
Role of C5aR1 and C5L2 Receptors in Ischemia-Reperfusion Injury

 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Cell Culture
2.2. Kidney Biopsies
Immunohistochemistry
2.3. Statistical Analysis
3. Results
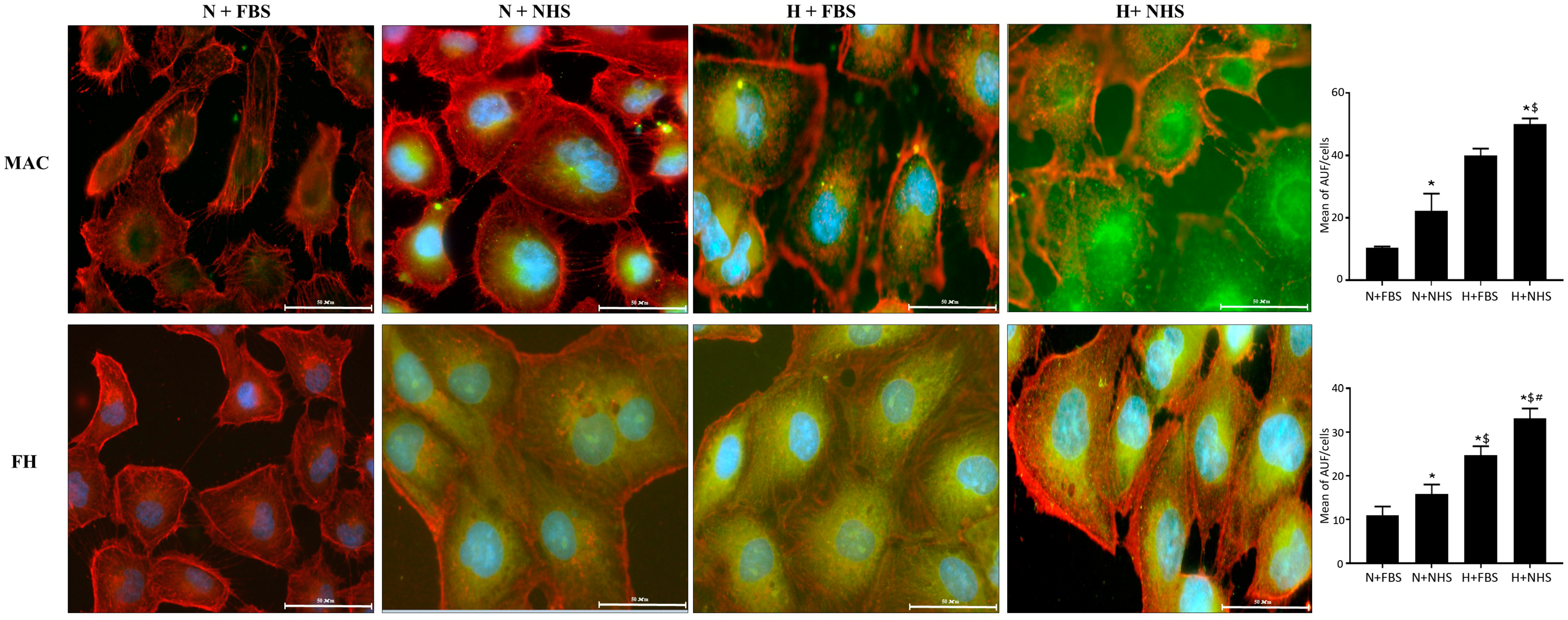
3.1. Expression of MAC and FH-Related Proteins in HK-2 Cells
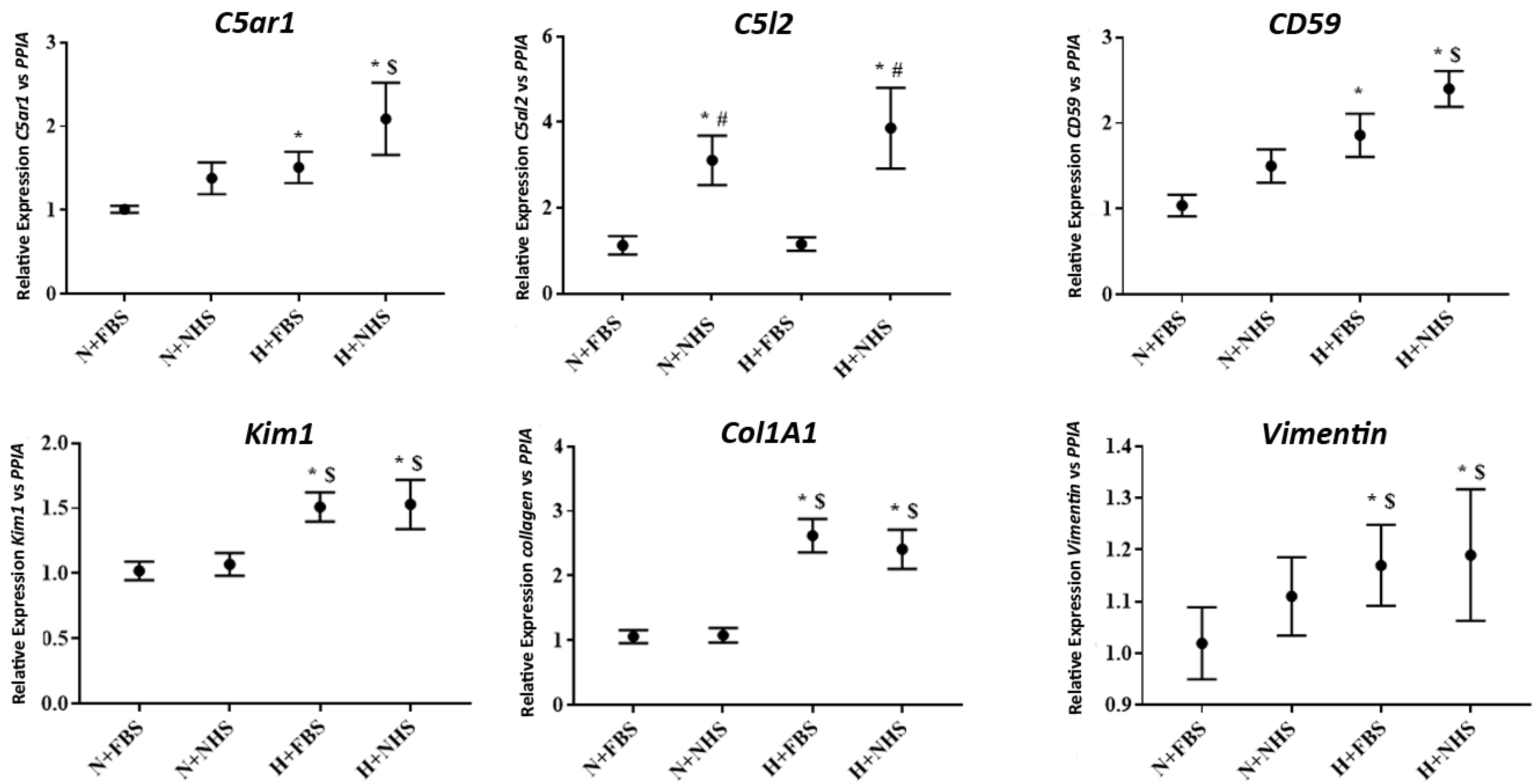
3.2. Gene Expression in HK-2 Cells
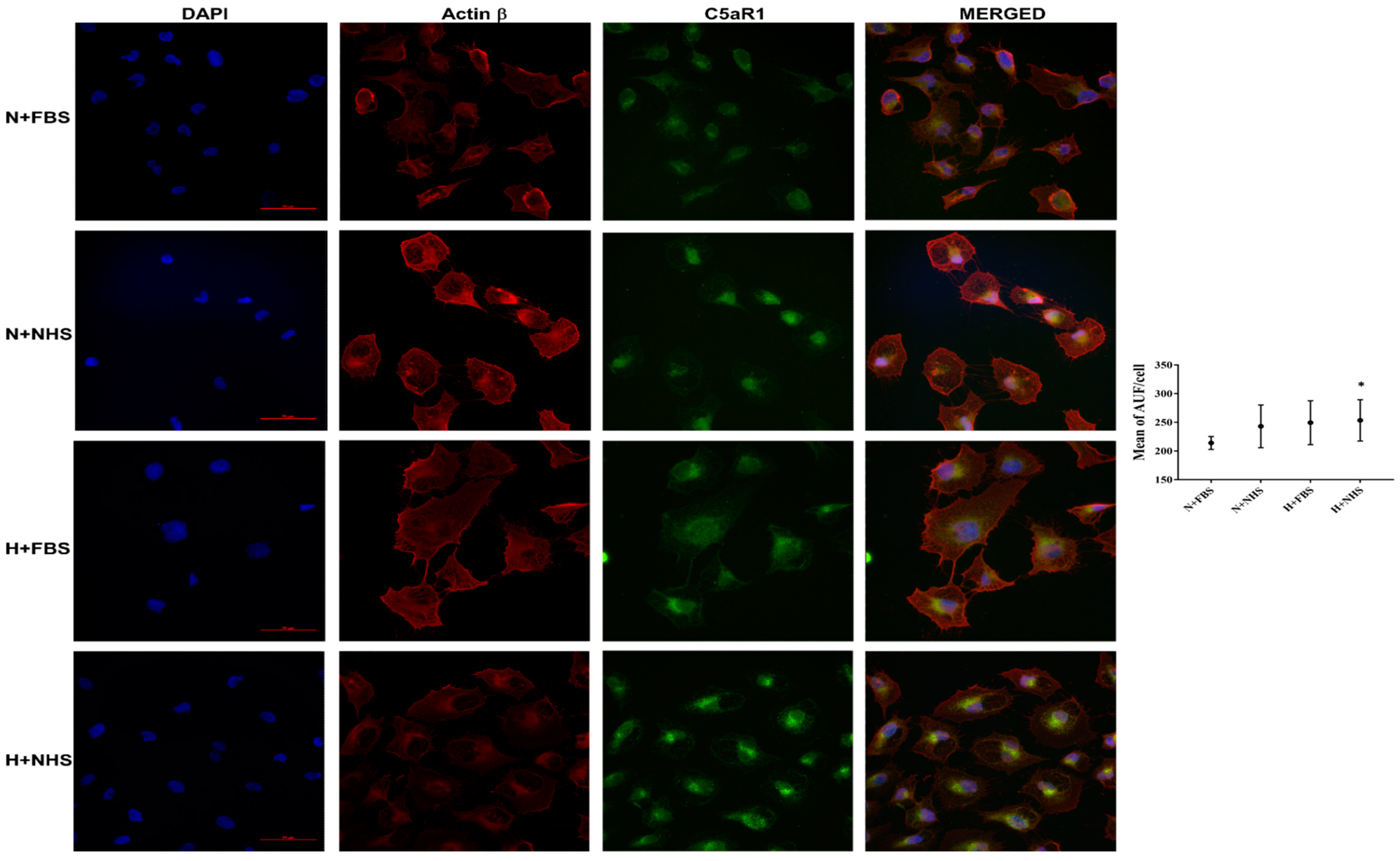
3.3. Expression of C5aR1 Protein in HK-2 Cells
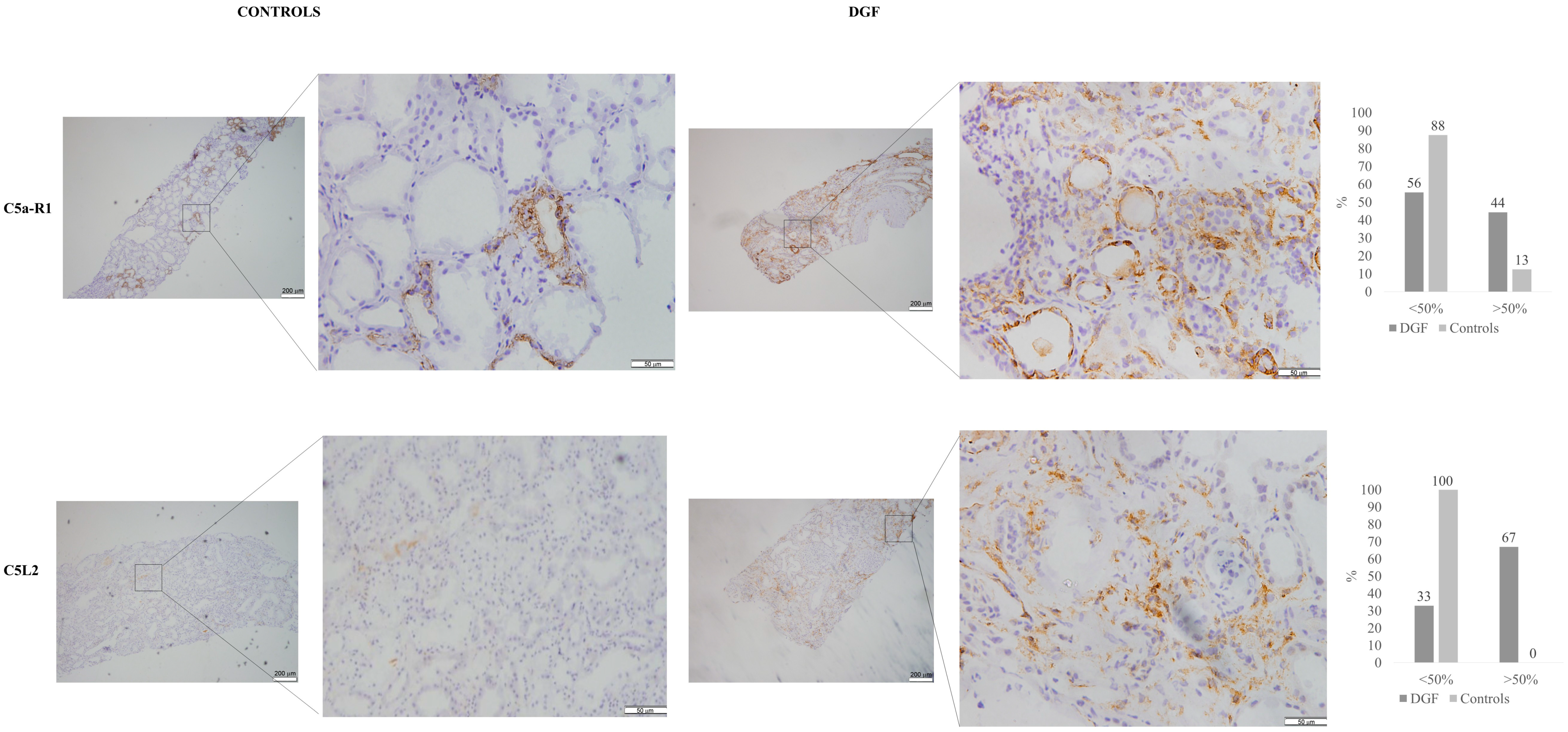
3.4. Immunohistochemistry for C5aR1 andC5L2 in Kidney Tissue Samples
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Thadhani, R.; Pascual, M.; Bonventre, J.V. Acute Renal Failure. N. Engl. J. Med. 1996, 334, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Yago, T.; Petrich, B.G.; Zhang, N.; Liu, Z.; Shao, B.; Ginsberg, M.H.; McEver, R.P. Blocking Neutrophil Integrin Activation Prevents Ischemia–Reperfusion Injury. J. Exp. Med. 2015, 212, 1267–1281. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of Mitochondrial Dynamics in Acute Kidney Injury in Cell Culture and Rodent Models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Damman, J.; Daha, M.R.; van Son, W.J.; Leuvenink, H.G.; Ploeg, R.J.; Seelen, M.A. Crosstalk between Complement and Toll-like Receptor Activation in Relation to Donor Brain Death and Renal Ischemia-Reperfusion Injury: Complement and TLRs in Kidney Transplantation. Am. J. Transplant. 2011, 11, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Le Dorze, M.; Legrand, M.; Payen, D.; Ince, C. The Role of the Microcirculation in Acute Kidney Injury. Curr. Opin. Crit. Care 2009, 15, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Farrar, C.A.; Asgari, E.; Schwaeble, W.J.; Sacks, S.H. Which Pathways Trigger the Role of Complement in Ischaemia/Reperfusion Injury? Front. Immun. 2012, 3, 341. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Miwa, T.; Liu, J.; Nangaku, M.; Song, W.-C. Critical Protection from Renal Ischemia Reperfusion Injury by CD55 and CD59. J. Immunol. 2004, 172, 3869–3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diepenhorst, G.M.P.; van Gulik, T.M.; Hack, C.E. Complement-Mediated Ischemia-Reperfusion Injury: Lessons Learned From Animal and Clinical Studies. Ann. Surg. 2009, 249, 889–899. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Skerka, C. Complement Regulators and Inhibitory Proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Damman, J.; Schuurs, T.A.; Ploeg, R.J.; Seelen, M.A. Complement and Renal Transplantation: From Donor to Recipient. Transplantation 2008, 85, 923–927. [Google Scholar] [CrossRef] [Green Version]
- Blogowski, W.; Dolegowska, B.; Salata, D.; Budkowska, M.; Domanski, L.; Starzynska, T. Clinical Analysis of Perioperative Complement Activity during Ischemia/Reperfusion Injury Following Renal Transplantation. Clin. J. Am. Soc. Nephrol. 2012, 7, 1843–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, D.K.; van der Pol, P.; van Anken, G.E.; van Gijlswijk, D.J.; Damman, J.; Lindeman, J.H.; Reinders, M.E.J.; Schaapherder, A.F.; van Kooten, C. Acute but Transient Release of Terminal Complement Complex After Reperfusion in Clinical Kidney Transplantation. Transplant. J. 2013, 95, 816–820. [Google Scholar] [CrossRef]
- Rodríguez, E.; Gimeno, J.; Arias-Cabrales, C.; Barrios, C.; Redondo- Pachón, D.; Soler, M.J.; Crespo, M.; Sierra-Ochoa, A.; Riera, M.; Pascual, J. Membrane Attack Complex and Factor H in Humans with Acute Kidney Injury. Kidney Blood Press. Res. 2018, 43, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Goetz, L.; Laskowski, J.; Renner, B.; Pickering, M.C.; Kulik, L.; Klawitter, J.; Stites, E.; Christians, U.; van der Vlag, J.; Ravichandran, K.; et al. Complement Factor H Protects Mice from Ischemic Acute Kidney Injury but Is Not Critical for Controlling Complement Activation by Glomerular IgM. Eur. J. Immunol. 2018, 48, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Renner, B.; Ferreira, V.P.; Cortes, C.; Goldberg, R.; Ljubanovic, D.; Pangburn, M.K.; Pickering, M.C.; Tomlinson, S.; Holland-Neidermyer, A.; Strassheim, D.; et al. Binding of Factor H to Tubular Epithelial Cells Limits Interstitial Complement Activation in Ischemic Injury. Kidney Int. 2011, 80, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.-F.; Ward, P.A. Role of C5a in Inflammatory Responses. Annu. Rev. Immunol. 2005, 23, 821–852. [Google Scholar] [CrossRef] [PubMed]
- de Vries, B.; Matthijsen, R.A.; Wolfs, T.G.A.M.; van Bijnen, A.A.J.H.M.; Heeringa, P.; Buurman, W.A. Inhibition of Complement Factor C5 Protects against Renal Ischemia-Reperfusion Injury: Inhibition of Late Apoptosis and Inflammation1. Transplantation 2003, 75, 375–382. [Google Scholar] [CrossRef]
- de Vries, B.; Kohl, J.; Leclercq, W.K.G.; Wolfs, T.G.A.M.; van Bijnen, A.A.J.H.M.; Heeringa, P.; Buurman, W.A. Complement Factor C5a Mediates Renal Ischemia-Reperfusion Injury Independent from Neutrophils. J. Immunol. 2003, 170, 3883–3889. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zhang, X.; Feng, B.; Sun, H.; Suzuki, M.; Ichim, T.; Kubo, N.; Wong, A.; Min, L.R.; Budohn, M.E.; et al. Gene Silencing of Complement C5a Receptor Using SiRNA for Preventing Ischemia/Reperfusion Injury. Am. J. Pathol. 2008, 173, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Garstka, M.A.; Li, K. The Controversial C5a Receptor C5aR2: Its Role in Health and Disease. J. Immunol. Res. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Poppelaars, F.; van Werkhoven, M.B.; Kotimaa, J.; Veldhuis, Z.J.; Ausema, A.; Broeren, S.G.M.; Damman, J.; Hempel, J.C.; Leuvenink, H.G.D.; Daha, M.R.; et al. Critical Role for Complement Receptor C5aR2 in the Pathogenesis of Renal Ischemia-Reperfusion Injury. FASEB J. 2017, 31, 3193–3204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorenz, A.; Derlin, K.; Schröder, C.; Dressler, L.; Vijayan, V.; Pradhan, P.; Immenschuh, S.; Jörns, A.; Echtermeyer, F.; Herzog, C.; et al. Enhanced Activation of Interleukin-10, Heme Oxygenase-1, and AKT in C5aR2-Deficient Mice Is Associated with Protection from Ischemia Reperfusion Injury-Induced Inflammation and Fibrosis. Kidney Int. 2018, 94, 741–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Werkhoven, M.B.; Damman, J.; Daha, M.R.; Krikke, C.; van Goor, H.; van Son, W.J.; Hillebrands, J.-L.; van Dijk, M.C.R.F.; Seelen, M.A.J. Novel Insights in Localization and Expression Levels of C5aR and C5L2 under Native and Post-Transplant Conditions in the Kidney. Mol. Immunol. 2013, 53, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Jayne, D.R.W.; Bruchfeld, A.N.; Harper, L.; Schaier, M.; Venning, M.C.; Hamilton, P.; Burst, V.; Grundmann, F.; Jadoul, M.; Szombati, I.; et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2017, 28, 2756–2767. [Google Scholar] [CrossRef] [Green Version]
- van der Pol, P.; Roos, A.; Berger, S.P.; Daha, M.R.; van Kooten, C. Natural IgM Antibodies Are Involved in the Activation of Complement by Hypoxic Human Tubular Cells. Am. J. Physiol. Ren. Physiol. 2011, 300, F932–F940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Márquez, E.; Riera, M.; Pascual, J.; Soler, M.J. Albumin inhibits the insulin-mediated ACE2 increase in cultured podocytes. Am. J. Physiol. Ren. Physiol. 2014, 306, F1327–F1334. [Google Scholar] [CrossRef] [Green Version]
- Guirado, R.; Carceller, H.; Castillo-Gómez, E.; Castrén, E.; Nacher, J. Automated analysis of images for molecular quantification in immunohistochemistry. Heliyon 2018, 4, e00669. [Google Scholar] [CrossRef]
- Solez, K.; Colvin, R.B.; Racusen, L.C.; Haas, M.; Sis, B.; Mengel, M.; Halloran, P.F.; Baldwin, W.; Banfi, G.; Collins, A.B.; et al. Banff 07 Classification of Renal Allograft Pathology: Updates and Future Directions. Am. J. Transplant. 2008, 8, 753–760. [Google Scholar] [CrossRef]
- Zhao, H.; Alam, A.; Soo, A.P.; George, A.J.T.; Ma, D. Ischemia-Reperfusion Injury Reduces Long Term Renal Graft Survival: Mechanism and Beyond. EBioMedicine 2018, 28, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Yarlagadda, S.G.; Coca, S.G.; Formica, R.N.; Poggio, E.D.; Parikh, C.R. Association between Delayed Graft Function and Allograft and Patient Survival: A Systematic Review and Meta-Analysis. Nephrol. Dial. Transplant. 2008, 24, 1039–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, J.; Pérez-Sáez, M.J.; Mir, M.; Crespo, M. Chronic Renal Allograft Injury: Early Detection, Accurate Diagnosis and Management. Transplant. Rev. 2012, 26, 280–290. [Google Scholar] [CrossRef]
- Arias-Cabrales, C.E.; Pérez-Sáez, M.J.; Redondo-Pachón, D.; Buxeda, A.; Burballa, C.; Duran, X.; Mir, M.; Crespo, M.; Pascual, J. Relevance of KDPI Value and Acute Rejection on Kidney Transplant Outcomes in Recipients with Delayed Graft Function—A Retrospective Study. Transpl. Int. 2020, 33, 1071–1077. [Google Scholar] [CrossRef]
- Peake, P.W.; O’Grady, S.; Pussell, B.A.; Charlesworth, J.A. C3a Is Made by Proximal Tubular HK-2 Cells and Activates Them via the C3a Receptor. Kidney Int. 1999, 56, 1729–1736. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Farrar, C.A.; Abe, K.; Pratt, J.R.; Marsh, J.E.; Wang, Y.; Stahl, G.L.; Sacks, S.H. Predominant Role for C5b-9 in Renal Ischemia/Reperfusion Injury. J. Clin. Investig. 2000, 105, 1363–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Miyazaki, M.; Koji, T.; Furusu, A.; Nakamura-Kurashige, T.; Nishino, T.; Ozono, Y.; Harada, T.; Sakai, H.; Kohno, S. Enhanced Expression of Complement C5a Receptor MRNA in Human Diseased Kidney Assessed by in Situ Hybridization. Kidney Int. 2001, 60, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueler, F.; Rong, S.; Gwinner, W.; Mengel, M.; Bröcker, V.; Schön, S.; Greten, T.F.; Hawlisch, H.; Polakowski, T.; Schnatbaum, K.; et al. Complement 5a Receptor Inhibition Improves Renal Allograft Survival. JASN 2008, 19, 2302–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamberg, C.E.; Mackay, C.R.; Lee, H.; Zahra, D.; Jackson, J.; Lim, Y.S.; Whitfeld, P.L.; Craig, S.; Corsini, E.; Lu, B.; et al. The C5a Receptor (C5aR) C5L2 Is a Modulator of C5aR-Mediated Signal Transduction. J. Biol. Chem. 2010, 285, 7633–7644. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Whitfeld, P.L.; Mackay, C.R. Receptors for Complement C5a. The Importance of C5aR and the Enigmatic Role of C5L2. Immunol. Cell Biol. 2008, 86, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Hirata, T.; Enomoto, M.; Araki, T.; Ishimaru, H.; Takahashi, T.A. A Putative Chemoattractant Receptor, C5L2, Is Expressed in Granulocyte and Immature Dendritic Cells, but Not in Mature Dendritic Cells. Mol. Immunol. 2000, 37, 407–412. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Clinical promise of next-generation complement therapeutics. Nat. Rev. Drug Discov. 2019, 18, 707–729. [Google Scholar] [CrossRef]
- Patschan, D.; Patschan, S.; Müller, G.A. Inflammation and Microvasculopathy in Renal Ischemia Reperfusion Injury. J. Transplant 2012, 2012, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodsky, S.V.; Yamamoto, T.; Tada, T.; Kim, B.; Chen, J.; Kajiya, F.; Goligorsky, M.S. Endothelial Dysfunction in Ischemic Acute Renal Failure: Rescue by Transplanted Endothelial Cells. Am. J. Physiol. Ren. Physiol. 2002, 282, F1140–F1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Tada, T.; Brodsky, S.V.; Tanaka, H.; Noiri, E.; Kajiya, F.; Goligorsky, M.S. Intravital Videomicroscopy of Peritubular Capillaries in Renal Ischemia. Am. J. Physiol. Ren. Physiol. 2002, 282, F1150–F1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls (n = 8) | DGF (n = 12) | p-Value | |
|---|---|---|---|
| Age at transplantation (y, mean ± SD) | 55.1 ± 9.97 | 60.9 ± 10.23 | 0.219 |
| Donor age | 56.0 ± 9.81 | 64.3 ± 11.55 | 0.185 |
| Recipient sex male (n, %) | 4 (50) | 11 (92) | 0.088 |
| History of DM (n, %) | 2 (25) | 6 (50) | 0.160 |
| History of hypertension (n, %) | 7 (87.5) | 12 (100) | 0.191 |
| Pretransplant hemodialysis (n, %) | 4 (50) | 10 (83) | 0.116 |
| Donor Type (n, %) | 0.260 | ||
| DBD | 6 (75) | 6 (50) | |
| DCD | 2 (25) | 6 (50) | |
| 12-month creatinine mg/dL (mean ± SD) | 1.14 ± 0.27 | 2.16 ± 1.11 | 0.004 |
| Cold ischemic time (hours, mean ± SD) | 10.6 ± 6.86 | 15.3 ± 6.81 | 0.076 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arias-Cabrales, C.; Rodriguez-Garcia, E.; Gimeno, J.; Benito, D.; Pérez-Sáez, M.J.; Redondo-Pachón, D.; Buxeda, A.; Burballa, C.; Crespo, M.; Riera, M.; et al. Role of C5aR1 and C5L2 Receptors in Ischemia-Reperfusion Injury. J. Clin. Med. 2021, 10, 974. https://doi.org/10.3390/jcm10050974
Arias-Cabrales C, Rodriguez-Garcia E, Gimeno J, Benito D, Pérez-Sáez MJ, Redondo-Pachón D, Buxeda A, Burballa C, Crespo M, Riera M, et al. Role of C5aR1 and C5L2 Receptors in Ischemia-Reperfusion Injury. Journal of Clinical Medicine. 2021; 10(5):974. https://doi.org/10.3390/jcm10050974
Chicago/Turabian StyleArias-Cabrales, Carlos, Eva Rodriguez-Garcia, Javier Gimeno, David Benito, María José Pérez-Sáez, Dolores Redondo-Pachón, Anna Buxeda, Carla Burballa, Marta Crespo, Marta Riera, and et al. 2021. "Role of C5aR1 and C5L2 Receptors in Ischemia-Reperfusion Injury" Journal of Clinical Medicine 10, no. 5: 974. https://doi.org/10.3390/jcm10050974