
NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations
by
 , ,
, ,
David Niederseer
1 ,
,
Bernhard Wernly
2,3,4,
Elmar Aigner
5,
Felix Stickel
6 and
Christian Datz
7,* 1
Department of Cardiology, University Heart Center Zurich, University Hospital Zurich, University of Zurich, 8091 Zurich, Switzerland
2
Department of Anaesthesiology, Perioperative Medicine and Intensive Care Medicine, Paracelsus Medical University Salzburg, 5020 Salzburg, Austria
3
Center for Public Health and Healthcare Research, Paracelsus Medical University Salzburg, 5020 Salzburg, Austria
4
Department of Cardiology, Paracelsus Medical University Salzburg, 5020 Salzburg, Austria
5
First Department of Medicine, Paracelsus Medical University, 5020 Salzburg, Austria
6
Department of Gastroenterology, University Hospital Zurich, University of Zurich, 8091 Zurich, Switzerland
7
Department of Internal Medicine, General Hospital Oberndorf, Teaching Hospital of the Paracelsus Medical University, 5110 Oberndorf, Austria
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2021, 10(3), 467; https://doi.org/10.3390/jcm10030467
Submission received: 30 December 2020
/
Revised: 21 January 2021
/
Accepted: 21 January 2021
/
Published: 26 January 2021
(This article belongs to the Special Issue Nonalcoholic Fatty Liver Disease (NAFLD): Updates and Future Directions)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Overwhelming evidence suggests an association of cardiovascular disease (CVD) with non-alcoholic fatty liver disease (NAFLD); however, the underlying mechanisms remain largely speculative. It is, however, likely that common mechanisms contribute to the development of CVD and NAFLD, with lifestyle factors such as smoking, sedentary lifestyle with poor nutrition habits and physical inactivity being major candidates. These behavioral factors, on a predisposing genetic background, trigger changes in gut microbiota, inflammation, dyslipidemia and oxidative stress, leading to metabolic syndrome, diabetes and obesity as well as atherosclerosis. Treatment options to counteract both the progression and development of CVD and NAFLD include lifestyle interventions, optimal medical therapy of comorbid conditions and, as final possibility, bariatric surgery. As no causal pharmacotherapy of NAFLD is available, further research is urgently needed to address the unmet need of a growing population with NAFLD and CVD.
Keywords:
diabetes; metabolic syndrome; lifestyle; atherosclerosis; NASH; liver; exercise; nutrition1. Introduction
During the last decade, strong evidence has demonstrated a significant interplay and multifaceted relationship between non-alcoholic fatty liver disease (NAFLD) and cardiovascular disease (CVD). Pathophysiological mechanisms associating NAFLD with CVD are incompletely understood, and the current literature on the role of NAFLD as an independent risk factor for CVD and excess CV mortality in NAFLD patients has yielded conflicting results and should therefore be interpreted with caution [1,2,3,4,5]. Nevertheless, NAFLD should be regarded as a systemic metabolic disease affecting extrahepatic tissues throughout the body via complex mechanisms, including diet-induced alterations of the gastrointestinal tract, adipose tissue inflammation and lipotoxicity. Here, we aim to summarize the available data on epidemiological links of NAFLD to CVD and the possible mechanisms as well as current and future treatment options.
2. Epidemiological Data Linking NAFLD to Cardiovascular Disease (CVD)
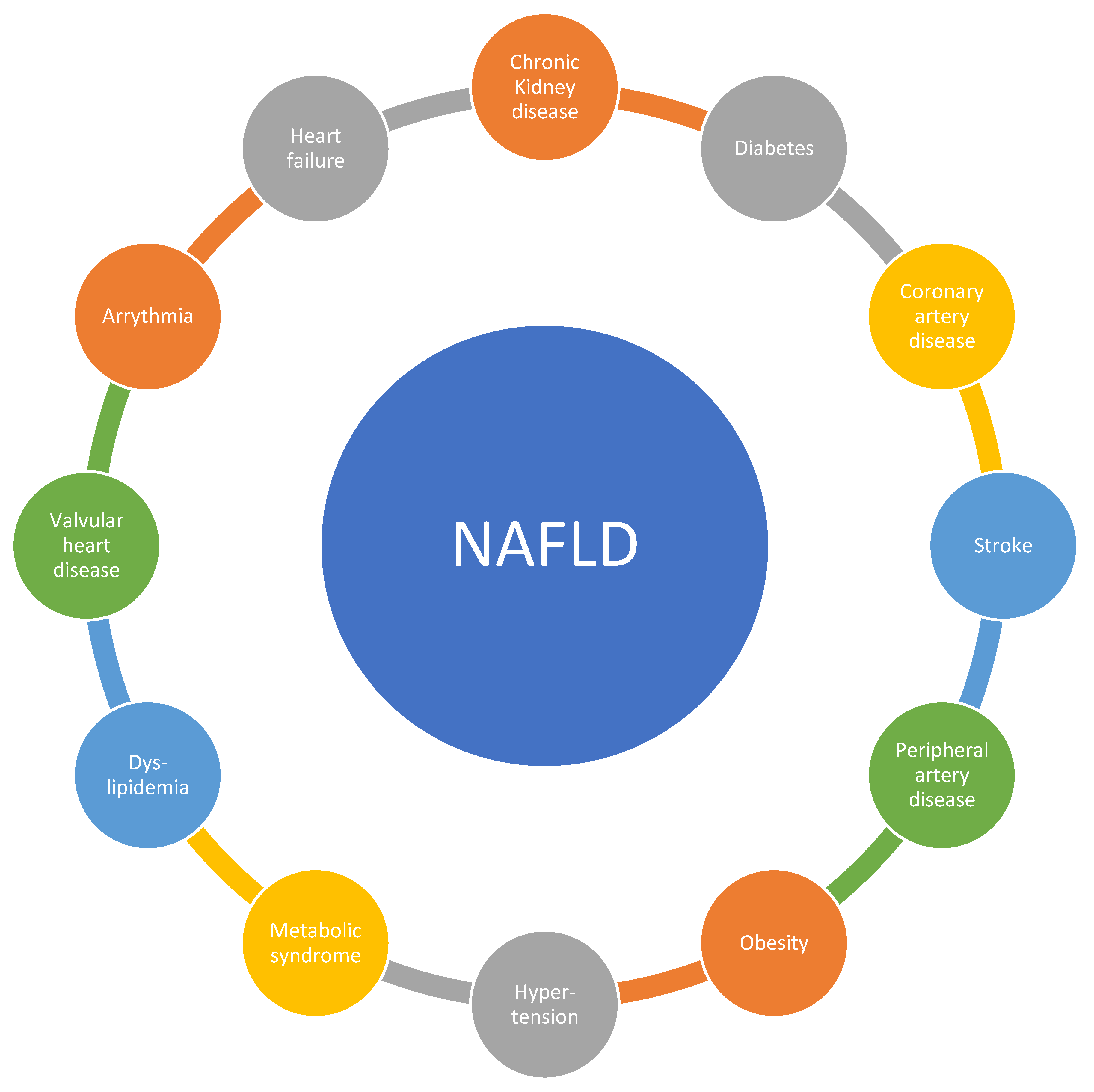
There are substantial epidemiological data that link NAFLD to CVD. We discuss vascular and metabolic disease but also other CV conditions (Figure 1).
2.1. Vascular Disease: Atherosclerosis Including Stroke, Peripheral Artery Disease, and Coronary Artery Disease
Although the understanding, prevention, and therapy of non-communicable diseases (NCDs) have improved during recent decades, NCDs constitute the primary cause (around 70%) of death worldwide [6]. Among NCDs, cardiovascular disease (CVD) is responsible for about half of the observed mortality [6]. Besides mortality, CVD leads to high morbidity and costs in Western societies [7,8,9]. CVD can be considered a phenotype with multiple interlapping mechanisms, which include arterial hypertension, obesity, diabetes, socioeconomic factors, and behavioral factors such as physical inactivity and poor nutrition as well as genetic factors, age, and sex [10,11].
NAFLD and CVD share several risk factors. Like CVD, NAFLD is highly prevalent in the total population of developed countries with a prevalence between 20% and 30% [12]. However, in obese patients and patients with diabetes, these proportions increase to up to 90% [13]. Both NAFLD and CVD are more common in men than women, although NAFLD progresses faster in women [13]. More than half of the patients diagnosed with NAFLD suffer from hyperlipidemia, an established independent risk factor for cardiovascular disease [10,11,14]. Furthermore, low-grade systemic inflammation was reported in both NAFLD and CVD [15]. Some experts consider NAFLD as the hepatic manifestation of metabolic syndrome, insulin resistance, and type 2 diabetes mellitus (T2DM) [16].
As both NAFLD and CVD are diseases with high prevalence, sharing several risk factors, it comes as no surprise that epidemiological data supporting NAFLD and CVD coincidence are abundant [1]. Although traditionally considered a hepatic disease, a growing body of evidence suggests that cardiovascular disease in patients with NAFLD determines the outcome rather than liver disease progression [17,18,19].
Observational studies suggest a link between NAFLD diagnosis and an increased risk for CVD and cardiovascular events, including death [4,20,21]. Markers of endothelial dysfunction such as increased intima media thickness and lower flow-mediated dilatation are more common in NAFLD patients [15,22,23]. The risk of stroke was higher in subjects with NAFLD than the general population [15]. Elevated brachial ankle pulse wave velocity and increased arterial stiffness are other surrogate parameters for subclinical atherosclerosis described in NAFLD [24,25].
NAFLD patients evidence higher cardiovascular risk according to the Framingham risk score [26]. Several meta-analyses support the association of NAFLD with increased risk for CVD, although the link between NAFLD and CVD is less robust than between NAFLD and metabolic syndrome [3,16,27]. While some studies have found associations between NAFLD surrogates and CVD, many of these analyses are limited by insufficient adjustment for established cardiovascular risk factors [28,29]. The crucial question of whether NAFLD is a risk factor for CVD independent of metabolic syndrome is, therefore, not yet completely clear. However, since the inclusion of NAFLD in risk calculations for CVD has so far brought no added value compared to models that only consider classical metabolic risk factors, it is, based on the available evidence, currently more likely that NAFLD has no independent impact on cardiovascular outcome beyond their common metabolic background [30,31,32].
2.2. Metabolic Disease: Metabolic Syndrome, Dyslipidemia, Diabetes, and Obesity
NAFLD is closely embedded into metabolic syndrome (MetS), which describes the typical common occurrence of established cardiovascular risk factors such as overweight/obesity, dyslipidemia, and disordered glucose homeostasis, as indicated by insulin resistance (IR) or T2DM and arterial hypertension [33]. The prevalence of NAFLD increases with the number of manifestations of metabolic syndrome components [34]. Most NAFLD subjects are obese, and the prevalence in the general population increases with progressive degrees of obesity, approximating almost 95% in subjects undergoing metabolic surgery [34,35]. Likewise, it approaches 60–70% in subjects with T2DM [36]. However, increasing BMI as an indicator of excess adipose tissue appears not to be the sole determining or sufficient factor for NAFLD development as 10% of lean subjects also have NAFLD, and a small proportion of even morbidly obese subjects maintain a healthy liver [37,38,39]. The finding of NAFLD in the absence of excess body weight is commonly referred to as “lean NAFLD” [40]. Further studies have elucidated the central relevance of adipose tissue distribution to NAFLD development as particularly in lean subjects or subjects with a low degree of overweight, substantial visceral fat accumulation is present and directly linked to the presence of a fatty liver [41,42], A meta-analysis by Young et al. demonstrated that lean NAFLD versus healthy subjects or lean controls had higher odds for abnormalities on their metabolic profile, including metabolic syndrome and its components, glycemic dysregulation, renal and liver function, and patatin-like phospholipase domain-containing protein 3 (PNPLA3) G allele, and their inflammatory profile, including uric acid and C-reactive protein [43]. Moreover, Golabi et al. have already demonstrated that lean NAFLD subjects have a higher risk of cardiovascular mortality and overall mortality than normal subjects [44]. In our own experience, lean NAFLD is associated with metabolic syndrome and increased cardiovascular risk as assessed by the Framingham Risk Score (Semmler et al., under review). Hence, limited healthy expandability of subcutaneous adipose tissue appears to represent a pivotal mechanism that triggers fat deposition in ectopic regions such as visceral fat depots and the liver. IR is the unifying underlying mechanism of metabolic complications such as CVD and NAFLD [45]. The importance of IR as a driver for metabolic complications is exemplified by the increasing morbidity and mortality risk with the progression of IR to T2DM [46,47]. This increase in morbidity and mortality in T2DM is true for liver-related complications (i.e., end-stage liver disease and hepatocellular carcinoma (HCC)) as well as for CVD (i.e., heart failure and ischemic events) [48,49]. Thus, the close interrelations are well confirmed epidemiologically between the various manifestations of insulin resistance or MetS on one side and NAFLD on the other; however, these frequently reciprocal relationships make it difficult to ascertain a specific contribution of NAFLD to CVD, as the biological co-evolution allows interpretation as shared risk factors, mutual augmentation, and confounding and may even be due to reverse causation, as genetic analyses indicate [50] (e.g., the possibility of increased prevalence of NAFLD in subjects with CVD due to less physical activity).
2.3. Other Cardiovascular Disease Manifestations
Several studies have reported epidemiological associations of NAFLD and valvular heart disease besides vascular and metabolic disease, i.e., mitral annulus calcification and aortic sclerosis. Subsequently, mitral regurgitation and aortic stenosis might develop, which increase the risk of heart failure on the basis of left ventricular dilation (in mitral regurgitation) and left ventricular hypertrophy (in aortic stenosis). Furthermore, arrhythmias have been reported in NAFLD, mainly atrial fibrillation (due to left atrial enlargement secondary to increased filling pressures and mitral regurgitation), increased QTc interval (corrected QT interval on electrocardiogram), and degenerative cardiac conduction disease (mainly first-degree atrioventricular block and left anterior hemiblock) [51,52]. It was recently suggested that NAFLD and non-alcoholic steatohepatitis (NASH) are associated with expansion of epicardial adipose tissue and the release of pro-inflammatory adipocytokines that cause microcirculatory dysfunction and fibrosis of the adjoining myocardium, resulting in atrial fibrillation as well as heart failure with a preserved ejection fraction (HFpEF). Inflammatory changes in the left atrium lead to electroanatomical remodeling; thus, NAFLD and NASH markedly increase the risk of atrial fibrillation. Simultaneously, patients with NAFLD or NASH commonly show diastolic dysfunction or latent HFpEF [53].
Finally, the prevalence of chronic kidney disease (CKD, estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73 m2, abnormal albuminuria, or overt proteinuria) is markedly increased in patients with NAFLD. The prevalence of CKD ranged from approximately 20% to 55% in patients with NAFLD as compared to 5% to 30% in patients without NAFLD, and the significant association of CKD and NAFLD mostly persisted after adjusting for confounding factors such as hypertension and T2DM [54,55].
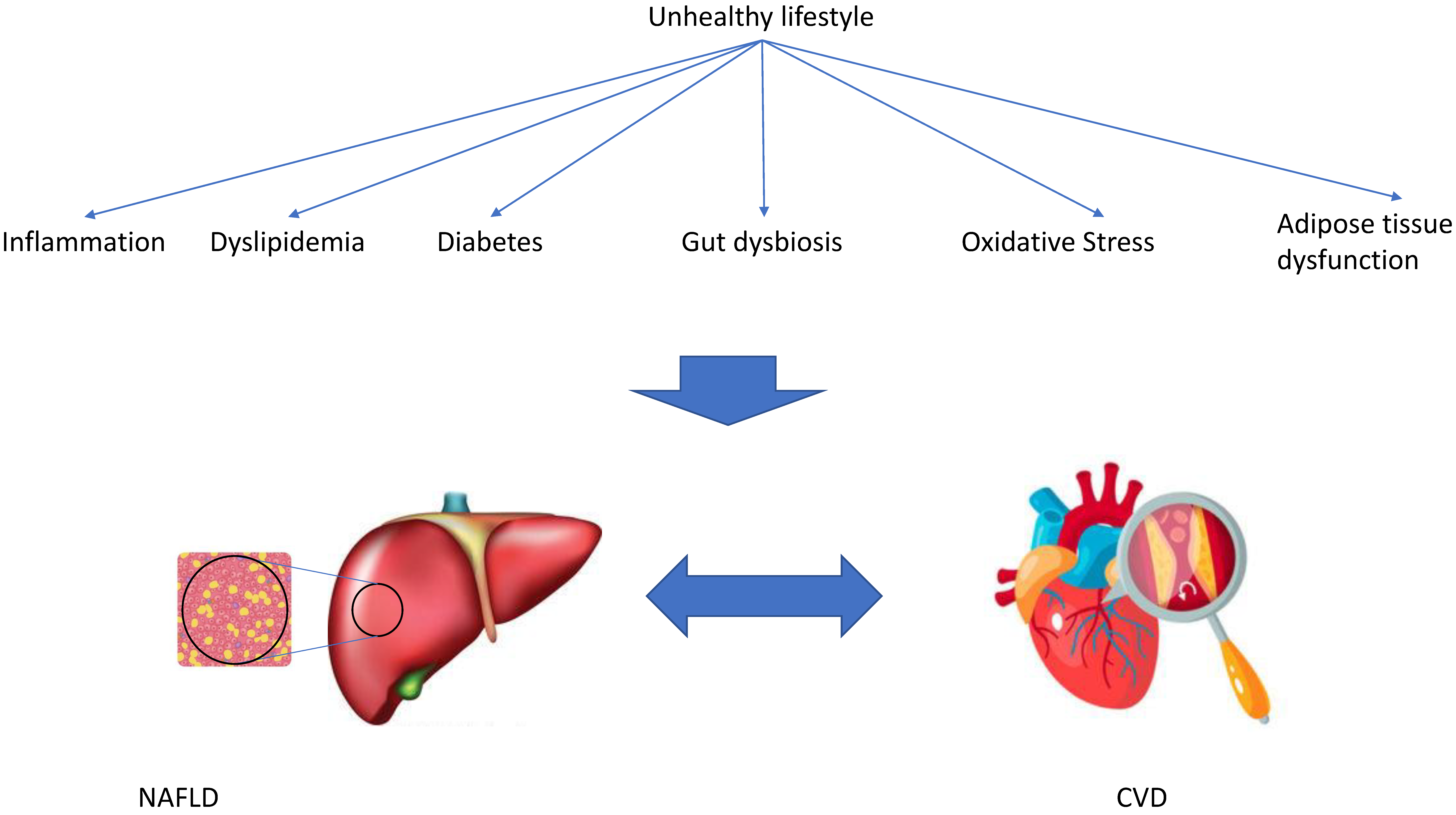
3. Possible Mechanisms That Might Explain the Association between NAFLD and CVD
Several possible mechanisms have been proposed that might explain the association between NAFLD and CVD, including inflammation and oxidative stress, gut microbiota, and dyslipidemia. All of these possible mechanisms are associated with lifestyle factors (Figure 2).
3.1. Inflammation and Oxidative Stress
Metabolic inflammation may be regarded as a sterile condition and is a crucial component in the pathogenesis of obesity, T2DM, CVD, and NAFLD, accompanied by increased levels of acute-phase proteins [56,57]. Metabolic inflammation is characterized by a systemic, low-grade inflammatory process in response to several non-infectious factors such as unhealthy dietary habits, especially a high-fat diet [58,59]. This so-called lipotoxicity activates inflammatory pathways and components of the immune system and has been observed in the liver, in arterial vessels, adipose tissue, muscle, pancreas, and the central nervous system. Various compartments such as the liver, the gastrointestinal tract, and adipose tissue are significant sources of pro-inflammatory drivers, including tumor necrosis factor (TNF), interleukin-6 (IL-6), interleukin-1β (IL-1β), C-reactive protein, fibrinogen, and fetuin A [60,61].
Dysregulation of lipid and glucose metabolism and oxidative processes play a key role in the pathogenesis of metabolic dysregulation in obesity, dysglycemia, NAFLD, and atherosclerosis [62,63,64,65,66]. Perturbation of endoplasmic reticulum (ER) homeostasis and oxidative stress have been shown to impact metabolic inflammation by inducing endothelial dysfunction, predisposing patients with NAFLD to develop CV events [67,68]. Persistent ER stress and mitochondrial dysfunction contribute to pathophysiological changes and play an important role in the progression from NALFD to NASH. Calcium ion (Ca2+), a critical and versatile intracellular secondary messenger, is involved in various cellular processes. The ER is known to be the most important intracellular Ca2+ store. The abnormal release of ER Ca2+ not only induces ER stress and mitochondrial dysfunction but also exacerbates hepatic cell lipotoxicity. The sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) pump, the main regulator of intracellular Ca2+, actively re-accumulates released Ca2+ back into the ER and therefore maintains Ca2+ homeostasis. SERCA activity is reduced in NALFD, while enhanced SERCA activity alleviates ER stress and apoptosis. Recent studies showed that the homeostasis of Ca2+ is closely related to the development of NALFD to NASH [69].
Expanded adipose tissue is a significant source of pro-inflammatory mediators since an estimated excess of 20–30 million macrophages accumulate with each kilogram of excess fat in humans, and adipose tissue hypoxia has a negative impact on the production of anti-inflammatory cytokines [70,71]. Dysfunctional adipocytes act as antigen-presenting cells and express pro-inflammatory cytokines such as TNF-alpha, IL-6, Il-1β, MCP4/CCL13, RANTES/CCL5, and MCP1/CCL2 [72,73,74,75]. The C-C motif Chemokine ligand-2 (CCL2) has been shown to be a key mediator of a cross-talk among adipocytes, macrophages, and endothelial cells potentially aggravating the inflammatory state, resulting in increased expression of pro-inflammatory cytokines, chemokines, adipokines, and angiogenic factors [76]. Genetic deficiency of chemokine receptor 2 (CCR2) and pharmacological antagonism of CCR2 in mice lowered macrophage content and the inflammatory profile of adipose tissue and ameliorated hepatic steatosis. Interestingly, the degree of liver inflammation in patients with NASH is correlated with increased expression of genes that regulate inflammation in adipose tissues [77]. Collectively, adipose tissue inflammation contributes to systemic inflammation, thereby promoting IR and CVD and potentially leading to deterioration of NAFLD [78,79].
Liver inflammation in NAFLD may be regarded as a multidirectional process where inflammatory stimuli may arise from extrahepatic tissues such as adipose tissue and the gut and inside the liver [80]. The role of inflammation in the progression from NAFLD to NASH, cirrhosis, and HCC is incompletely understood and has been somewhat disregarded in recent years as long-term outcomes of patients with NAFLD, including cardiovascular mortality, have been correlated with liver fibrosis [81]. It is well documented that liver inflammation mostly precedes or drives fibrosis [80]. Steatohepatitis may be a highly dynamic process of chronic relapsing or intermittent nature characterized by progression or “spontaneous” fibrosis regression. NAFLD, as a chronic inflammatory condition, may contribute via complex pathways to the systemic metabolic inflammation associated with atherosclerosis and impaired cardiac function [60,82,83]. Hepatic necro-inflammation is likely to be an independent proatherogenic mechanism that could possibly lead to an increased risk of CVD in patients with NASH compared with patients lacking histological features of liver inflammation [84,85]. In contrast, therapeutic antagonism of pro-inflammatory cytokines leads to improvement of hepatic steatosis, liver inflammation, and fibrosis [86,87]. Based on the insights gained from the significance of systemic metabolic dysregulation and its role in disease progression, several consensus statements and experts have recently proposed to rename NAFLD as metabolic-associated fatty liver disease (MAFLD) [88,89,90,91,92]. In order to underpin the importance of metabolic inflammation in the pathophysiology of this disease, highly sensitive C-reactive protein levels have been included as one crucial metabolic risk factor for the disease definition [89,93]. Although this new definition is a big step forward in the nomenclature, several authors have already raised their concerns about this definition; Younossi et al. [94] highlight that the heterogenous nature of NAFLD might not be fully covered by the new MAFLD criteria. In a study by Lin et al. (2020) [95], 620 of 4347 NAFLD patients did not meet the MAFLD criteria (14.3%) [96]. Although metabolic parameters were lower or less frequent in these patients, some had severe steatosis and/or advanced fibrosis (assessed non-invasively) [97].
However, it is unclear whether the new definition can identify patients at increased risk for cardiovascular events or liver-related morbidity and how patients who do not meet these criteria should be managed.
Similarly to differences in the metabolic characterization, differences in the prevalence of MAFLD might be predominantly explained by the use of a more sensitive tool to diagnose NAFLD (i.e., liver stiffness measurement, including controlled attenuation parameter), which probably allowed for identifying patients either at an earlier stage of disease or with a milder phenotype. Thus, associated metabolic disturbances are lower.
To sum up, although the new nomenclature of MAFLD can facilitate diagnosis and patient education, the utility of this new definition still needs to be confirmed.
A conceptual framework of proposed molecular and cellular mechanisms contributing to the metabolic burden of NAFLD on atherosclerosis has recently been published elsewhere [98].
In summary, future studies should be aiming at unraveling the role of hepatic inflammation as a major contributor to systemic metabolic inflammation in the pathogenesis of atherosclerosis and other cardiovascular impairments.
3.2. Gut Microbiota
A plethora of microorganisms such as bacteria, archaea, fungi, phages, and viruses colonize the gastrointestinal tract. The microbial community has a strong mutualistic relationship with its host and plays a vital role in digestion, immunity, and human metabolism [99,100]. Tremendous efforts have been made over the last years in elucidating the role of the gut microbiota in fueling metabolic inflammation and unraveling the impact of intestinal microbial composition on the development and disease progression of NAFLD and CVD [101,102,103,104]. The vast majority of intestinal microbiome-related studies assess compositional changes in the fecal microbial community, leading to overwhelming associative data, but are often limited when it comes to causality. Most of these studies were performed in animals; research in humans has only just started to describe microbiome-associated alterations in metabolic diseases. Several studies identified microbiome signatures linked to obesity and metabolic diseases such as T2DM, NAFLD, and cardiometabolic disorders [105,106,107,108,109,110]. A potential link between a specifically dysregulated gut microbiome in NAFLD, NASH, NAFLD-cirrhosis, and HCC discriminating these patients from healthy individuals has been recently presented [102,111,112,113,114,115]). Interestingly, significant changes in the gut microbiome signature have been observed in lean NAFLD [116,117]. The most compelling evidence for a specific intestinal microbiome signature in fibrotic NASH and NAFLD-cirrhosis comes from two recently published studies [102,113]. In their first study, the authors identified an association of advanced fibrosis with abundance of Proteobacteria and Escherichia coli and a decrease in Firmicutes such as Faecalibacterium prausnitzii. In a second study, they described a core gut microbiome signature that can identify patients with cirrhosis across different geographical backgrounds, independent of the effects of environmental factors and host genetics. Interestingly, several Veillonella species were commonly altered in the cirrhosis groups. In addition, profound changes in the intestinal virome signature have recently been identified in patients with NAFLD [118]. These findings have enormous potential to develop a diagnostic panel that could serve as a non-invasive biomarker for liver disease diagnosis and disease progression.
The close relationship between the gut, the liver, and (visceral) adipose tissue (AT) anatomically and functionally has led to intriguing concepts termed “gut–liver axis” and “AT-liver axis”, as excellently summarized recently [79]. These concepts demonstrate that any change in the gut microbial community not only triggers intestinal disorders but also impacts distant organs and causes associated diseases. It is obvious that gut microbial variations and the development of metabolic diseases such as NAFLD and CVD are tightly interrelated via shared common pathways, including metabolic inflammation [119].
Obesity and related disorders are characterized by profound alterations of the intestinal microbiota composition and functional capacities, raising the concept of intestinal dysbiosis, which, in conjunction with a defective and impaired mucosal barrier, may lead to the translocation of bacteria, bacterial metabolites, bile acids, lipopolysaccharides (LPS), endogenously generated ethanol, and endotoxins into the blood stream, which may result in systemic low-grade inflammation [101,120,121,122,123].
An imbalance of the intestinal microbial composition impacts hepatic fat accumulation via decreased fasting-induced adipocyte factor (FIAF) and increased energy delivery in the form of short-chain fatty acids (SCFAs) produced by the gut microbiota [107,124].
Dietary components such as carnitine, choline, and phosphatidylcholine are converted by gut bacteria to trimethylamine (TMA), which is metabolized in the liver to TMA N-oxide (TMAO). TMAO impacts the liver through modulating glucose and lipid homeostasis and triggering adipose tissue inflammation [125]. TMAO also triggers platelet dysfunction, and circulating TMAO levels are associated with the severity of NAFLD and atherosclerosis, ventricular arrhythmias, and clinical outcomes [126,127,128,129,130]. It should be noted that TMAO may be regarded a crucial microbiota-derived biomarker of CVD and potentially also in the cross-talk between NAFLD and CVD; however, TMA/TMAO metabolism not only relies on an imbalance of the intestinal microbiota composition but also on dietary factors, co-metabolism, and host genetics [131].
Although suggested in several studies, the intestinal microbiota’s role in inducing inflammation and atherogenesis is incompletely understood. A recently published study demonstrated that introducing pro-inflammatory gut microbiota into mice enhances systemic inflammation and accelerates atherosclerosis [132]. In this context, it is intriguing that statins, independently of their effect in lowering low-density lipoprotein (LDL), may contribute to clinical benefits by their anti-inflammatory actions. In addition, it has recently been demonstrated that the use of statins is associated with a lower prevalence of gut microbiota dysbiosis, thereby potentially attenuating systemic inflammation [133].
Significant alterations in the intestinal microbiota were found in patients with coronary heart disease (CHD) complicated by NAFLD, showing a decrease in Colinsella and Parabacterioides as a potential explanation for the worse clinical outcome and disease progression compared to CHD without NAFLD [134].
It seems plausible that the gastrointestinal tract may be regarded as a site of origin of systemic inflammatory changes, thereby playing a crucial role in metabolic diseases such as NAFLD and CVD. Targeting metabolic inflammation may not only be attractive in combating atherosclerosis but may also become a central component in the management of other metabolic disorders in the future [87]. Although in its infancy, future studies focusing on causal interactions between inhabitants of the gut, including phages, viruses, and fungi, and metabolism may allow the development of new therapeutic agents for the treatment of NAFLD and CVD.
3.3. Dyslipidemia and Adipose Tissue (Dys-)Function
The mechanisms associating CVD with NAFLD are complex. It is the main challenge to identify where a distinct contribution of NAFLD occurs independently from the cardiovascular risk environment already constituted by MetS. However, several lines of evidence suggest that an affected liver may have an additional role in promoting CVD. From one aspect, this pertains to the dyslipidemia of NAFLD. Dyslipidemia is well established as closely linked to CVD, and low-density lipoprotein (LDL) cholesterol is accepted as a key driver of atherosclerosis [135]. The typical dyslipidemia of NAFLD is characterized by low high-density lipoprotein (HDL) cholesterol and high triglycerides, with moderately increased LDL cholesterol [136]. Lipid homeostasis is affected on several levels in NAFLD. While the liver is the central organ of lipid turnover, it does not store lipids in a healthy state [137]. In NAFLD, excess hepatic lipids derive from adipose tissue lipolysis with increased fatty acid flux to the liver, increased dietary uptake, increased de novo lipogenesis, and impaired export of newly synthesized VLDL (very low density lipoprotein) particles [138]. While typically in early stages, an increase in mitochondrial oxidation compensates, at least partially, for the excess in hepatocellular lipids [139], this becomes impaired at later stages. Normally, large VLDL particles lose their triglyceride moiety once secreted into circulation via lipoprotein lipase activity, turning into an equal number of atherogenic LDL particles with a dominant cholesterol content. LDL particles are taken up into the liver via the LDL receptor pathway [137]. The overproduction of VLDL particles in NAFLD triggers a series of lipoprotein abnormalities which result in the characteristic atherogenic dyslipidemia pattern that includes a predominance of particularly atherogenic, small dense LDL particles [140,141]. The severity of these lipid abnormalities appears to increase with more severe and advanced stages of liver disease [142,143]. These small dense LDL particles have a particular propensity to penetrate the vascular endothelium into the subendothelial space, which serves as an initiating event of atherosclerotic plaque formation. LDL cholesterol in the vascular wall is further subjected to oxidation and elicits an innate immune response via Toll-like receptors (TLRs) [144].
Atherosclerosis development is mainly mediated by apo-B-containing particles. Thus, besides LDL cholesterol-mediated risk for atherosclerosis, triglyceride-rich lipoproteins (TRLs) such as VLDL and IDL (intermediate density lipoprotein) also contribute to increased risk for CVD [145]. These particles also contain apo-C3, which likely has a role in activating TLRs and, therefore, the inflammasome reaction [146,147]. This, in turn, activates interleukin (IL)-1β family via caspase-1, inducing the prototypical pro-inflammatory mediators IL-1, IL-6, and CRP in the vascular wall [148]. This constitutes a link between lipid deposition and propagated inflammation in the vascular wall as the mechanism to CVD. This pathway may further be activated by saturated fatty acids, which are particularly increased in de novo lipogenesis of the steatotic liver and have been associated with increased cardiovascular risk such as palmitic acid C16:0 [149].
Besides its impact on lipids as a risk factor for CVD, NAFLD, like CVD, is an inflammatory disease, and pro-inflammatory cytokines such as TNF-α, interleukin-1β, or high-sensitive C-reactive protein (hs-CRP) are elevated in MetS-associated diseases [150]. This inflammatory milieu reflects pro-inflammatory mechanisms which are active in adipose tissue, the vascular wall, circulating immune competent cells, and also the liver. The liver is the physiological source for most acute-phase reactants, orchestrating the extent and duration of any immune response. In NAFLD, the liver becomes an additional permanent autonomous source of pro-inflammatory and pro-coagulant mediators [151,152,153]. Although the specific contribution of these additional NAFLD-derived factors to cardiovascular endpoints has not been demonstrated conclusively, from a pathophysiological point of view, it is plausible to consider the steatotic and inflamed liver as an enhancer of the inflammatory reaction in the vascular wall and as a catalyst of a prothrombotic coagulation state, favoring thrombus formation.
The gene polymorphisms of adiponectin rs266729 [154], adiponectin-encoding gene (ADIPOQ), leptin receptor (LEPR), apolipoprotein C3 (APOC3), peroxisome proliferator-activated receptors (PPAR), sterol regulatory element-binding proteins (SREBP), transmembrane 6 superfamily member 2 (TM6SF2), microsomal triglyceride transfer protein (MTTP), tumor necrosis factor-alpha (TNF-α), and manganese superoxide dismutase (MnSOD) have been reported to be related to NAFLD and coronary artery disease [155].
3.4. Endothelial Dysfunction
Endothelial dysfunction precedes atherosclerosis. In NAFLD, elevated levels of asymmetric dimethyl arginine, an endogenous antagonist of nitric oxide synthase, are observed [156,157]. Increased serum homocysteine levels are seen in NAFLD, primarily due to changes in methionine metabolism, which changes the production and catabolism of homocysteine in the liver. Hyperhomocysteinemia is associated with increased intrahepatic vascular resistance, which impairs nitric oxide formation, and increased oxidative stress activates platelets. Furthermore, systemic inflammation (Section 3.1) increases endothelial dysfunction, alters vascular tone, and enhances vascular plaque formation. These mechanisms are supported by the clinical findings in a study of NAFLD patients that found significantly reduced flow-mediated vasodilation compared with age- and sex-matched control subjects [158]. As endothelial dysfunction is the main mechanism of erectile dysfunction in men, erectile dysfunction was shown to be a relevant issue in patients with NAFLD [159].
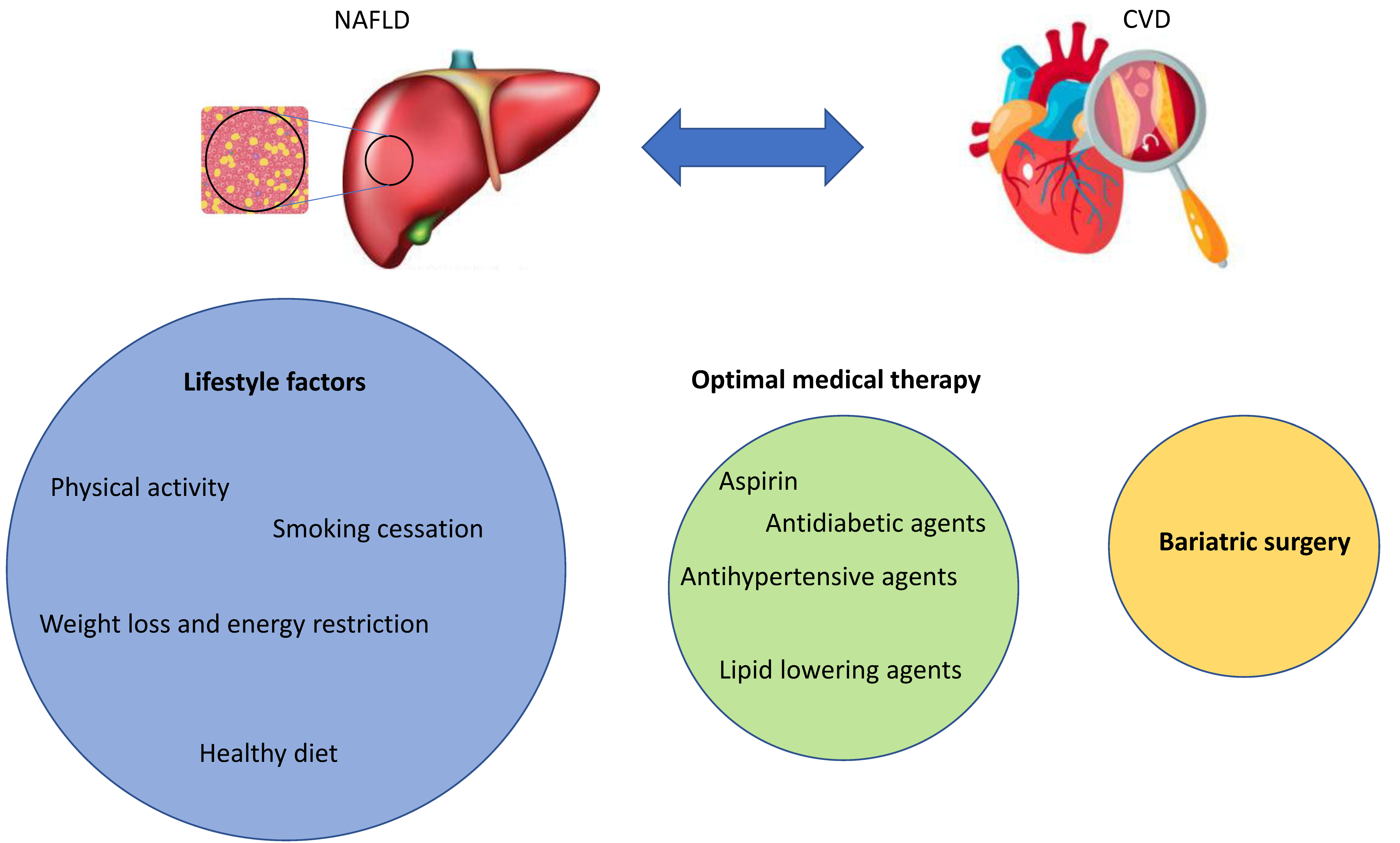
4. Current and Future Treatment Options for NAFLD Complicated by CVD
Besides lifestyle intervention and, for some patients, surgery, a number of established and emerging CV medications might play a role in the treatment of NAFLD complicated by CVD (Figure 3).
4.1. Lifestyle Interventions
There are currently no Food and Drug Administration (FDA)-approved pharmacological therapies for NAFLD, and lifestyle interventions including smoking cessation, weight loss, healthy nutrition, and exercise remain the cornerstones for treatment [160].
4.1.1. Smoking Cessation
There is associative evidence available that smoking contributes to the development of NAFLD via direct and indirect mechanisms [161]. As smoking is an established cardiovascular risk factor and smoking cessation is central to the prevention of cardiovascular events, also in subjects with NAFLD, smoking cessation is desirable.
4.1.2. Weight Loss and Energy Restriction
Energy restriction induces weight loss and the reduction of liver fat, independent of the macronutrient composition of the diet [162]. Consequently, a reduction of 500–1000 kcal of daily energy intake might induce a weight loss of 500–1000 g/week. A total of 7–10% weight loss is desirable in overweight and obese patients with NAFLD, as weight loss leads to significant reduction in hepatic steatosis [163]. Cognitive–behavioral treatment, a combination of exercise with energy restriction, and long-term maintenance approaches delay vascular and metabolic complications in NAFLD and the onset of T2DM [164].
4.1.3. Healthy Nutrition
Although the main aspect in healthy nutrition is energy restriction, irrespective of macronutrient composition, low-to-moderate fat and moderate-to-high carbohydrate, low-carbohydrate ketogenic, or high-protein diets have been suggested by some studies to be beneficial beyond the effect of sole energy restriction. As evidence on macronutrient composition and the best diet to prevent or treat NAFLD is scarce and conflicting, energy restriction should be the main aspect to focus on in evidence-based nutrition counselling [160]. Guidelines for both the treatment of NAFLD and cardiovascular disease prevention currently recommend adaption of the macronutrient composition to the pattern of the Mediterranean diet. This, in brief, involves a vegetable-based diet with plant oils (olive oil) as the main dietary fat source, whole grains as the main dietary source of carbohydrates, and predominant consumption of plant protein such as nuts and legumes with avoidance of processed red meat and limited amounts of dairy products and fish or white meat. From a cardiovascular perspective, the additional restriction of salt consumption (DASH diet, dietary approach to stop hypertension) complements the dietary pattern currently considered healthy. Avoiding fructose-containing beverages and foods, however, has been shown to be associated with lower prevalence of NAFLD and appears to be helpful for CV risk reduction [165]. Alcohol intake should be moderated and limited to 30 g in men and 20 g in women, as alcohol intake below this “risk threshold” was associated with a lower prevalence of NAFLD, NASH, and even lower fibrosis at histology [166,167,168]. However, absolute alcohol abstinence is mandatory in advanced fibrosis and NASH-cirrhosis to reduce the risk of worsening liver dysfunction and HCC [169]. Coffee was shown to be protective in NAFLD, as in liver disease of other etiologies, reducing histological severity and liver-related outcomes [170].
4.1.4. Exercise
Exercise and physical activity are universally recommended for the promotion of metabolic and vascular health, i.e., in healthy or at-risk subjects and patients [171]. Hence, moderate-intensity aerobic physical activities in 3–5 sessions (a total of 150–300 min/week) are advised. Resistance training in two sessions per week has additional effects and promotes musculoskeletal fitness, with effects on metabolic risk factors, mainly linked to augmented muscular glucose utilization. A sedentary lifestyle induces fatigue and daytime sleepiness and reduces compliance with exercise recommendations. Physical activity is dose-dependent and vigorous (running) rather than moderate exercise (brisk walking) carries the full benefit, including for NASH and fibrosis [172,173]. Any engagement in physical activity or increase over previous levels is better than continuing inactivity. Long-term adherence is a major limitation for exercise and physical activity to unfold its full potential [174].
4.2. Medication
4.2.1. Aspirin
Acetylsalicylic acid (ASA) still constitutes a cornerstone in antiplatelet therapy once a definitive diagnosis of atherosclerotic disease is established [10,11]. There are data from basic and small observational studies suggesting an effect of ASA on the progression of NAFLD [175]. The intake of ASA could be associated with a reduction in liver fibrosis, and several observational studies showed a lower incidence of liver fibrosis and evidence of slower fibrosis progression in patients taking ASA [176,177]. However, these data remain speculative, and there is no randomized evidence supporting the use of ASA in patients with NAFLD without manifestation of atherosclerosis [178]. On the other hand, the safety profile of ASA is good even in patients with manifestation of liver cirrhosis without esophageal varices [179]. ASA should, therefore, be administered in NAFLD patients based on the recommendations aiming at the prevention of cardiovascular events [10,11]. From a mechanistic perspective, ASA intake via reduced platelet activation and sinusoidal microthrombus formation could limit local hypoxia and, thus, fibrogenesis in a similar fashion to the action of low-molecular-weight heparin [180,181].
4.2.2. Lipid-Lowering Agents
Lipid-lowering drugs, among which statins are the most widely prescribed and potent substances with the best evidence, are an essential component in the drug modification of cardiovascular risk [10,11,182].
Although statins have hepatic toxicity as a rare side effect, statins should not be withheld from patients with NAFLD for primary and secondary prevention of cardiovascular events [183]. This is of high clinical relevance as statins should also be administered to patients with elevated transaminases (due to their NAFLD), but this is often not practiced in daily reality. In a study that analyzed 261 patients with NAFLD, only 13% received statin therapy, which may be considered too low given the high coincidence of NAFLD, CVD, and metabolic syndrome [184,185]. In both the GREACE study and the IDEAL study, atorvastatin was shown to reduce risk compared to a less potent lipid-lowering therapy with adequate safety, with a particular cardiovascular benefit for subjects with elevated transaminases [186,187,188]. Furthermore, in several studies, surrogate parameters for a reduction in the progression of the liver disease NAFLD could be detected in addition to a cardiovascular risk reduction [189,190]. Therefore, statin therapy is not contraindicated in patients with NAFLD but is likely particularly beneficial from both a cardiological and a hepatological perspective.
Similarly, the use of ezetimibe, another lipid-lowering agent supported by guidelines, is considered to be safe and effective for the prevention of cardiovascular events in NAFLD [191]. However, the use of ezetimibe in NAFLD patients beyond the indication supported by the lipid-lowering evidence is currently speculative, as is a possible effect of ezetimibe on liver-related endpoints [10,11,182,191].
The PCSK9 inhibitor (PCSK9i) represents a relatively new substance class in lipid-lowering therapy which has an excellent potency to lower LDL cholesterol especially [192,193,194]. PCSK9i has a relatively narrow indication in cardiological patients with very high cardiovascular risk, statin intolerance, or persistently high LDL concentrations despite statin therapy [195]. Basic science and clinical studies suggest that high intrahepatic or circulating levels of PCSK9 play an important role in muscle and liver lipid storage and may contribute significantly to the pathogenesis and progression of NAFLD [196]. Conversely, PCSK9 inhibition increases LDL via the LDL receptor-mediated pathway and, therefore, lipid content of hepatocytes. Based on these data, it is speculative whether PCSK9i has an impact on the cardiovascular and hepatological complications of NAFLD, and several indirect studies evaluating, e.g., berberine suggest a beneficial effect [197,198]. No liver-related signals were observed in large PCSK9 Fourier and Odyssey outcome studies, and thus, it appears to be safe in the target population with regard to liver disease. However, since randomized outcome studies in NAFLD patients investigating the efficacy and safety of PCSK9i are still pending, this assumption remains speculative [196]. Therefore, based on the available evidence, NAFLD diagnosis should not restrict PCSK9i therapy when indicated for primary or secondary prevention.
4.2.3. Antihypertensive Agents
Among NAFLD patients, the prevalence of arterial hypertension varies from 40% to 70%, and evidence from prospective studies from France and Germany has shown that NAFLD is associated with a 2–3-fold increase in the incidence of arterial hypertension over time [199,200]. In a population-based cohort of hypertensive (n = 433) and normotensive (n = 457) subjects from Finland, fatty liver was associated with significantly higher ambulatory daytime and nighttime systolic blood pressure levels [201].
Furthermore, the severity of progressive NASH tends to increase in patients with hypertension, although this association does not appear to be as strong as that with T2DM.
Despite the strong association between NAFLD and arterial hypertension, sufficient evidence on the comparative benefit of certain antihypertensive agents in NASH is lacking. Thus, antihypertensive management in patients with NAFLD is routinely performed along with current guidelines irrespective of this diagnosis, although several pharmaceutical agents harbor theoretical advantages over others. Betablockers are highly effective at lowering systolic and diastolic blood pressure but may also adversely affect physical activity in some patients with NAFLD and, thus, counteract calorie expenditure. Furthermore, betablockers suppress stress reactions to hypoglycemia in NAFLD patients concomitantly treated for T2DM. For these reasons, angiotensin-converting enzyme inhibitors (ACE-I) and angiotensin receptor blockers (ARB) are favored groups of agents. Another reason is their nephroprotective potential, bearing in mind the high prevalence of T2DM and consecutive kidney dysfunction among NAFLD patients [202]. Additionally, both groups of agents target myofibroblasts and hepatic stellate cells in experimental models of liver fibrosis and counteract portal hypertension [203], but current evidence suggests exercising caution in the extended use of ACE-I/ARB solely for the purpose of managing fibrosis in NAFLD patients [204]. Once cirrhosis with significant portal hypertension or signs of hepatic dysfunction evolve, treatment with both ACE-I and ARB should be reevaluated and a risk–benefit analysis should be performed due to a higher incidence of complications, including kidney failure and sodium and potassium derangements.
4.2.4. Antidiabetic Agents
The relevance of T2DM in patients with NAFLD is twofold: First, over half of all NAFLD patients reveal T2DM, and second, the presence of T2DM promotes the progression of NAFLD and is a predictor of advanced fibrosis and mortality [205]. T2DM is not only associated with NAFLD but is also a significant risk factor of progressive liver disease per se. Based on data from a recent meta-analysis including 22.8 million individuals followed up for a median of 10 years, T2DM is associated with an increased risk of incident severe liver disease events (adjusted HR (hazard ratio) 2.25, 95% CI 1.83–2.76, p < 0.001) [206]. Conversely, patients with NAFLD have an up to twofold increased risk of subsequent incident T2DM [202]. Therefore, patients with NAFLD should be screened and surveilled for T2DM and, vice versa, diabetic patients should be assessed for possible NAFLD.
Management of T2DM in NASH patients must take into account coexisting comorbidities and consider its impact on underlying cofactors of NAFLD. This is particularly true for overweight. Metformin is the first choice of treatment for T2DM and also in NAFLD [160], although the evidence for an improvement in histological changes such as liver fat or fibrosis is weak. Euglycemic control with metformin is satisfactory in many patients at relatively low costs, and metformin causes no weight gain. Moreover, some retrospective human data point to an anti-tumor effect of metformin on HCC [207], which may also support its use in NAFLD, a disease with a substantial risk of HCC also in non-cirrhotic patients.
In patients who do not achieve adequate glycemic control with metformin, therapeutic escalation can be undertaken with the following agents:
- Thiazolidinediones (TZDs): TZDs are selective and potent PPARγ agonists by which they act as insulin sensitizers and, thus, synergistically with circulating insulin. Through this effect, TZD overcomes insulin resistance as a hallmark of NAFLD. Of the three compounds introduced to the market, only pioglitazone currently remains available after troglitazone was withdrawn due to severe hepatotoxicity and rosiglitazone was linked to adverse cardiovascular incidents. Pioglitazone was compared to vitamin E and a placebo in the PIVENS trial and given for 96 months at a dose of 30 mg daily. Elevated serum liver enzymes were reduced with both agents, and both were associated with significant reductions in hepatic steatosis (p = 0.005 for vitamin E and p < 0.001 for pioglitazone) and lobular inflammation (p = 0.02 for vitamin E and p = 0.004 for pioglitazone). However, improvements in fibrosis scores were observed for neither active treatment. For the latter, however, a meta-analysis of five randomized controlled trials with pioglitazone in NAFLD patients [208] found an association with improved advanced fibrosis (OR (odds ratio), 3.15; 95% CI, 1.25–7.93; p = 0.01), fibrosis of any stage (OR, 1.66; 95% CI, 1.12–2.47; p = 0.01), and NASH resolution (OR, 3.22; 95% CI, 2.17–4.79; p < 0.001), even when given to subjects without diabetes [209]. Relevant disadvantages of pioglitazone in the treatment of T2DM in NAFLD patients include significant weight gain, fluid retention, and some evidence from observational studies demonstrating an increased risk of bladder cancer [210]. Pioglitazone is the current treatment of choice for NAFLD in subjects with T2DM.
- Sodium-glucose cotransporter-2 (SGLT-2) inhibitors: SGLT2 inhibitors are compounds that inhibit SGLT-2 proteins located in the renal tubules of the kidneys, which are responsible for the reabsorption of glucose, thereby increasing the loss of glucose via urinary excretion. There are three SGLT-2-selective inhibitors approved by the Food and Drug Administration (FDA), namely canagliflozin, dapagliflozin, and empagliflozin. In a recent systematic review of studies investigating SGLT-2 inhibitors in NAFLD patients, seven studies were reviewed, of which six demonstrated certain improvements by SGLT-2 inhibitors in NAFLD. Meanwhile, five studies showed a reduction in steatosis, and only one study suggested an improvement of NASH histology features [211]. Beyond these liver-related endpoints, SGLT-2 inhibitors also favorably impact weight, cardiovascular, and nephrological outcomes, rendering them a satisfactory and increasingly popular, albeit not ideal, group of agents to treat T2DM in NAFLD. Urinary tract infections limit their use in a relevant number of patients [212].
- Glucagon-like peptide 1 (GLP-1) agonists. GLP-1 is an antihyperglycemic hormone that induces pancreatic β cells to secrete insulin, and it was demonstrated that patients with NAFLD reveal insufficient GLP-1 secretion [213]. Thus, pharmaceuticals that offset this imbalance to improve glucose homeostasis are attractive candidates for treating T2DM in the context of NAFLD. Indeed, GLP-1 agonists have recently gained much attention based on clinical data from trials that provide strong evidence for a powerful effect of GLP-1 agonism on hepatic endpoints in patients with NAFLD and also a reduction in CV endpoints.
In the LEAN study, 26 patients with biopsy-proven NASH were randomly assigned to receive liraglutide at 1.8 mg daily and 26 were assigned to a placebo as part of a multicenter, double-blinded, randomized, placebo-controlled, 48-week phase 2 trial [214]. Premier findings were an improvement of NASH histology in 39% of NASH patients on liraglutide compared to only 9% of NASH patients receiving the placebo who revealed improved histology. In addition, worsening of fibrosis occurred in 36% of patients on the placebo but only 9% in those receiving liraglutide. Tolerability was acceptable overall, with the expected gastrointestinal side-effects from liraglutide including diarrhea, constipation, loss of appetite, and nausea. It can be assumed that some of the improvement in liver-related endpoints can be ascribed to weight loss in a high proportion of patients. Similar observations were recently made with semaglutide, another GLP-1 agonist available for both subcutaneous injection and orally, which caused a significant drop in elevated serum liver enzyme levels and markers of inflammation in patients treated for T2DM and/or overweight [215]. Comparable observations have also been recorded for lixisenatide [216] and dulaglutide [217].
Importantly, with regard to NAFLD patients with cardiovascular comorbidities, GLP-1 agonists have been shown to reduce cardiovascular and all-cause mortality and reduce heart failure and the progression of chronic kidney disease (CKD).
Overall, GLP-1 agonists appear to be the most pertinent group of therapeutic agents since they have the potential to reduce body weight as a crucial clinical condition in patients with NAFLD, and the availability of an orally administered compound can be considered a true turning point in the treatment of NAFLD. Their specific use to treat NAFLD, however, is not licensed, and additional studies with liver-specific primary endpoints are required to decipher their actual value in the treatment of NAFLD.
Other antidiabetic drugs including insulin, sulfonylurea agents, and dipeptidyl peptidase IV inhibitors are either less studied in patients with NAFLD, reveal significant disadvantages (weight gain and worsening of sarcopenia), or show no benefit for the underlying liver disease. Their use is therefore either discouraged (insulin) or, at best, a second-line option, should more favorable compounds fail or prove intolerant.
Furthermore, currently ongoing NASH trials are frequently focused on liver-specific outcomes only, and the effect of CV outcomes will need to be studied further, particularly in light of available data of SGLT2 inhibitors and GLP-1 agonists which appear to improve both. Additionally, it will be interesting to see whether treatment of liver disease via liver targets will have an impact on cardiovascular disease outcomes as this may provide important insights into the causality relation of liver and vascular disease in the future. A practical and comprehensive overview on the use of antidiabetic drugs in NAFLD is provided elsewhere [218].
4.3. Surgery
If lifestyle intervention and adequate treatment of comorbidities are not sufficient to reduce the overall risk, bariatric surgery in NAFLD is a final option. Bariatric surgery may induce weight loss and improve the detrimental effects of MetS and T2DM. Recent data demonstrated significant effects of bariatric surgery on GLP-1 and other gut hormones and important beneficial changes in lipid, metabolic, and inflammatory abnormalities in NAFLD. Bariatric surgery may, therefore, reverse some of the pathological liver changes in NAFLD and NASH patients and reduce CV outcomes [219].
5. Conclusions
In conclusion, overwhelming evidence suggests an association of CVD with NAFLD; however, the related mechanisms remain largely speculative. It is, however, likely that identical mechanisms contribute to the development of CVD and NAFLD, with lifestyle factors such as smoking, sedentary lifestyle with poor nutrition habits, and physical inactivity being major aspects. These behavioral factors, among others, trigger changes in the gut microbiota, inflammation, dyslipidemia, and oxidative stress, leading to metabolic syndrome, diabetes, and obesity as well as atherosclerosis. Treatment options to counteract both the progression and development of CVD and NAFLD include lifestyle interventions, optimal medical therapy of comorbid conditions, and, as final possibility, bariatric surgery (Figure 3). As no causal pharmacotherapy of NAFLD is available, further research is urgently needed to address the unmet needs of a growing population with NAFLD and CVD.
Author Contributions
Conceptualization, D.N., E.A., B.W., F.S. and C.D.; resources, D.N., E.A., B.W., F.S. and C.D.; writing—original draft preparation, D.N., E.A., B.W., F.S. and C.D.; writing—review and editing, D.N., E.A., B.W., F.S. and C.D.; visualization, D.N.; supervision, C.D.; project administration, D.N. All authors have read and agreed to the published version of the manuscript.
Funding
C.D. was funded by SPAR Austria AG.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Targher, G.; Day, C.P.; Bonora, E. Risk of Cardiovascular Disease in Patients with Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Association of non-alcoholic fatty liver disease with major adverse cardiovascular events: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 33386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söderberg, C.; Stål, P.; Askling, J.; Glaumann, H.; Lindberg, G.; Marmur, J.; Hultcrantz, R. Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology 2010, 51, 595–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanova, M.; Younossi, Z.M. Independent Association between Nonalcoholic Fatty Liver Disease and Cardiovascular Disease in the US Population. Clin. Gastroenterol. Hepatol. 2012, 10, 646–650. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Health Estimates 2016: Disease Burden by Cause, Age, Sex, by Country and by Region, 2000–2016; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease. Circulation 2019, 74, 1376–1414. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics—2019 update: A report from the American heart association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [Green Version]
- Bellentani, S.; Marino, M. Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD). Ann. Hepatol. 2009, 8, S4–S8. [Google Scholar] [CrossRef]
- Scorletti, E.; Calder, P.C.; Byrne, C.D. Non-alcoholic fatty liver disease and cardiovascular risk: Metabolic aspects and novel treatments. Endocrinology 2011, 40, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Lentz, J.S. Fatty liver hepatitis (steatohepatitis) and obesity: An autopsy study with analysis of risk factors. Hepatology 1990, 12, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Wojcik-Cichy, K.; Koslinska-Berkan, E.; Piekarska, A. The influence of NAFLD on the risk of atherosclerosis and cardiovascular diseases. Clin. Exp. Hepatol. 2018, 4, 1–6. [Google Scholar] [CrossRef]
- Morrison, A.E.; Zaccardi, F.; Khunti, K.; Davies, M.J. Causality between non-alcoholic fatty liver disease and risk of cardiovascular disease and type 2 diabetes: A meta-analysis with bias analysis. Liver Int. 2019, 39, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, G.; Moscatiello, S.; Di Domizio, S.; Forlani, G. Obesity-associated liver disease. J. Clin. Endocrinol. Metab. 2008, 93, S74–S80. [Google Scholar] [CrossRef]
- Kotronen, A.; Yki-Jarvinen, H. Fatty liver: A novel component of the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 27–38. [Google Scholar] [CrossRef]
- De Alwis, N.M.; Day, C.P. Non-alcoholic fatty liver disease: The mist gradually clears. J. Hepatol. 2008, 48, S104–S112. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.; Wong, G.L.; Yeung, J.C.; Fung, C.Y.; Chan, J.K.; Chang, Z.H.; Kwan, C.T.; Lam, H.W.; Limquiaco, J.; Chim, A.M.; et al. Long-term clinical outcomes after fatty liver screening in patients undergoing coronary angiogram: A prospective cohort study. Hepatology 2016, 63, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Treeprasertsuk, S.; Leverage, S.; Adams, L.A.; Lindor, K.D.; St Sauver, J.; Angulo, P. The Framingham risk score and heart disease in nonalcoholic fatty liver disease. Liver Int. 2012, 32, 945–950. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.C.; Kim, D.J.; Huh, K.B. Association between nonalcoholic fatty liver disease and carotid intima-media thickness according to the presence of metabolic syndrome. Atherosclerosis 2009, 204, 521–525. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: A systematic review. J. Hepatol. 2008, 49, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Salvi, P.; Ruffini, R.; Agnoletti, D.; Magnani, E.; Pagliarani, G.; Comandini, G.; Praticò, A.; Borghi, C.; Benetos, A.; Pazzi, P. Increased arterial stiffness in nonalcoholic fatty liver disease: The Cardio-GOOSE study. J. Hypertens. 2010, 28, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, A.; Motoyama, M.; Matsuda, T.; Miyanishi, T. Brachial-ankle Pulse Wave Velocity in Japanese University Students. Intern. Med. 2005, 44, 696–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motamed, N.; Rabiee, B.; Poustchi, H.; Dehestani, B.; Hemasi, G.R.; Khonsari, M.R.; Maadi, M.; Saeedian, F.S.; Zamani, F. Non-alcoholic fatty liver disease (NAFLD) and 10-year risk of cardiovascular diseases. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ampuero, J.; Gallego-Duran, R.; Romero-Gomez, M. Association of NAFLD with subclinical atherosclerosis and coronary-artery disease: Meta-analysis. Rev. Esp. Enferm. Dig. 2015, 107, 10–16. [Google Scholar] [PubMed]
- Ekstedt, M.; Franzén, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, N.; Preiss, D.; Sattar, N. Liver enzymes, nonalcoholic fatty liver disease, and incident cardiovascular disease: A narrative review and clinical perspective of prospective data. Hepatology 2010, 52, 1156–1161. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Coupland, C.; Robson, J.; Brindle, P. Derivation, validation, and evaluation of a new QRISK model to estimate lifetime risk of cardiovascular disease: Cohort study using QResearch database. BMJ 2010, 341, c6624. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.; Loomis, A.K.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Non-alcoholic fatty liver disease and risk of incident acute myocardial infarction and stroke: Findings from matched cohort study of 18 million European adults. BMJ 2019, 367, l5367. [Google Scholar] [CrossRef] [Green Version]
- Sattar, N.; Forrest, E.; Preiss, D. Non-alcoholic fatty liver disease. BMJ 2014, 349, g4596. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Forlani, G.; Cerrelli, F.; Manini, R.; Natale, S.; Baraldi, L.; Ermini, G.; Savorani, G.; Zocchi, D.; Melchionda, N. WHO and ATPIII proposals for the definition of the metabolic syndrome in patients with Type 2 diabetes. Diabetes Med. 2004, 21, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Jung, H.S.; Cho, J.; Zhang, Y.; Yun, K.E.; Lazo, M.; Pastor-Barriuso, R.; Ahn, J.; Kim, C.W.; Rampal, S.; et al. Metabolically Healthy Obesity and the Development of Nonalcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2016, 111, 1133–1140. [Google Scholar] [CrossRef]
- Chen, F.; Esmaili, S.; Rogers, G.B.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A Distinct Entity Shaped by Differential Metabolic Adaptation. Hepatology 2020, 71, 1213–1227. [Google Scholar] [CrossRef]
- Iacobini, C.; Pugliese, G.; Blasetti Fantauzzi, C.; Federici, M.; Menini, S. Metabolically healthy versus metabolically unhealthy obesity. Metabolism 2019, 92, 51–60. [Google Scholar] [CrossRef]
- Younes, R.; Bugianesi, E. NASH in Lean Individuals. Semin. Liver Dis. 2019, 39, 086–095. [Google Scholar] [CrossRef] [Green Version]
- Gentile, C.L.; Weir, T.L.; Cox-York, K.A.; Wei, Y.; Wang, D.; Reese, L.; Moran, G.; Estrada, A.; Mulligan, C.; Pagliassotti, M.J.; et al. The role of visceral and subcutaneous adipose tissue fatty acid composition in liver pathophysiology associated with NAFLD. Adipocyte 2015, 4, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Graffy, P.M.; Pickhardt, P.J. Quantification of hepatic and visceral fat by CT and MR imaging: Relevance to the obesity epidemic, metabolic syndrome and NAFLD. Br. J. Radiol. 2016, 89, 20151024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.; Tariq, R.; Provenza, J.; Satapathy, S.K.; Faisal, K.; Choudhry, A.; Friedman, S.L.; Singal, A.K. Prevalence and Profile of Nonalcoholic Fatty Liver Disease in Lean Adults: Systematic Review and Meta-Analysis. Hepatol. Commun. 2020, 4, 953–972. [Google Scholar] [CrossRef] [PubMed]
- Golabi, P.; Paik, J.; Fukui, N.; Locklear, C.T.; de Avilla, L.; Younossi, Z.M. Patients with Lean Nonalcoholic Fatty Liver Disease Are Metabolically Abnormal and Have a Higher Risk for Mortality. Clin. Diabetes 2018, 37, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stols-Gonçalves, D.; Hovingh, G.K.; Nieuwdorp, M.; Holleboom, A.G. NAFLD and Atherosclerosis: Two Sides of the Same Dysmetabolic Coin? Trends Endocrinol. Metab. 2019, 30, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.P.; Rawal, K.; Bagchi, A.K.; Akolkar, G.; Bernardes, N.; Dias, D.D.S.; Gupta, S.; Singal, P.K. Insulin resistance: An additional risk factor in the pathogenesis of cardiovascular disease in type 2 diabetes. Heart Fail. Rev. 2016, 21, 11–23. [Google Scholar] [CrossRef]
- Balakumar, P.; Maung-U, K.; Jagadeesh, G. Prevalence and prevention of cardiovascular disease and diabetes mellitus. Pharmacol. Res. 2016, 113, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.-W.; Lin, C.-L.; Liao, K.-F. Association between diabetes mellitus and hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2019, 31, 898–899. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Lauridsen, B.K.; Stender, S.; Kristensen, T.S.; Kofoed, K.F.; Køber, L.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Liver fat content, non-alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta-analysis of 279,013 individuals. Eur. Heart J. 2018, 39, 385–393. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Mantovani, A.; Tilg, H.; Targher, G. Risk of cardiomyopathy and cardiac arrhythmias in patients with nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 425–439. [Google Scholar] [CrossRef]
- Mantovani, A.; Dauriz, M.; Sandri, D.; Bonapace, S.; Zoppini, G.; Tilg, H.; Byrne, C.D.; Targher, G. Association between non-alcoholic fatty liver disease and risk of atrial fibrillation in adult individuals: An updated meta-analysis. Liver Int. 2019, 39, 758–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M. Atrial Fibrillation and Heart Failure with Preserved Ejection Fraction in Patients with Nonalcoholic Fatty Liver Disease. Am. J. Med. 2020, 133, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Cohney, S.; De Michieli, F.; Pinach, S.; Saba, F.; Gambino, R. Fatty Liver and Chronic Kidney Disease: Novel Mechanistic Insights and Therapeutic Opportunities. Diabetes Care 2016, 39, 1830–1845. [Google Scholar] [CrossRef] [Green Version]
- Targher, G.; Chonchol, M.; Byrne, C.D. CKD and Nonalcoholic Fatty Liver Disease. Am. J. Kidney Dis. 2014, 64, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-Reactive Protein and Other Markers of Inflammation in the Prediction of Cardiovascular Disease in Women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef]
- Thorand, B.; Löwel, H.; Schneider, A.; Kolb, H.; Meisinger, C.; Fröhlich, M.; Koenig, W. C-reactive protein as a predictor for incident diabetes mellitus among middle-aged men: Results from the MONICA Augsburg cohort study, 1984–1998. Arch. Intern. Med. 2003, 163, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Kubes, P.; Mehal, W.Z. Sterile Inflammation in the Liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Tosello-Trampont, A.C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer Cells Trigger Nonalcoholic Steatohepatitis Development in Diet-induced Mouse Model through Tumor Necrosis Factor-α Production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Macrophages in Nonalcoholic Fatty Liver Disease: A Role Model of Pathogenic Immunometabolism. Semin. Liver Dis. 2017, 37, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, K.; McKenzie, A.L.; Kränkel, N.; Von Schacky, C.; Worm, N.; Nixdorff, U.; Lechner, B.; Scherr, J.; Weingärtner, O.; Krauss, R.M. High-Risk Atherosclerosis and Metabolic Phenotype: The Roles of Ectopic Adiposity, Atherogenic Dyslipidemia, and Inflammation. Metab. Syndr. Relat. Disord. 2020, 18, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef] [Green Version]
- Hirase, T.; Node, K. Endothelial dysfunction as a cellular mechanism for vascular failure. Am. J. Physiol. Circ. Physiol. 2012, 302, H499–H505. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Guo, S.; Zhang, S.; Liu, A.; Shi, L.; Zhang, Y. Matrine attenuates endoplasmic reticulum stress and mitochondrion dysfunction in nonalcoholic fatty liver disease by regulating SERCA pathway. J. Transl. Med. 2018, 16, 319. [Google Scholar] [CrossRef]
- O’Rourke, R.W.; Metcalf, M.D.; White, A.E.; Madala, A.; Winters, B.R.; Maizlin, I.I.; Jobe, B.A.; Roberts, C.T., Jr.; Slifka, M.K.; Marks, D.L. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int. J. Obes. 2009, 33, 978–990. [Google Scholar]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Tourniaire, F.; Romier-Crouzet, B.; Lee, J.H.; Marcotorchino, J.; Gouranton, E.; Salles, J.; Malezet, C.; Astier, J.; Darmon, P.; Blouin, E.; et al. Chemokine Expression in Inflamed Adipose Tissue is Mainly Mediated by NF-κB. PLoS ONE 2013, 8, e66515. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar] [PubMed]
- Lagathu, C.; Yvan-Charvet, L.; Bastard, J.P.; Maachi, M.; Quignard-Boulangé, A.; Capeau, J.; Caron, M. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia 2006, 49, 2162–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, J.; Kiefer, F.W.; Zeyda, M.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; Zlabinger, G.J.; Stulnig, T.M. CC Chemokine and CC Chemokine Receptor Profiles in Visceral and Subcutaneous Adipose Tissue Are Altered in Human Obesity. J. Clin. Endocrinol. Metab. 2008, 93, 3215–3221. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Du Plessis, J.; van Pelt, J.; Korf, H.; Mathieu, C.; van der Schueren, B.; Lannoo, M.; Oyen, T.; Topal, B.; Fetter, G.; Nayler, S.; et al. Association of Adipose Tissue Inflammation with Histologic Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 635–648.e14. [Google Scholar] [CrossRef] [Green Version]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in NAFLD—Revisited After a Decade. Hepatology 2020. [Google Scholar] [CrossRef]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Fricker, Z.P.; Pedley, A.; Massaro, J.M.; Vasan, R.S.; Hoffmann, U.; Benjamin, E.J.; Long, M.T. Liver Fat is Associated with Markers of Inflammation and Oxidative Stress in Analysis of Data From the Framingham Heart Study. Clin. Gastroenterol. Hepatol. 2019, 17, 1157–1164.e4. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, M.; Simons, N.; Stehouwer, C.D.A.; Isaacs, A. Non-alcoholic fatty liver disease and cardiovascular disease: Assessing the evidence for causality. Diabetology 2020, 63, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismaiel, A.; Dumitraşcu, D.L. Cardiovascular Risk in Fatty Liver Disease: The Liver-Heart Axis—Literature Review. Front. Med. 2019, 6, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouvas, N.; Kontogiannis, C.; Georgiopoulos, G.; Spartalis, M.; Tsilimigras, D.I.; Spartalis, E.; Kapelouzou, A.; Kosmopoulos, M.; Chatzidou, S. The complex crosstalk between inflammatory cytokines and ventricular arrhythmias. Cytokine 2018, 111, 171–177. [Google Scholar] [CrossRef]
- Widjaja, A.A.; Singh, B.K.; Adami, E.; Viswanathan, S.; Dong, J.; D’Agostino, G.A.; Ng, B.; Lim, W.; Tan, J.; Paleja, B.S.; et al. Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology 2019, 157, 777–792.e14. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Eslam, M.; Sarin, S.K.; Wong, V.W.-S.; Fan, J.-G.; Kawaguchi, T.; Ahn, S.H.; Zheng, M.-H.; Shiha, G.; Yilmaz, Y.; Gani, R.; et al. The Asian Pacific Association for the Study of the Liver clinical practice guidelines for the diagnosis and management of metabolic associated fatty liver disease. Hepatol. Int. 2020, 14, 1–31. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Shiha, G.; Alswat, K.; Al Khatry, M.; I Sharara, A.; Örmeci, N.; Waked, I.; Benazzouz, M.; Al-Ali, F.; Hamed, A.E.; Hamoudi, W.; et al. Nomenclature and definition of metabolic-associated fatty liver disease: A consensus from the Middle East and north Africa. Lancet Gastroenterol. Hepatol. 2021, 6, 57–64. [Google Scholar] [CrossRef]
- Shiha, G.; Korenjak, M.; Eskridge, W.; Casanovas, T.; Velez-Moller, P.; Högström, S.; Richardson, B.; Munoz, C.; Sigurðardóttir, S.; Coulibaly, A.; et al. Redefining fatty liver disease: An international patient perspective. Lancet Gastroenterol. Hepatol. 2021, 6, 73–79. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Arrese, M.; Gadano, A.; Oliveira, C.P.; Fassio, E.; Arab, J.P.; Chávez-Tapia, N.C.; Dirchwolf, M.; Torre, A.; Ridruejo, E.; et al. The Latin American Association for the Study of the Liver (ALEH) position statement on the redefinition of fatty liver disease. Lancet Gastroenterol. Hepatol. 2021, 6, 65–72. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.; Harrison, S.A.; Brunt, E.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a premature change in terminology. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. 2020, 40, 2082–2089. [Google Scholar] [CrossRef]
- Targher, G. Concordance of MAFLD and NAFLD diagnostic criteria in “real-world” data. Liver. Int. 2020, 40, 2879–2880. [Google Scholar] [CrossRef]
- Huang, J.; Kumar, R.; Wang, M.; Zhu, Y.; Lin, S. MAFLD criteria overlooks a number of patients with severe steatosis: Is it clinically relevant? J. Hepatol. 2020, 73, 1265–1267. [Google Scholar] [CrossRef]
- Zhang, L.; She, Z.-G.; Li, H.-L.; Zhang, X.-J. Non-alcoholic fatty liver disease: A metabolic burden promoting atherosclerosis. Clin. Sci. 2020, 134, 1775–1799. [Google Scholar] [CrossRef]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef]
- Hasan, N.; Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rodriguez, E.; Egea-Zorrilla, A.; Plaza-Díaz, J.; Jerónimo, A.-V.; Muñoz-Quezada, S.; Tercedor-Sánchez, L.; Abadía-Molina, F. The Gut Microbiota and Its Implication in the Development of Atherosclerosis and Related Cardiovascular Diseases. Nutrition 2020, 12, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.W.; Bäckhed, F.; Landmesser, U.; Hazen, S.L. Intestinal Microbiota in Cardiovascular Health and Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2089–2105. [Google Scholar] [CrossRef] [PubMed]
- Indias, I.E.; Queipo-Ortuño, M.I.; Tinahones, F.J.; Queipo-Ortuno, M.I. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front. Microbiol. 2014, 5, 190. [Google Scholar]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [Green Version]
- Henao-Mejia, J.; Elinav, E.; Jin, C.-C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nat. Cell Biol. 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.; Rivera, L.; Furness, L.R.J.B.; Angus, C.L.P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Clément, K. The gut microbiome, diet, and links to cardiometabolic and chronic disorders. Nat. Rev. Nephrol. 2015, 12, 169–181. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernández-Real, J.M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Sharpton, S.R.; Ajmera, V.; Loomba, R. Emerging Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease: From Composition to Function. Clin. Gastroenterol. Hepatol. 2019, 17, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.G.; Kim, S.M.; Caussy, C.; Fu, T.; Guo, J.; Bassirian, S.; Singh, S.; Madamba, E.V.; Bettencourt, R.; Richards, L.; et al. A Universal Gut-Microbiome-Derived Signature Predicts Cirrhosis. Cell. Metab. 2020, 32, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, H.E.; Teterina, A.; Comelli, E.M.; Taibi, A.; Arendt, B.M.; Fischer, S.E.; Lou, W.; Allard, J.P. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Lang, S.; Demir, M.; Martin, A.; Jiang, L.; Zhang, X.; Duan, Y.; Gao, B.; Wisplinghoff, H.; Kasper, P.; Roderburg, C.; et al. Intestinal Virome Signature Associated with Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 159, 1839–1852. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J. Endocr. Soc. 2020, 4, bvz039. [Google Scholar] [CrossRef] [Green Version]
- Verdugo, J.P.A.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 321–350. [Google Scholar]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Arslan, N. Obesity, fatty liver disease and intestinal microbiota. World J. Gastroenterol. 2014, 20, 16452–16463. [Google Scholar] [CrossRef]
- Turnbaugh, P.J. Microbiology: Fat, bile and gut microbes. Nature 2012, 487, 47–48. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Zhou, X.; Wang, M.; Zhou, L.; Wang, Z.; Wang, M.; Deng, J.; Wang, Y.; Zhou, Z.; Zhang, Y.; et al. Gut microbe-derived metabolite trimethylamine N-oxide activates the cardiac autonomic nervous system and facilitates ischemia-induced ventricular arrhythmia via two different pathways. EBioMedicine 2019, 44, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-M.; Liu, Y.; Zhou, R.-F.; Chen, X.-L.; Wang, C.; Tan, X.-Y.; Wang, L.-J.; Zheng, R.-D.; Zhang, H.-W.; Ling, W.-H.; et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 2016, 6, 19076. [Google Scholar] [CrossRef]
- Tan, X.; Liu, Y.; Long, J.; Chen, S.; Liao, G.; Wu, S.; Li, C.; Wang, L.; Ling, W.; Zhu, H. Trimethylamine N-Oxide Aggravates Liver Steatosis through Modulation of Bile Acid Metabolism and Inhibition of Farnesoid X Receptor Signaling in Nonalcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900257. [Google Scholar] [CrossRef]
- Arias, N.; Arboleya, S.; Allison, J.; Kaliszewska, A.; Higarza, S.G.; Gueimonde, M.; Arias, J.L. The Relationship between Choline Bioavailability from Diet, Intestinal Microbiota Composition, and Its Modulation of Human Diseases. Nutrition 2020, 12, 2340. [Google Scholar]
- Brandsma, E.; Kloosterhuis, N.J.; Koster, M.; Dekker, D.C.; Gijbels, M.J.; Van Der Velden, S.; Ríos-Morales, M.; Van Faassen, M.J.; Loreti, M.G.; De Bruin, A.; et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ. Res. 2019, 124, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Vieira-Silva, S.; Falony, G.; Belda, E.; Nielsen, T.; Aron-Wisnewsky, J.; Chakaroun, R.; Forslund, S.K.; Assmann, K.; Valles-Colomer, M.; Nguyen, T.T.D.; et al. Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nat. Cell Biol. 2020, 581, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Wang, X.; Ren, X.; Liu, Y. Changes of intestinal bacterial microbiota in coronary heart disease complicated with nonalcoholic fatty liver disease. BMC Genom. 2019, 20, 862. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halcox, J.P.J.; Banegas, J.R.; Roy, C.; Dallongeville, J.; De Backer, G.G.; Guallar, E.; Perk, J.; Hajage, D.; Henriksson, K.M.; Borghi, C. Prevalence and treatment of atherogenic dyslipidemia in the primary prevention of cardiovascular disease in Europe: EURIKA, a cross-sectional observational study. BMC Cardiovasc. Disord. 2017, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.G. Hepatic glucose and lipid metabolism. Diabetologia 2016, 59, 1098–1103. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725.e6. [Google Scholar] [CrossRef]
- Patterson, R.E.; Kalavalapalli, S.; Williams, C.M.; Nautiyal, M.; Mathew, J.T.; Martinez, J.; Reinhard, M.K.; McDougall, D.J.; Rocca, J.R.; Yost, R.A.; et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E484–E494. [Google Scholar] [CrossRef] [Green Version]
- Chatrath, H.; Vuppalanchi, R.; Chalasani, N. Dyslipidemia in Patients with Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2012, 32, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.W.; Yu, J.H.; Jin, Y.J.; Suh, Y.J.; Lee, J.W. Correlation between the small dense LDL level and nonalcoholic fatty liver disease: Possibility of a new biomarker. Medicine 2020, 99, e21162. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Fuchs, M.; Idowu, M.O.; Luketic, V.A.; Boyett, S.; Sargeant, C.; Stravitz, R.T.; Puri, P.; Matherly, S.; Sterling, R.K.; et al. Severity of Nonalcoholic Fatty Liver Disease and Progression to Cirrhosis Are Associated with Atherogenic Lipoprotein Profile. Clin. Gastroenterol. Hepatol. 2015, 13, 1000–1008.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imajo, K.; Hyogo, H.; Yoneda, M.; Honda, Y.; Kessoku, T.; Tomeno, W.; Ogawa, Y.; Taguri, M.; Mawatari, H.; Nozaki, Y.; et al. LDL-Migration Index (LDL-MI), an Indicator of Small Dense Low-Density Lipoprotein (sdLDL), Is Higher in Non-Alcoholic Steatohepatitis than in Non-Alcoholic Fatty Liver: A Multicenter Cross-Sectional Study. PLoS ONE 2014, 9, e115403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, M.; Berneis, K.; Corrado, E.; Novo, S. The significance of low-density-lipoproteins size in vascular diseases. Int. Angiol. 2006, 25, 4–9. [Google Scholar] [PubMed]
- Saeed, A.; Feofanova, E.V.; Yu, B.; Sun, W.; Virani, S.S.; Nambi, V.; Coresh, J.; Guild, C.S.; Boerwinkle, E.; Ballantyne, C.M.; et al. Remnant-Like Particle Cholesterol, Low-Density Lipoprotein Triglycerides, and Incident Cardiovascular Disease. J. Am. Coll. Cardiol. 2018, 72, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Dissociation between APOC3 variants, hepatic triglyceride content and insulin resistance. Hepatology 2010, 53, 467–474. [Google Scholar] [CrossRef]
- Dittrich, J.; Beutner, F.; Teren, A.; Thiery, J.; Burkhardt, R.; Scholz, M.; Ceglarek, U. Plasma levels of apolipoproteins C-III, A-IV, and E are independently associated with stable atherosclerotic cardiovascular disease. Atherosclerosis 2019, 281, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Zewinger, S.; Reiser, J.; Jankowski, V.; Al Ansary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 2019, 21, 30–41. [Google Scholar] [CrossRef]
- Afonso, M.S.; Lottenberg, A.M.; Koike, M.K.; Cintra, D.E.; Ferreira, F.D.; Nunes, V.S.; Castilho, G.; Gioielli, L.A.; Bombo, R.P.; Catanozi, S.; et al. Dietary interesterified fat enriched with palmitic acid induces atherosclerosis by impairing macrophage cholesterol efflux and eliciting inflammation. J. Nutr. Biochem. 2016, 32, 91–100. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F.; Zhu, S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, S.; Panera, N.; Mina, M.; Gnani, D.; De Stefanis, C.; Crudele, A.; Rychlicki, C.; Petrini, S.; Bruscalupi, G.; Agostinelli, L.; et al. LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 2015, 6, 41434–41452. [Google Scholar] [CrossRef] [Green Version]
- Verrijken, A.; Francque, S.; Mertens, I.; Prawitt, J.; Caron, S.; Hubens, G.; Van Marck, E.; Staels, B.; Michielsen, P.; Van Gaal, L. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2014, 59, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.N.W.; Treskes, P.; Martin, S.F.; Manning, J.R.; Dunbar, D.R.; Rogers, S.M.; Le Bihan, T.; Lockman, K.A.; Morley, S.D.; Hayes, P.C.; et al. Fibrinogen production is enhanced in an in-vitro model of non-alcoholic fatty liver disease: An isolated risk factor for cardiovascular events? Lipids Heal. Dis. 2015, 14, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.-J.; Wang, P.W.; Hu, T.H. Association of Adiponectin Gene Polymorphism with Nonalcoholic Fatty Liver Disease in Taiwanese Patients with Type 2 Diabetes. PLoS ONE 2015, 10, e0127521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-L.; Sui, J.-Q.; Lu, L.-L.; Zhang, N.-N.; Xu, X.; Dong, Q.-Y.; Xin, Y.; Xuan, S. Gene polymorphisms associated with non-alcoholic fatty liver disease and coronary artery disease: A concise review. Lipids Health Dis. 2016, 15, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, E.P.; Dhindsa, D.S.; Lee, S.K.; Sandesara, P.B.; Chalasani, N.P.; Sperling, L.S. Nonalcoholic Fatty Liver Disease and the Heart: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 948–963. [Google Scholar] [CrossRef]
- Francque, S.M.; van der Graaff, D.; Kwanten, W.J. Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications. J. Hepatol. 2016, 65, 425–443. [Google Scholar] [CrossRef] [Green Version]
- Villanova, N.; Moscatiello, S.; Ramilli, S.; Bugianesi, E.; Magalotti, D.; Vanni, E.; Zoli, M.; Marchesini, G. Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology 2005, 42, 473–480. [Google Scholar] [CrossRef]
- Hawksworth, D.J.; Burnett, A.L. Nonalcoholic Fatty Liver Disease, Male Sexual Dysfunction, and Infertility: Common Links, Common Problems. Sex. Med. Rev. 2020, 8, 274–285. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Jung, H.S.; Chang, Y.; Kwon, M.J.; Sung, E.; Yun, K.E.; Cho, Y.K.; Shin, H.; Ryu, S. Smoking and the Risk of Non-Alcoholic Fatty Liver Disease: A Cohort Study. Am. J. Gastroenterol. 2019, 114, 453–463. [Google Scholar] [CrossRef]
- Boden, G. High- or Low-Carbohydrate Diets: Which is better for Weight Loss, Insulin Resistance, and Fatty Livers? Gastroenterology 2009, 136, 1490–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perreault, L.; Pan, Q.; Mather, K.J.; Watson, K.E.; Hamman, R.F.; Kahn, S.E. Effect of regression from prediabetes to normal glucose regulation on long-term reduction in diabetes risk: Results from the Diabetes Prevention Program Outcomes Study. Lancet 2012, 379, 2243–2251. [Google Scholar] [CrossRef] [Green Version]
- Lazo, M.; Solga, S.F.; Horska, A.; Bonekamp, S.; Diehl, A.M.; Brancati, F.L.; Wagenknecht, L.E.; Pi-Sunyer, F.X.; Kahn, S.E.; Clark, J.M.; et al. Effect of a 12-Month Intensive Lifestyle Intervention on Hepatic Steatosis in Adults with Type 2 Diabetes. Diabetes Care 2010, 33, 2156–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera, F.; George, J. The Role of Diet and Nutritional Intervention for the Management of Patients with NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef]
- Dunn, W.; Sanyal, A.J.; Brunt, E.M.; Unalp-Arida, A.; Donohue, M.; McCullough, A.J.; Schwimmer, J.B. Modest alcohol consumption is associated with decreased prevalence of steatohepatitis in patients with non-alcoholic fatty liver disease (NAFLD). J. Hepatol. 2012, 57, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.K.; Greenson, J.K.; Conjeevaram, H.S. Effect of lifetime alcohol consumption on the histological severity of non-alcoholic fatty liver disease. Liver Int. 2013, 34, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Liangpunsakul, S.; Chalasani, N. What Should We Recommend to Our Patients with NAFLD Regarding Alcohol Use? Am. J. Gastroenterol. 2012, 107, 976–978. [Google Scholar] [CrossRef] [Green Version]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.-R.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Saab, S.; Mallam, D.; Cox, G.A.; Tong, M.J. Impact of coffee on liver diseases: A systematic review. Liver Int. 2014, 34, 495–504. [Google Scholar] [CrossRef]
- Pelliccia, A.; Sharma, S.; Gati, S.; Bäck, M.; Börjesson, M.; Caselli, S.; Collet, J.-P.; Corrado, D.; Drezner, J.A.; Halle, M. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease: The Task Force on sports cardiology and exercise in patients with cardiovascular disease of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 42, 17–96. [Google Scholar]
- Keating, S.E.; Hackett, D.A.; George, J.; Johnson, N.A. Exercise and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 57, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, E.; Negri, C.; Targher, G.; Faccioli, N.; Lanza, M.; Zoppini, G.; Zanolin, E.; Schena, F.; Bonora, E.; Moghetti, P. Both resistance training and aerobic training reduce hepatic fat content in type 2 diabetic subjects with nonalcoholic fatty liver disease (the RAED2 Randomized Trial). Hepatology 2013, 58, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Roman, J.H.; Patel, S.S. Why Do Lifestyle Recommendations Fail in Most Patients with Nonalcoholic Fatty Liver Disease? Gastroenterol. Clin. N. Am. 2020, 49, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Ismaiel, A.; Dumitrascu, D.L. How to Reduce Cardiovascular Risk in Nonalcoholic Fatty Liver Disease. Am. J. Ther. 2020. [Google Scholar] [CrossRef]
- Jiang, Z.G.; Feldbrügge, L.; Tapper, E.B.; Popov, Y.; Ghaziani, T.; Afdhal, N.; Robson, S.C.; Mukamal, K.J. Aspirin use is associated with lower indices of liver fibrosis among adults in the United States. Aliment. Pharmacol. Ther. 2016, 43, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Henson, J.; Osganian, S.; Masia, R.; Chan, A.T.; Chung, R.T.; Corey, K.E. Daily Aspirin Use Associated with Reduced Risk For Fibrosis Progression in Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 2776–2784.e4. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Nozaki, Y.; Wada, K.; Yoneda, M.; Endo, H.; Takahashi, H.; Iwasaki, T.; Inamori, M.; Abe, Y.; Kobayashi, N.; et al. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut 2008, 57, 1583–1591. [Google Scholar] [CrossRef]
- Russo, M.W.; Pierson, J.; Narang, T.; Montegudo, A.; Eskind, L.; Gulati, S. Coronary Artery Stents and Antiplatelet Therapy in Patients with Cirrhosis. J. Clin. Gastroenterol. 2012, 46, 339–344. [Google Scholar] [CrossRef]
- Li, C.-J.; Yang, Z.-H.; Shi, X.-L.; Liu, D.-L. Effects of aspirin and enoxaparin in a rat model of liver fibrosis. World J. Gastroenterol. 2017, 23, 6412–6419. [Google Scholar] [CrossRef]
- Leonardi, F.; Maria, N.; Villa, E. Anticoagulation in cirrhosis: A new paradigm? Clin. Mol. Hepatol. 2017, 23, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Cholesterol Treatment Trialists. Efficacy and safety of LDL-lowering therapy among men and women: Meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015, 385, 1397–1405. [Google Scholar] [CrossRef]
- Björnsson, E.S.; Jacobsen, E.I.; Kalaitzakis, E. Hepatotoxicity associated with statins: Reports of idiosyncratic liver injury post-marketing. J. Hepatol. 2012, 56, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Labenz, C.; Huber, Y.; Kalliga, E.; Nagel, M.; Ruckes, C.; Straub, B.K.; Galle, P.R.; Wörns, M.-A.; Anstee, Q.M.; Schuppan, D.; et al. Predictors of advanced fibrosis in non-cirrhotic non-alcoholic fatty liver disease in Germany. Aliment. Pharmacol. Ther. 2018, 48, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Labenz, C.; Prochaska, J.H.; Huber, Y.; Nagel, M.; Straub, B.K.; Wild, P.; Galle, P.R.; Schattenberg, J.M. Cardiovascular Risk Categories in Patients with Nonalcoholic Fatty Liver Disease and the Role of Low-Density Lipoprotein Cholesterol. Hepatol. Commun. 2019, 3, 1472–1481. [Google Scholar] [CrossRef] [Green Version]
- Athyros, V.G.; Tziomalos, K.; Gossios, T.D.; Griva, T.; Anagnostis, P.; Kargiotis, K.; Pagourelias, E.D.; Theocharidou, E.; Karagiannis, A.; Mikhailidis, D.P. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: A post-hoc analysis. Lancet 2010, 376, 1916–1922. [Google Scholar] [CrossRef]
- Athyros, V.G.; Kakafika, A.I.; Papageorgiou, A.A.; Tziomalos, K.; Skaperdas, A.; Pagourelias, E.; Pirpasopoulou, A.; Karagiannis, A.; Mikhailidis, D.P. Atorvastatin decreases triacylglycerol-associated risk of vascular events in coronary heart disease patients. Lipids 2007, 42, 999–1009. [Google Scholar] [CrossRef]
- Tikkanen, M.J.; Fayyad, R.; Faergeman, O.; Olsson, A.G.; Wun, C.-C.; Laskey, R.; Kastelein, J.J.; Holme, I.; Pedersen, T.R. Effect of intensive lipid lowering with atorvastatin on cardiovascular outcomes in coronary heart disease patients with mild-to-moderate baseline elevations in alanine aminotransferase levels. Int. J. Cardiol. 2013, 168, 3846–3852. [Google Scholar] [CrossRef]
- Kargiotis, K.; Athyros, V.G.; Giouleme, O.; Katsiki, N.; Katsiki, E.; Anagnostis, P.; Boutari, C.; Doumas, M.; Karagiannis, A.; Mikhailidis, D.P. Resolution of non-alcoholic steatohepatitis by rosuvastatin monotherapy in patients with metabolic syndrome. World J. Gastroenterol. 2015, 21, 7860–7868. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Mannisto, V.; Mancina, R.M.; Pipitone, R.M.; Karja, V.; Maggioni, M.; Kakela, P.; Wiklund, O.; Mozzi, E.; et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J. Hepatol. 2015, 63, 705–712. [Google Scholar] [CrossRef]
- Averna, M. The effect of ezetimibe on NAFLD. Atheroscler. Suppl. 2015, 17, 27–34. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and Safety of Evolocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, D.M.; Morris, P.B.; Ballantyne, C.M.; Birtcher, K.K.; Daly, D.D., Jr.; DePalma, S.M.; Minissian, M.B.; Orringer, C.E.; Smith, S.C., Jr. 2016 ACC Expert Consensus Decision Pathway on the Role of Non-Statin Therapies for LDL-Cholesterol Lowering in the Management of Atherosclerotic Cardiovascular Disease Risk: A Report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents. J. Am. Coll. Cardiol. 2016, 68, 92–125. [Google Scholar] [PubMed]
- Theocharidou, E.; Papademetriou, M.; Reklou, A.; Sachinidis, A.; Boutari, C.; Giouleme, O. The Role of PCSK9 in the Pathogenesis of Non-alcoholic Fatty Liver Disease and the Effect of PCSK9 Inhibitors. Curr. Pharm. Des. 2019, 24, 3654–3657. [Google Scholar] [CrossRef]
- Cameron, J.; Ranheim, T.; Kulseth, M.A.; Leren, T.P.; Berge, K.E. Berberine decreases PCSK9 expression in HepG2 cells. Atheroscler. 2008, 201, 266–273. [Google Scholar] [CrossRef]
- Yan, H.-M.; Xia, M.-F.; Wang, Y.; Chang, X.-X.; Yao, X.-Z.; Rao, S.-X.; Zeng, M.-S.; Tu, Y.-F.; Feng, R.; Jia, W.-P.; et al. Efficacy of Berberine in Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0134172. [Google Scholar] [CrossRef] [Green Version]
- Aneni, E.C.; Oni, E.T.; Martin, S.S.; Blaha, M.J.; Agatston, A.S.; Feldman, T.; Veledar, E.; Conçeicao, R.D.; Carvalho, J.A.; Santos, R.D.; et al. Blood pressure is associated with the presence and severity of nonalcoholic fatty liver disease across the spectrum of cardiometabolic risk. J. Hypertens. 2015, 33, 1207–1214. [Google Scholar] [CrossRef]
- Ryoo, J.-H.; Suh, Y.J.; Shin, H.C.; Cho, Y.K.; Choi, J.-M.; Park, S.K. Clinical association between non-alcoholic fatty liver disease and the development of hypertension. J. Gastroenterol. Hepatol. 2014, 29, 1926–1931. [Google Scholar] [CrossRef]
- Vasunta, R.-L.; Kesäniemi, Y.A.; Ylitalo, A.S.; Ukkola, O. High ambulatory blood pressure values associated with non-alcoholic fatty liver in middle-aged adults. J. Hypertens. 2012, 30, 2015–2019. [Google Scholar] [CrossRef]
- Muthiah, M.D.; Sanyal, A.J. Current management of non-alcoholic steatohepatitis. Liver Int. 2020, 40, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisseleva, T.; A Brenner, D. Anti-fibrogenic strategies and the regression of fibrosis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 305–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, H.; Wu, W.; Ye, J.; Fang, D.; Shi, D.; Li, L. Clinical application of angiotensin receptor blockers in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. Oncotarget 2018, 9, 24155–24167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Jarvis, H.; Craig, D.; O Barker, R.; Spiers, G.; Stow, D.; Anstee, Q.M.; Hanratty, B. Metabolic risk factors and incident advanced liver disease in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of population-based observational studies. PLoS Med. 2020, 17, e1003100. [Google Scholar] [CrossRef] [PubMed]
- Schulte, L.; Scheiner, B.; Voigtländer, T.; Koch, S.; Schweitzer, N.; Marhenke, S.; Ivanyi, P.; Manns, M.P.; Rodt, T.; Hinrichs, J.B.; et al. Treatment with metformin is associated with a prolonged survival in patients with hepatocellular carcinoma. Liver Int. 2019, 39, 714–726. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern. Med. 2017, 177, 633–640. [Google Scholar] [CrossRef]
- Bril, F.; Kalavalapalli, S.; Clark, V.C.; Lomonaco, R.; Soldevila-Pico, C.; Liu, I.-C.; Orsak, B.; Tio, F.; Cusi, K. Response to Pioglitazone in Patients with Nonalcoholic Steatohepatitis With vs. Without Type 2 Diabetes. Clin. Gastroenterol. Hepatol. 2018, 16, 558–566.e2. [Google Scholar] [CrossRef]
- Ripamonti, E.; Azoulay, L.; Abrahamowicz, M.; Platt, R.W.; Suissa, S. A systematic review of observational studies of the association between pioglitazone use and bladder cancer. Diabet. Med. 2019, 36, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, J.A.; Guirguis, E.; Thornby, K.-A. A Systematic Review of Newer Antidiabetic Agents in the Treatment of Nonalcoholic Fatty Liver Disease. Ann. Pharmacother. 2021, 55, 65–79. [Google Scholar] [CrossRef]
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019, 42, S90–S102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernsmeier, C.; Meyer-Gerspach, A.C.; Blaser, L.S.; Jeker, L.; Steinert, R.E.; Heim, M.H.; Beglinger, C. Glucose-Induced Glucagon-Like Peptide 1 Secretion Is Deficient in Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2014, 9, e87488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Newsome, P.N.; Francque, S.; Harrison, S.; Ratziu, V.; Van Gaal, L.; Calanna, S.; Hansen, M.; Linder, M.; Sanyal, A.J. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment. Pharmacol. Ther. 2019, 50, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluud, L.L.; Knop, F.K.; Vilsbøll, T. Effects of lixisenatide on elevated liver transaminases: Systematic review with individual patient data meta-analysis of randomised controlled trials on patients with type 2 diabetes. BMJ Open 2014, 4, e005325. [Google Scholar] [CrossRef]
- Cusi, K.; Sattar, N.; García-Pérez, L.-E.; Pavo, I.; Yu, M.; Robertson, K.E.; Karanikas, C.A.; Haupt, A. Dulaglutide decreases plasma aminotransferases in people with Type 2 diabetes in a pattern consistent with liver fat reduction: A post hoc analysis of the AWARD programme. Diabet. Med. 2018, 35, 1434–1439. [Google Scholar] [CrossRef]
- Tacelli, M.; Celsa, C.; Magro, B.; Giannetti, A.; Pennisi, G.; Spatola, F.; Petta, S. Antidiabetic Drugs in NAFLD: The Accomplishment of Two Goals at Once? Pharmacy 2018, 11, 121. [Google Scholar] [CrossRef] [Green Version]
- Laursen, T.L.; Hagemann, C.A.; Wei, C.; Kazankov, K.; Thomsen, K.L.; Knop, F.K.; Grønbæk, H. Bariatric surgery in patients with non-alcoholic fatty liver disease—From pathophysiology to clinical effects. World J. Hepatol. 2019, 11, 138–149. [Google Scholar] [CrossRef]
Figure 1.
Substantial epidemiological data that link non-alcoholic fatty liver disease (NAFLD) to cardiovascular disease (CVD).
Figure 1.
Substantial epidemiological data that link non-alcoholic fatty liver disease (NAFLD) to cardiovascular disease (CVD).

Figure 2.
Several possible mechanisms have been proposed that might explain the association between non-alcoholic fatty liver disease (NAFLD) and cardiovascular disease (CVD). All of these possible mechanisms are associated with lifestyle factors.
Figure 2.
Several possible mechanisms have been proposed that might explain the association between non-alcoholic fatty liver disease (NAFLD) and cardiovascular disease (CVD). All of these possible mechanisms are associated with lifestyle factors.

Figure 3.
Besides lifestyle intervention and, for some patients, surgery, a number of established and emerging CV medications might play a role in the treatment of NAFLD complicated by CVD.
Figure 3.
Besides lifestyle intervention and, for some patients, surgery, a number of established and emerging CV medications might play a role in the treatment of NAFLD complicated by CVD.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Niederseer, D.; Wernly, B.; Aigner, E.; Stickel, F.; Datz, C. NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations. J. Clin. Med. 2021, 10, 467. https://doi.org/10.3390/jcm10030467
AMA Style
Niederseer D, Wernly B, Aigner E, Stickel F, Datz C. NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations. Journal of Clinical Medicine. 2021; 10(3):467. https://doi.org/10.3390/jcm10030467
Chicago/Turabian StyleNiederseer, David, Bernhard Wernly, Elmar Aigner, Felix Stickel, and Christian Datz. 2021. "NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations" Journal of Clinical Medicine 10, no. 3: 467. https://doi.org/10.3390/jcm10030467
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.