates geographic location by continent. RMS incidence in Africa is presented as two countries per region, which illustrate the minimum and maximum values of the range by region, as reported by Stefan et al. [27].

Figure 3.
The Ras signaling pathway and proteins encoded by genes that are implicated in each of the cancer predisposition RASopathies: Costello syndrome, neurofibromatosis type I, and Noonan syndrome.
Figure 3.
The Ras signaling pathway and proteins encoded by genes that are implicated in each of the cancer predisposition RASopathies: Costello syndrome, neurofibromatosis type I, and Noonan syndrome.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Martin-Giacalone, B.A.; Weinstein, P.A.; Plon, S.E.; Lupo, P.J. Pediatric Rhabdomyosarcoma: Epidemiology and Genetic Susceptibility. J. Clin. Med. 2021, 10, 2028. https://doi.org/10.3390/jcm10092028
AMA Style
Martin-Giacalone BA, Weinstein PA, Plon SE, Lupo PJ. Pediatric Rhabdomyosarcoma: Epidemiology and Genetic Susceptibility. Journal of Clinical Medicine. 2021; 10(9):2028. https://doi.org/10.3390/jcm10092028
Chicago/Turabian StyleMartin-Giacalone, Bailey A., P. Adam Weinstein, Sharon E. Plon, and Philip J. Lupo. 2021. "Pediatric Rhabdomyosarcoma: Epidemiology and Genetic Susceptibility" Journal of Clinical Medicine 10, no. 9: 2028. https://doi.org/10.3390/jcm10092028
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.