Preliminary Characterization of a Ni2+-Activated and Mycothiol-Dependent Glyoxalase I Enzyme from Streptomyces coelicolor



1
Department of Chemistry, University of Waterloo, 200 University Avenue West, Waterloo, ON N2L 3G1, Canada
2
Institute of Nutrition, Mahidol University, 25/25 Phutthamonthon 4 Rd., Salaya, Phutthamonthon, Nakhon Pathom 73170, Thailand
*
Author to whom correspondence should be addressed.
Inorganics 2019, 7(8), 99; https://doi.org/10.3390/inorganics7080099
Submission received: 6 July 2019
/
Revised: 1 August 2019
/
Accepted: 9 August 2019
/
Published: 14 August 2019
(This article belongs to the Special Issue Bioinorganic Chemistry of Nickel)
Abstract
:The glyoxalase system consists of two enzymes, glyoxalase I (Glo1) and glyoxalase II (Glo2), and converts a hemithioacetal substrate formed between a cytotoxic alpha-ketoaldehyde, such as methylglyoxal (MG), and an intracellular thiol, such as glutathione, to a non-toxic alpha-hydroxy acid, such as d-lactate, and the regenerated thiol. Two classes of Glo1 have been identified. The first is a Zn2+-activated class and is exemplified by the Homo sapiens Glo1. The second class is a Ni2+-activated enzyme and is exemplified by the Escherichia coli Glo1. Glutathione is the intracellular thiol employed by Glo1 from both these sources. However, many organisms employ other intracellular thiols. These include trypanothione, bacillithiol, and mycothiol. The trypanothione-dependent Glo1 from Leishmania major has been shown to be Ni2+-activated. Genetic studies on Bacillus subtilis and Corynebacterium glutamicum focused on MG resistance have indicated the likely existence of Glo1 enzymes employing bacillithiol or mycothiol respectively, although no protein characterizations have been reported. The current investigation provides a preliminary characterization of an isolated mycothiol-dependent Glo1 from Streptomyces coelicolor. The enzyme has been determined to display a Ni2+-activation profile and indicates that Ni2+-activated Glo1 are indeed widespread in nature regardless of the intracellular thiol employed by an organism.
1. Introduction
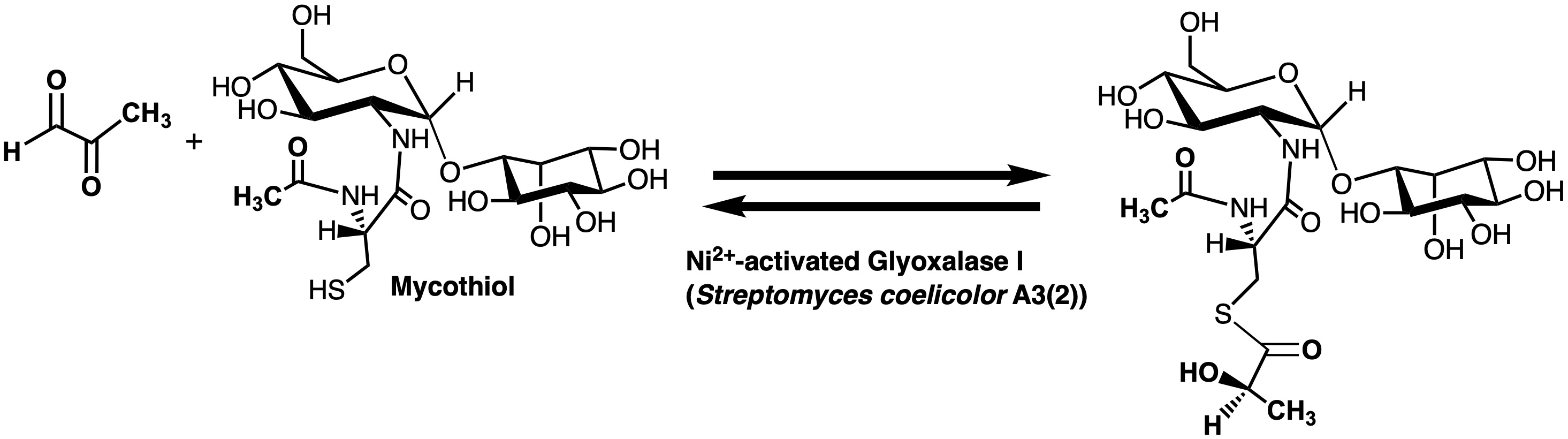
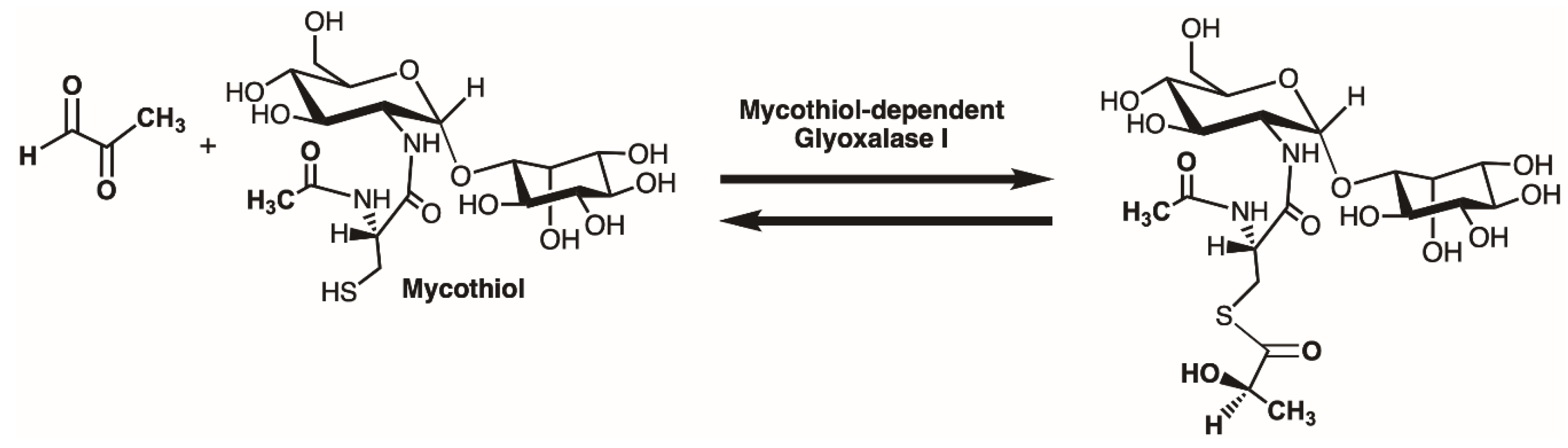
Intracellularly generated cytotoxic alpha-ketoaldehydes, such as methylglyoxal (MG; Figure 1), are highly reactive electrophiles that can inhibit protein synthesis, form adducts with protein, DNA and RNA, and promote Advanced Glycation End-products (AGEs) [1,2,3,4,5,6,7,8]. Detoxification of these molecules is a critical cellular process. The enzymes Glyoxalase I (Glo1) and Glyoxalase II (Glo2) work in tandem and convert the hemithioacetal, formed between the cytotoxic α-ketoaldehyde and an intracellular thiol, such as glutathione (GSH), into a non-toxic alpha-hydroxy acid, such as d-lactate (in the case of MG), and the regenerated thiol (Figure 1) [9,10,11,12,13]. Reduction of cellular toxicity is also dependent upon S-lactoylglutathione levels as this product of the Glo1 reaction can also control bacterial potassium efflux pumps and cytosolic acidification [14]. The metalloenzyme Glo1 catalyzes the isomerization reaction resulting in the production of the thioester product. Hydrolysis of the thioester is catalyzed by the second metalloenzyme, Glo2. An additional type of glyoxalase enzyme, Glo3, utilizes an active site cysteine to convert MG to d-lactate directly [15,16,17,18].
Glo1 is a metalloenzyme that can be divided into two different metal-activation classes [19]. The Zn2+-activated class is exemplified by the enzyme from Homo sapiens (homodimeric), and the Ni2+-activated Glo1 class is exemplified by the enzyme from Escherichia coli (homodimeric) [20,21,22,23,24,25]. In the case of the E. coli enzyme, the Ni2+-active form (Protein Database (PDB): 1F9Z) is situated in an octahedral ligand arrangement (residues from chain A: His5 and Glu56; residues from chain B: His74 and Glu122) with two water molecules as non-proteinaceous ligands, but the inactive Zn2+-bound complex (PDB: 1FA5) has a trigonal bipyramidal ligand arrangement and has only one water molecule as a non-proteinaceous ligand (Figure 2) [26,27]. A similar octahedral ligand arrangement (residues from chain A: Gln34 and Glu100; residues from chain B: His127 and Glu173) is observed for the catalytically active Zn2+-bound form of the H. sapiens Glo1 (PDBN: 1QIN) [28,29]. Thus, only octahedral metal environments appear to be active for this enzyme, regardless of the metal-activation class the Glo1 belongs to, and this arrangement appears critical to the enzyme’s catalytic mechanism [30,31,32].
Structural studies have been reported on several Glo1, and these reports have provided additional insight into nature’s control of active site geometry. As mentioned, the H. sapiens and E. coli enzymes are homodimeric, and each of their two active sites is formed by residues contributed by each of the two subunits. The molecular structure of the Clostridium acetobutylicum Glo1 (Ni2+-activation class) is also homodimeric, but each of the two active sites is formed by protein residues solely from a single subunit [34]. Furthermore, larger single chain Glo1 are also known [35,36,37,38,39,40,41], and the recent report on the X-ray structure of the maize enzyme shows two metal-binding sites formed by the single protein chain with one site being catalytically active [42]. For the homodimeric Glo1, as well as the maize enzyme, detailed studies employing metal activation, NMR and X-ray experiments have provided unambiguous evidence that only a single active site is required for maximal activity [42,43,44]. In addition, deletional mutagenesis experiments have recently provided insight into the underlying structural factors involved in metal selectivity among the Glo1 enzymes [45].
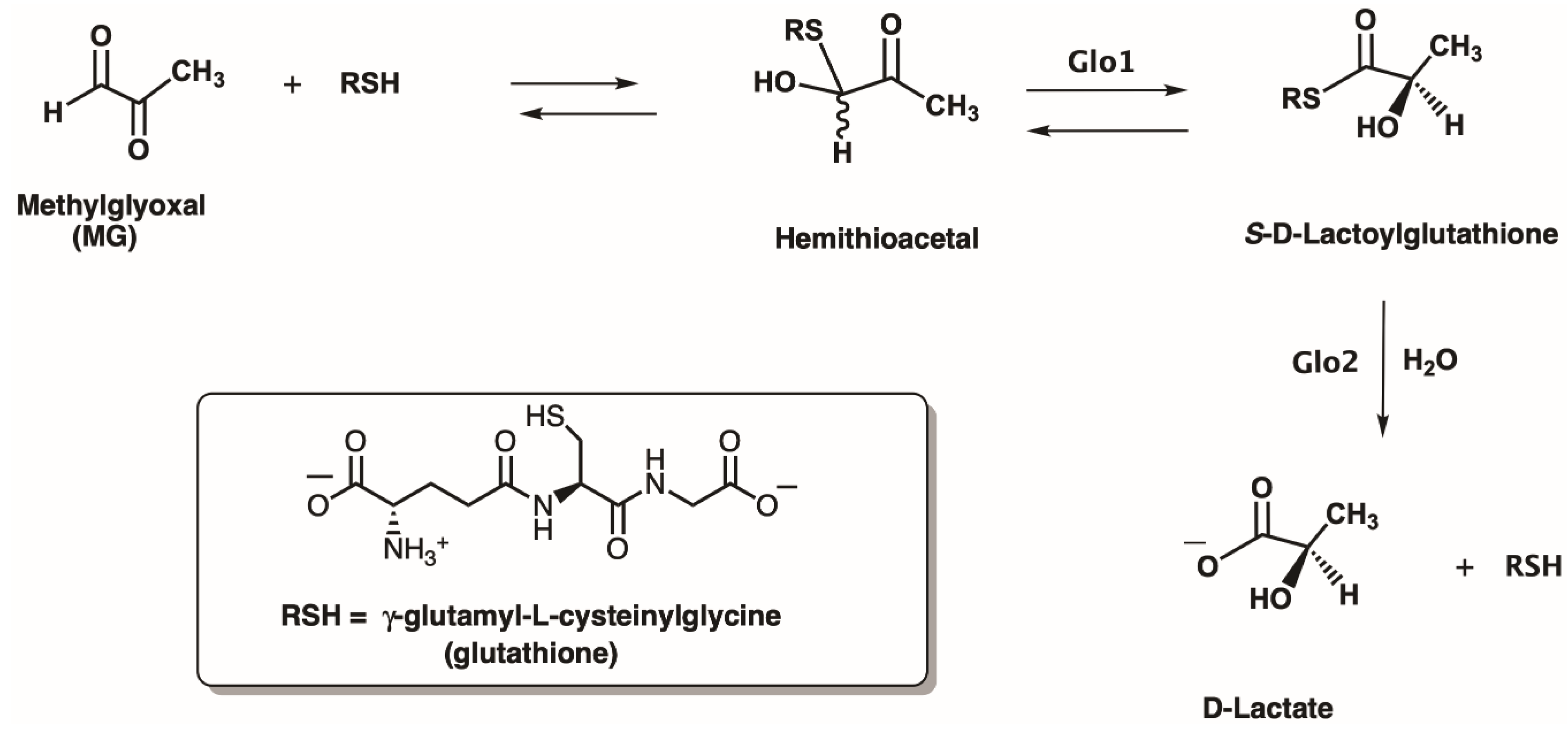
To further add to the complex but fascinating biochemistry of Glo1, it has become clear that Glo1 enzymes have evolved to employ whatever major intracellular thiol [46,47,48,49,50] is available to them within a particular organism (Figure 3). In addition to glutathione (GSH), which is present in eukaryotes and most Gram-negative bacteria, protozoans, such as Leishmania and Trypanosoma employ N1,N8-bis (l-γ-glutamyl-l-hemicystinyl-glycyl) spermidine, trypanothione, for their cellular biochemistry, which includes their Glo1 enzymes [51,52,53]. The Leishmania major Glo1 has been determined to be a Ni2+-activated class enzyme and lacks catalytic activity with Zn2+ [51,54]. The Glo1 from Trypanosoma cruzi has also been determined to be Ni2+-activated enzymes [55]. The intracellular thiol, (2S)-2-[[2-(l-cysteinylamino)-2-deoxy-α-d-glucopyranosyl]oxy]succinic acid, known as bacillithiol (Figure 3), is present in bacilli, such as Bacillus subtilis and B. anthracis, and has also been identified in Staphylococcus aureus, as well as Deinococcus radiodurans [49,56,57,58,59]. Genetic studies on Bacillus subtilis have indicated the likely existence of a Glo1 enzyme employing bacillithiol, although no reports on the isolation and characterization of a bacillithiol-dependent Glo1 have been reported [60].
Actinomycetes and mycobacteria biosynthesize the thiol, 1-d-myo-inositol-2-(N-acetyl-l-cysteinyl) amido-2-deoxy-α-d-glucopyranoside, mycothiol (Figure 3) [61,62,63,64]. Although substantial information is now available on the biochemical pathways employing mycothiol, and the functions of mycothiol and its metabolic pathways appear to parallel those of glutathione, to date no reports have appeared on the characterization of a mycothiol-dependent Glo1. However, mycothiol-null mutants in Corynebacterium glutamicum have been reported to endow this organism with MG sensitivity, indicating the possible existence of a mycothiol-dependent Glo1 [63].
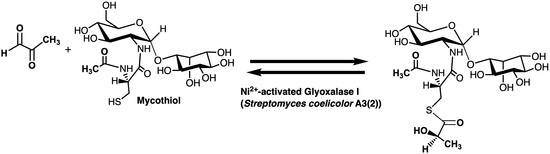
In order to extend our knowledge of the Glo1 metalloenzymes, an investigation was undertaken to identify and provide a preliminary characterization of a mycothiol-dependent Glo1 (Figure 4), if it indeed existed. The current investigation reports on the identification of such an enzyme in Streptomyces coelicolor and provides a preliminary characterization of the isolated Glo1. The Glo1 from Streptomyces coelicolor has been determined to exhibit a homodimeric quaternary structure and to be a member of the Ni2+-activation class of glyoxalase I enzymes.
2. Results and Discussion
2.1. Sequence Analysis
As mycothiol is the key intracellular thiol in Streptomyces, sequence searching of a number of sequence databases, including the Streptomyces Annotation Server (http://streptrdb.streptomyces.org.uk), for a possible Glo1 enzyme employing various known Glo1 sequences was undertaken for the organism Streptomyces coelicolor A3(2) [65,66]. The search revealed two genes of interest with annotations of a putative dioxygenase/glyoxalase/ bleomycin resistance family gene product (SCO1970; EMBLCAC42744; NCBI reference sequence: NP_626233.1), which was termed putative dioxygenase (PDO), and a putative lyase (SCO2237; EMBLCAC37263; NCBI reference sequence: NP_626487.1), which was termed putative lyase (PLA). Multiple sequence alignments of PDO and PLA with other Glo1 from various sources, including Gram-negative bacteria (E. coli [24], Yersinia pestis [19], Neisseria meningitides [19,67], Pseudomonas aeruginosa GloA2 [68], P. aeruginosa GloA3 [68], and P. putida [22]), protozoa (Leishmania major [51]), human (H. sapiens [23]), and the previously identified gene [60] correlated with possible bacillithiol-dependent Glo1 activity in Bacillus subtilis are shown in Figure 5. In this group, the known metal-specificities of the Glo1 are as follows: Zn2+-dependent (P. putida, H. sapiens, P. aeruginosa GloA3) and Ni2+-dependent (E. coli, Y. pestis, N. meningitidis, P. aeruginosa GloA2, L. major). The metal specificity of the B. subtilis enzyme is unknown as the protein has not been isolated nor characterized. PDO possesses four potential metal-binding residues (His15, Asp63, His89, Glu142) corresponding to metal-binding residue positions in other Glo1 enzymes (astericks in Figure 5). The corresponding residues in PLA (Val7, Asp62, Tyr75, Gln127) do not map as well, and the alignment suggests that PDO would be the more likely candidate to demonstrate Glo1 activity, although both gene products were investigated. Based on the alignment, PDO has a 21.7% identity and 20.2% identity to the N. meningitidis Glo1 and the B. subtilis gene product associated with the bacillithiol-dependent Glo1 activity, respectively (Figure S1). Based on previous sequence alignments and deletional mutagenesis experiments, it has been suggested that shorter Glo1 sequences tend to be Ni2+-activated class enzymes [19]. In the case of PDO, if indeed a Glo1, a Ni2+-activated class enzyme was hypothesized.
2.2. Overproduction, Isolation and Characterization of PDO and PLA
The putative dioxygenase gene (pdo) and putative lyase gene (pla) from chromosomal DNA from S. coelicolor A3(2) (NC_003888) were cloned into pET-28b(+) expression vectors utilizing NdeI and BamHI restriction endonuclease enzymes and polymerase chain reaction (PCR) to generate the purified proteins with an N-terminal His-tag followed by a thrombin protease cleavage site. The protein purification of PDO and PLA made use of the N-terminal His-tag, which was eventually removed by thrombin protease, resulting in three extra amino acids at the N-terminus (Gly-Ser-His) fused to the N-terminal Met residues. These modifications changed the predicted molecular weights of PDO and PLA to 16569.5 Da and 16749.9 Da, respectively, which were confirmed by electrospray ionization mass spectrometry (ESI-MS) analysis (Figures S2 and S3). No post-translational modification of either purified protein was observed. Analysis of a gel permeation chromatographic separation suggested a homodimeric quaternary structure for both PDO (1.5 mg mL−1) and PLA (0.6 mg mL−1) in 50 mM Tris (pH 8.0) and 150 mM KCl with a molecular weight of approximately 27.51 ± 2.46 kDa and 33.15 ± 2.89 kDa, respectively (Figure S4). The thermal stabilities of PDO and PLA as isolated were performed using differential scanning calorimetry (DSC) analysis, which suggested the estimated Tm of PDO (3 mg mL−1) and PLA (1.2 mg mL−1) in 50 mM MOPS (pH 7.0) and 10% v/v glycerol to be 57.8 and 58.5 °C, respectively (Figure S5). Their secondary structures were investigated by circular dichroism (CD) analysis which showed that both PDO (5.6 mg mL−1) and PLA (3.6 mg mL−1) in buffer containing 50 mM potassium phosphate buffer (KPB; pH 7.0) and 200 mM KCl possessed a negative maximum at 222 nm and a small shoulder at 208 nm, suggesting the presence of predominantly β-sheet structures (Figure S6), which are consistent with the previous reports on the X-ray crystallographic structures of other Glo1. The K2D3 secondary structural prediction program (http://cbdm-01.zdv.uni-mainz.de/~andrade/k2d3/) also estimated the secondary structural contents of PDO to be 1–2% α-helix, 37–41% β-sheet and 58–62% random coil and of PLA to be 2–6% α-helix, 34–40% β-sheet and 57–62% random coil. Other external factors, including pH (5–9), types of buffer (HEPES, KPB, Tris, MOPS and MOPSO), ionic strength (0–500 mM NaCl) and additive (0–30% v/v glycerol) did not significantly influence the secondary structures of either protein (data not shown).
2.3. Enzyme Assay for Thiol Cofactors
Optimization of the Glo1 enzyme assays employed the simpler and more readily available [69] truncated form of mycothiol (tMSH; des-myo-inositol mycothiol) (Figure 3) [70]. This analog has been employed in the study of mycothiol-utilizing enzymes [71]. Mycothiol was also employed at various stages in this study, especially with respect to final enzyme kinetic studies [70]. Experiments to study the details of the non-enzymatic formation of the hemithioacetal upon MG and tMSH reaction in solution were undertaken initially and included enzyme kinetic assays and 1H NMR time course studies, benchmarked to previously studied MG and glutathione adduct formation studies (Figures S7–S10 and Tables S1 and S2) [24,72,73,74,75,76,77,78,79]. Analysis of the results of these experiments indicated that a 30 min equilibration time would be appropriate for the MG-tMSH hemithioacetal equilibrium to be attained, a time somewhat longer than that for the equilibrium to be reached for the MG-GSH hemithioacetal equilibrium to be reached (15 min).
The dissociation constant (Kd) of the hemithioacetal (MG-tSH) was also determined, due to its significance in the calculation of the exact concentration of the substrate used in the Glo1 kinetic assays. After allowing the hemithioacetal to reach its equilibrium (15 min for MG-GSH and 30 min for MG-tMSH), the dissociation constant could be measured using different concentrations of MG with a fixed concentration of thiols, an approach previously employed in the literature for MG-GSH [24,72,73,74,75,76,77]. The dissociation profile of MG-GSH suggested its Kd of approximately 3.19 ± 0.29 mM (Figure S10A), which was in excellent agreement to the literature value (Kd of 3.1 mM). The Kd of the hemithioacetal forming non-enzymatically between MG and tMSH was determined similarly and estimated to be 3.33 ± 0.41 mM (Figure S10B).
The increase in UV absorbance at 240 nm observed during the reaction of Glo1 is due to the formation of the product, S-d-lactolyglutathione, from the Glo1 reaction using MG-GSH as a substrate. The investigation on the optimum detection wavelength for the reaction using MG-tMSH substrate was performed similarly, and the results suggested that the detection at 240 nm could also be employed to detect the thioester product from the MG-tMSH substrate (Figure S11). Furthermore, the expected product, S-d-lactoyl-des-myo-inositol mycothiol, produced from the Glo1 catalyzed reaction using Ni2+-reconstituted PDO (25 μg in 600 μL assay) with MG-tMSH (5 mM, Kd of 3.3 mM and equilibrium time of 30 min), was isolated using reverse phase C18 HPLC (Figure S12) and was identified by ESI-MS analysis (Figure S13), consistent with PDO serving as a mycothiol-dependent Glo1. It should be noted however that although the mass of the product is consistent with the chemical structure of the expected product, the exact determination of the stereochemistry of the lactate as d-lactate has not been shown, although it is highly likely given the commonality of the stereochemistry found in the product from various Glo1 enzymes.
Based on the information obtained on hemithioacetal formation as stated above, the substrate specificity of the Glo1 reaction was investigated in relation to metal-incorporation and the presence of PDO, PLA or commercial yeast Glo1 (1.5–3.0 μg for PDO and PLA and 0.05 μg for yeast Glo1 in 200 μL assay). The detection of the substrates, including MG, GSH, tMSH, hemithioacetal of MG-GSH and hemithioacetal of MG-tMSH did not show any significant increase in signal at 240 nm without the corresponding enzyme. Neither was product formation detected for any of these substrates in the presence of added metal ions (without the addition of the enzyme), suggesting that substrates with and without additional metal ions do not interfere with the detection of the enzyme reaction, if any—nor do they contribute any significant non-enzyme background activity. No Glo1 activity was observed for reactions containing only MG, only GSH, only tMSH, or only MG-GSH with Ni2+-reconstituted PDO. Neither were any reactions observed to occur in the reactions of apo-PDO and denatured PDO (boiled for 10 min) with hemiacetal MG-tMSH. However, trace activity was obtained in the reaction of isolated PDO (purified PDO without any additional metals) with MG-tMSH, while high activity was observed in the reaction of Ni2+-reconstituted PDO with the same substrate. These observations suggested that the incorporated metal into PDO is significant for the Glo1 reaction to occur and that the “as isolated” PDO is not entirely in its apo-form, and this was confirmed by inductively coupled mass spectrometry (ICP-MS) analysis for the presence of metal ions in the PDO protein (Table S3). The isolated PDO enzyme might accept some metals from the organism’s growth environment during the expression and purification processes undertaken in the laboratory. Additionally, the reaction catalyzed by the PDO enzyme is specific to the hemithioacetal substrate, MG-tMSH, while the substrate of the GSH-dependent Glo1 reaction, MG-GSH (5 mM), did not act as a substrate for PDO (data not shown). PLA, on the other hand, did not exhibit any glutathione or tMSH-dependent Glo1 activity under the above conditions. The enzymatic reaction catalyzed by yeast Glo1 is specific to its substrate, MG-GSH, in which it was observed to exhibit no activity in the presence of the MG-tMSH hemithioacetal.
2.4. Metal Characterization and Kinetic Studies
The metal analysis determined by ICP-MS (ALS Laboratory Group, Waterloo, ON, Canada), undertaken utilizing protocols [24,25,34,45,68] previously employed, suggested that the purified “as isolated” (no exogenously added metal ions) PDO (0.088 mg/mL) binds to copper, cobalt, nickel and zinc ions (Tables S3 and S4). The metal per enzyme ratios of nickel-and zinc-bound PDO were approximately 0.16–0.17, while those of cobalt and copper-bound enzyme were approx. 0.02. Thus, the ratios of Ni2+ and Zn2+ contents were approximately ten times higher than those of Co2+ and Cu2+, which might suggest that PDO has a higher metal-binding affinity for Ni2+ and Zn2+ than Co2+ and Cu2+ ions, or could reflect the availability of these metal ions in E. coli during protein production and isolation steps. The concentrations of these incorporated metals in the isolated protein were low and appeared almost completely in a de-metallated form. The ICP-MS analysis on this form of the PDO indicated that the enzyme contained no iron, a common metal found in dioxygenase enzymes, but it did contain ions, such as zinc and nickel bound to the isolate protein. Both Zn2+ and Ni2+, as previously mentioned, are metals that are usually associated with Glo1 enzymes. These results suggest that PDO may bind either of these metals in intact S. coelicolor, although, of course, these metals could have been picked up by the protein in steps associated with the protein isolation.
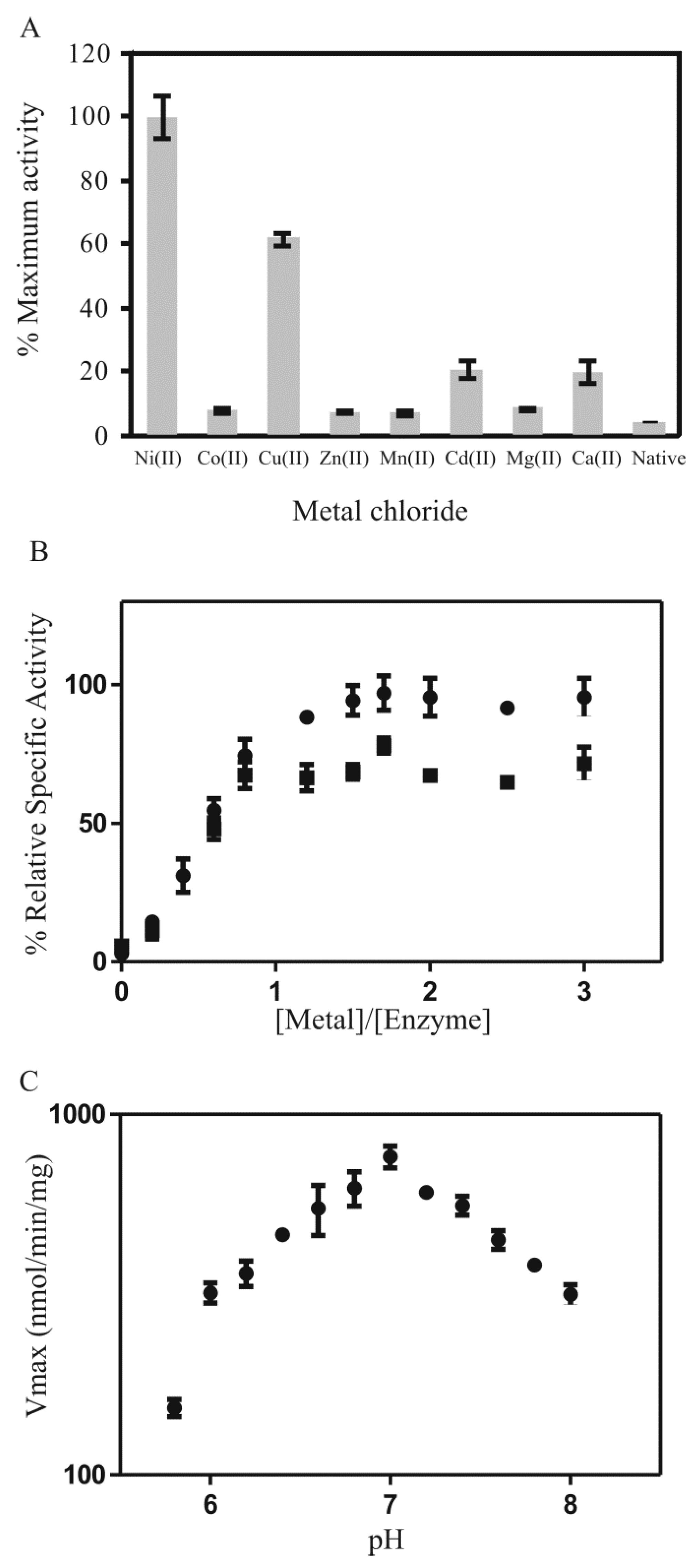
Analysis of the metal activation profile for PDO suggested that the enzyme was activated in the presence of Ni2+, Cu2+, and to a much lower extent by Cd2+ and Ca2+ (activity of PDO: Ni2+ > Cu2+ > Cd2+ > Ca2+) (Figure 6A). The enzyme with incorporated Ni2+ exhibited the highest activity of conversion of the MG-tMSH hemithioacetal into its thioester, while that of Cu2+-reconstitution gave approximately 60% compared to that of the Ni2+-reconstituted enzyme. The activities with Cd2+ and Ca2+-were low, while other metal ions (Zn2+, Mg2+, Co2+, Mn2+) were not activating. Metal titration studies of PDO with Ni2+ and Cu2+ indicated that 1 mole of metal per mole dimeric enzyme could optimize enzymatic activity (Figure 6B), suggesting a tight binding of the metal, as well as one functional active site (possibly two active sites per dimeric enzyme as PDO exhibits homodimeric quaternary structure as previously mentioned). These data are consistent with previous reports on other Glo1 (such as E. coli Glo1, P. aeruginosa GloA1, GloA2, and many others), which also find that the metal per dimeric enzyme ratio is approximately one [19,24,44,68]. The pH dependency of PDO activity was also determined (Figure 6C).
The kinetics of Ni2+-reconstituted PDO with MG-tMSH yielded a calculated Vmax of 5.8 ± 1.0 μmol/min/mg, Km of 1.25 ± 0.13 mM and kcat of 3.2 s−1 (Table 1). The Km is somewhat higher compared to other Km values for the MG-GSH substrate processed by other Glo1 [24,34,68,80,81], which is probably due to the structure of the MG-tMSH hemithioacetal lacking the inositol moiety. This hypothesis was confirmed by the kinetics of Ni2+-reconstituted PDO with the hemithioacetal of MG-MSH which yielded a calculated Vmax of 11.5 ± 1.8 μmol/min/mg, Km of 0.61 ± 0.06 mM and kcat of 6.4 s−1 (Table 1).
Metal analysis performed on the “as isolated” PLA exhibited similar results, where copper, nickel and zinc were detected bound to various copies of the protein (Tables S3 and S5). The metal per dimeric enzyme ratios of nickel-and zinc-bound PLA were approx. 0.11–0.16, while the ratio of copper bound enzyme was 0.08. The concentrations of these metals were low, which indicated that PLA is isolated in predominantly apo-form. Glo1 activity of PLA, on the other hand, was undetectable under the studied conditions. The enzyme exhibited no activity with MG-tMSH nor MG-GSH (data not shown). Neither activity was observed in the presence of different divalent metals (ZnCl2, NiCl2, CoCl2, CuCl2, MnCl2, MgCl2, CaCl2 and CdCl2), suggesting that PLA does not function as a Glo1.
Glo1 is a member of the βαβββ superfamily of proteins, consisting of fosfomycin resistance protein (FosA), methylmalonyl-CoA epimerase (MMCE), extradiol dioxygenase (DIOX), mitomycin C resistance protein (MRP) and bleomycin resistance protein (BRP) [23,34,40,82,83,84,85,86,87,88]. Among these proteins, Glo1 and MMCE share high structural similarity, thiol cofactor (GSH), presence of divalent metal and tetradentated metal-binding protein ligands [34,85]. Due to these similarities, an investigation of possible MMCE activity in PDO and PLA with selected incorporated metals (Ni2+, Co2+, Cu2+, and Zn2+) was performed, similar to those previously reported for studies on the C. acetobutylicum Glo1 [34]. Ni2+, Co2+ and Zn2+ are activating metal ions for particular Glo1 enzymes, while Co2+ is normally found in MMCE enzymes. Cu2+ was also chosen for our studies, due to its observed activation of PDO in the Glo1 reaction with Mg-tMSH. However, neither enzyme exhibited any MMCE activity understudied conditions (data not shown), and no divalent metal could activate these enzymes to accomplish the MMCE activity. Thus, PDO only functions as a Glo1 and not as an MMCE, while PLA is neither a Glo1 nor an MMCE. Previous investigations on the MMCE activity in GSH-dependent Glo1 from Clostridium acetobutylicum suggest that the enzyme is highly specific to its natural substrate (MG-GSH) even though its structure is more likely to resemble that of MMCE than other Glo1 [34]. These results suggest that Glo1 does not possess cross function with MMCE regardless of its different types of thiol cofactors that might be used across nature.
PDO is activated with Ni2+ and Cu2+ atoms. The unusual metal activation by Cu2+ in PDO may be anomalous and not related to any physiological role of this metal in the organism. However, it is interesting to speculate that it may be an important metal for this enzyme in the organism, since cytosolic copper has been found to be a major modulator of Streptomyces coelicolor germination, development and secondary metabolism [89]. It is also interesting to note that mycothiol has been found to be important in copper resistance in C. glutamicum [63], and perhaps there may be an advantage to mycothiol-dependent Glo1 to be able to utilize Cu2+ as an activating metal. However, this requires further research to confirm or dispute.
It had been found earlier that the isolated mammalian GloI exhibited high activity in the presence of Mg2+ and was thought to be a magnesium activated enzyme [21,90,91,92]. However, the naturally bound metal was later discovered to be zinc, which incorporated in the ratio of 1 mole metal per mole enzyme subunit [20]. The magnesium atoms were believed to be recovered from protein preparation processes and had no correlation with enzymatic activity. The analysis by electron paramagnetic resonance (EPR) of Co2+-bound human Glo1, however, suggested octahedral metal coordination with four metal-binding protein residues and two water molecules for this ion, as well as other activating metals, such as magnesium [29]. Yeast Glo1 was also found to be partially activated by the presence of Mg2+ and Ca2+ [20,36,93]. It was possible that these metals could reactivate the enzyme, but play a major role in protein stability. As well, X-ray crystallographic structure of E. coli Glo1 with bound Cd2+ (PDB ID: 1FA7) suggested that the enzyme could be activated with this metal and its metal coordination forms an octahedral geometry [27]. However, the activity was low compared to the enzyme with Ni2+ and Co2+, which might be related to the size of the metal that fits into the active site of the enzyme. Thus, the low activity observed in PDO with Ca2+ and Cd2+ might be explained in a similar fashion.
It is hoped that this preliminary characterization of a mycothiol-dependent Glo1 will provide important information for future studies on Glo1 enzymes from other microorganisms employing mycothiol, as well as bacillithiol, a closely related intracellular thiol.
3. Materials and Methods
3.1. DNA Cloning and Manipulation
All DNA manipulations and purifications were performed according to the protocols by Sambrook and Russell [94]. Putative dioxygenase gene (pdo) and putative lyase gene (pla) from S. coelicolor chromosome (NC_003888) were cloned into the pET-28b(+) expression vector utilizing NdeI and BamHI restriction endonuclease enzymes and the polymerase chain reaction (PCR) to generate the protein with the N-terminal His-tag followed by thrombin protease cleavage site. The forward and reverse primers (Sigma Genosys, Oakville, Ontario, Canada) were designed as follows: (+) 5′-CCGAAGCTTCATATGAGCCTGGGAGCC-3′ and (−) 5′-GGCGAATTCGGATCC TACTCGTAGTG CCGG-3′ for pdo cloning, and (+) 5′-CCGAAGCTTCATATGGACTTCACGCTCG-3′ and (−) 5′-GGCGAATTCGGATCCTAGGCCTTGTGCCGG-3′ for pla cloning. The plasmid was heat shock transformed into the competent E. coli DH5α cells. Its DNA sequence was verified (Molecular Biology Core Facility, University of Waterloo, Waterloo, ON, USA) followed by heat shock transforming into E. coli BL21 (DE3) cells for protein expression purposes. Sequence alignments and percent identities were computed using CLC Main Workbench (version 8.1.2) (http://www.qiagenbioinformatics.com).
3.2. Protein Induction, Expression, and Purification
Bacterial cultures (1 L) containing kanamycin (30 μg/mL LB) were grown and shaken in an air incubator (220 rpm) at 37 °C until an OD600 of 0.6 was reached. Proteins were then induced with 0.5 mM IPTG for 4 h. Cell pellets were harvested by centrifugation at 6000× g for 10 min and flash frozen in liquid nitrogen before storing at −80 °C. The purifications of both PDO and PLA were performed as previously reported for the C. acetobutylicum Glo1 [34] using HisTrap HP Ni2+-affinity (1 mL) and HiTrap Benzamidine FF affinity (1 mL) columns (GE Healthcare, Piscataway, NJ, USA). The protein concentration was determined by the Bradford Assay using bovine serum albumin (BSA) as a standard. Apo-enzyme preparation and metal analysis by ICP-MS were performed as described previously for other Glo1 and other enzymes [25,95,96,97]. The existence and the molecular weights of the denatured proteins were confirmed by the analysis of SDS-PAGE and ESI-MS data using a Micromass Q-TOF Global Ultima mass spectrometer (Mass Spectrometry Facility, University of Waterloo, Waterloo, ON, USA) using handling approaches as previously reported [68,78]. The molecular weight of the native protein was determined by gel permeation chromatography (Superose6 10/300 GL column) utilizing 50 mM Tris buffer (pH 8.0) and 150 mM KCl with a flow rate of 0.5 mL/min. A standard curve was prepared using Bio-Rad protein standards (Bio-Rad Laboratories, Hercules, CA, USA) containing γ-globulin (158 kDa), BSA (66 kDa), ovalbumin (44 kDa), carbonic anhydrase (29 kDa), myoglobulin (17 kDa) and vitamin B12 (1.35 kDa).
3.3. Protein Secondary Structure and Stability Experiments
CD experiments were performed on a Jasco J-715 spectropolarimeter from Jasco Inc. (Easton, MD, USA) and a Jasco J-700 Standard Analysis Program. The CD experiments with variation in protein concentration, pH, ionic strength and various additives were performed as previously reported. Prediction of protein secondary structure from CD spectra employed the K2D3 program as previously mentioned [98]. The transition midpoint (Tm) determined by DSC analysis was performed on a MicroCal VP-DSC microcalorimeter with cell volumes of 0.5 mL and self-contained pressurizing system of 0–30 p.s.i. for scanning solutions above boiling points to prevent any degassing during heating. The protein was run against 50 mM MOPS (pH 7.0) and 10% v/v glycerol over a temperature range of 10–80 °C with a scanning rate of 1 °C min−1. The Origin, scientific plotting software package, supplied by MicroCal was used for baseline subtraction and Tm calculation by the integration of the heat capacity (Cp) versus temperature (t) curve.
3.4. Preparation of Hemithioacetal Substrate
tMSH was synthesized according to the protocol developed by Unson et al. [70]. Purified MSH was a kind gift from Dr. Gerald Newton (UCSD, San Diego, CA, USA) [70]. Two factors involved in the hemithioacetal substrate formation, including equilibrium time and dissociation constant (Kd) of MG-GSH, were previously reported, but none for MG-tMSH and MG-MSH. We assumed that the hemithioacetal formations of MG-tMSH and MG-MSH were in a similar fashion, thus only equilibrium time and Kd of MG-tMSH were determined as detailed in the Supplementary Materials using the protocols reported for MG-GSH. 1H NMR experiments were performed on a Bruker (300 MHz) spectrometer (Bruker Ltd., Milton, ON, Canada). Predicted chemical shifts and integrations for substrates and hemithioacetals were estimated using ChemBioDraw Ultra 12.0 (http://www.cambridgesoft.com) software (version 12.0, PerkinElmer, Austin, TX, USA).
3.5. Enzyme Assays
An enzymatic assay was performed in 50 mM KPB (pH 6.6 or stated otherwise) using the hemithioacetal, a non-enzymatic product of MG and thiol cofactors (GSH, tMSH and MSH), as substrates utilizing previous Glo1 assay protocols [24,78]. The enzyme activity was measured as an increase in absorption at 240 nm (ε240 = 2860 M−1 cm−1) for the formation of S-d-lactoylglutathione using a 96-well UV-visible plate on a plate reader. Since the product of the mycothiol dependent Glo1 reaction is unknown, its maximum absorbance and identification were investigated to confirm the existence of the reaction product thioester (Supplementary Materials). Enzyme kinetics were evaluated using the initial rate that was fitted by the Michaelis-Menten equation with least squares fit parameters using GraphPad Prism software version 5.00 (GraphPad Software, Inc., La Jolla, CA, USA). Typical conditions for Glo1 activity studies on PDO were: PDO (1.5–3.0 µg in 200 µL assay) using two different hemithioacetal substrates (0.08–1 mM, Kd = 3.3 mM, 30 min equilibrium time) that formed non-enzymatically between methylglyoxal and thiol cofactors, including isolated MSH and tMSH, in 50 mM KPB (pH 6.6) at 25 °C. The protein was prepared in its apo-form and incubated with five equivalents of NiCl2 overnight at 4 °C prior to performing the assay.
Additionally, the investigation on the function of MMCE, a closely structural related protein to Glo1 in the same βαβββ superfamily, was performed using metal-substituted PDO and PLA (1 μg in 500 μL assay) in 20 mM Tris (pH 7.0) and 150 mM NaCl with the presence of 5 equivalents of Ni2+, Co2+, Cu2+ and Zn2+ as previously described for another Glo1 enzyme study [34].
4. Conclusions
Identification and isolation of a glyoxalase I (Glo1) enzyme from Streptomyces coelicolor A3(2) was accomplished in this study. A preliminary investigation of several properties of the protein, termed PDO (thermal stability, pH dependency on activity, secondary structure) was undertaken. PDO was found to be catalytically active as a glyoxalase I enzyme in the presence of Ni2+, and converted the hemithioacetals formed from methylglyoxal and des-myo-inositol mycothiol (tMSH) to the identified thioester product. No enzymatic activity was observed using glutathione, suggesting PDO is specific to tMSH and after further experimentation, mycothiol itself. From the metal activation profile, PDO functions as a mycothiol-dependent Glo1 of the Ni2+-activated class of glyoxalase I enzymes. This is the first protein characterization of an MSH-dependent Glo1.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/7/8/99/s1, Figure S1: Percent identities calculated for various Glo1, Figure S2: SDS-PAGE and ESI mass spectrum of purified PDO, Figure S3: SDS-PAGE and ESI mass spectrum of purified PLA, Figure S4: Gel permeation chromatographic profile for PDO and PLA, Figure S5: DSC plots for PDO and PLA, Figure S6: CD plots for PDO and PLA, Figure S7: Enzymatic assay using various incubation times, Figure S8: 1H NMR for various time incubations of MG and GSH, Table S1: Chemical Shifts and Integrations of 1H NMR signals (experimental and calculated) for GSH and MG-GSH, Figure S9: 1H NMR for various time incubations of MG and tMSH, Table S2: Chemical Shifts and Integrations of 1H NMR signals (experimental and calculated) for tMSH and MG-tMSH, Figure S10: Plots for the determination of Kd for MG-GSH and MG-tMSH hemithioacetals, Figure S11: UV detection of MG-tMSH thioester product, Figure S12: HPLC chromatogram of products of MG-tMSH Glo1 activity by PDO, Figure S13: ESI mass spectrum of isolated thioester product, Table S3–S5: ICP-MS element analyses of “as isolated” PDO and PLA.
Author Contributions
Conceptualization: J.F.H.; Investigation and draft writing: U.S. and J.F.H.; writing—review and editing: J.F.H. and U.S.
Funding
This research was funded by NSERC (Canada) (JH) and the University of Waterloo (JH), and the Government of Thailand for a graduate scholarship (US).
Acknowledgments
The authors would like to thank Richard Smith for electrospray ionization MS analysis of the protein sample, Elisabeth Daub for Streptomycetes handling and David Ward, Michele Cossette and Vincent Azhikannickal for support of tMSH synthesis and enzyme purification. As well, we thank Zhengding Su, Christine Hand and Nicole Sukdeo for sharing their knowledge and suggestions. Gerard Wright (McMaster University) is gratefully acknowledged for supplying the Streptomyces coelicolor A3(2) strain. The authors gratefully acknowledge Gerald Newton (University of California Sand Diego) for the kind gift of purified mycothiol used in this investigation.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Thornalley, P.J. Dietary AGEs and ALEs and risk to human health by their interaction with the receptor for advanced glycation endproducts (RAGE)—An introduction. Mol. Nutr. Food Res. 2007, 51, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Krautwald, M.; Munch, G. Advanced glycation end products as biomarkers and gerontotoxins—A basis to explore methylglyoxal-lowering agents for Alzheimer’s disease? Exp. Gerontol. 2010, 45, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalkwijk, C.G. Vascular AGE-ing by methylglyoxal: The past, the present and the future. Diabetologia 2015, 58, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Honek, J.F. Glyoxalase biochemistry. Biomol. Concepts 2015, 6, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems—Role in ageing and disease. Drug Metabol. Drug Interact. 2008, 23, 125–150. [Google Scholar] [CrossRef] [PubMed]
- Trellu, S.; Courties, A.; Jaisson, S.; Gorisse, L.; Gillery, P.; Kerdine-Romer, S.; Vaamonde-Garcia, C.; Houard, X.; Ekhirch, F.P.; Sautet, A.; et al. Impairment of glyoxalase-1, an advanced glycation end-product detoxifying enzyme, induced by inflammation in age-related osteoarthritis. Arthritis Res. Ther. 2019, 21, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bari, L.; Atlante, A.; Armeni, T.; Kalapos, M.P. Synthesis and metabolism of methylglyoxal, S-d-lactoylglutathione and d-lactate in cancer and Alzheimer’s disease. Exploring the crossroad of eternal youth and premature aging. Ageing Res. Rev. 2019, 53, 100915. [Google Scholar] [CrossRef] [PubMed]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: The first hundred years... and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sukdeo, N.; Honek, J.F. Microbial glyoxalase enzymes: Metalloenzymes controlling cellular levels of methylglyoxal. Drug Metabol. Drug Interact. 2008, 23, 29–50. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, N.; Thornalley, P.J. Glyoxalase Centennial conference: Introduction, history of research on the glyoxalase system and future prospects. Biochem. Soc. Trans. 2014, 42, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Glyoxalase I—Structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Ozyamak, E.; Black, S.S.; Walker, C.A.; Maclean, M.J.; Bartlett, W.; Miller, S.; Booth, I.R. The critical role of S-lactoylglutathione formation during methylglyoxal detoxification in Escherichia coli. Mol. Microbiol. 2010, 78, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Misra, K.; Banerjee, A.B.; Ray, S.; Ray, M. Glyoxalase III from Escherichia coli: A single novel enzyme for the conversion of methylglyoxal into d-lactate without reduced glutathione. Biochem. J. 1995, 305 Pt 3, 999–1003. [Google Scholar] [CrossRef]
- Choi, D.; Kim, J.; Ha, S.; Kwon, K.; Kim, E.H.; Lee, H.Y.; Ryu, K.S.; Park, C. Stereospecific mechanism of DJ-1 glyoxalases inferred from their hemithioacetal-containing crystal structures. FEBS J. 2014, 281, 5447–5462. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Su, Y.; Wang, Z.; Chen, C.; Wu, T.; Huang, Y. Identification of glutathione (GSH)-independent glyoxalase III from Schizosaccharomyces pombe. BMC Evol. Biol. 2014, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Subedi, K.P.; Choi, D.; Kim, I.; Min, B.; Park, C. Hsp31 of Escherichia coli K-12 is glyoxalase III. Mol. Microbiol. 2011, 81, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Sukdeo, N.; Clugston, S.L.; Daub, E.; Honek, J.F. Distinct classes of glyoxalase I: Metal specificity of the Yersinia pestis, Pseudomonas aeruginosa and Neisseria meningitidis enzymes. Biochem. J. 2004, 384, 111–117. [Google Scholar] [CrossRef]
- Aronsson, A.C.; Marmstal, E.; Mannervik, B. Glyoxalase I, a zinc metalloenzyme of mammals and yeast. Biochem. Biophys. Res. Commun. 1978, 81, 1235–1240. [Google Scholar] [CrossRef]
- Han, L.P.; Schimandle, C.M.; Davison, L.M.; Vander Jagt, D.L. Comparative kinetics of Mg2+-, Mn2+-, Co2+-, and Ni2+-activated glyoxalase I. Evaluation of the role of the metal ion. Biochemistry 1977, 16, 5478–5484. [Google Scholar] [CrossRef] [PubMed]
- Saint-Jean, A.P.; Phillips, K.R.; Creighton, D.J.; Stone, M.J. Active monomeric and dimeric forms of Pseudomonas putida glyoxalase I: Evidence for 3D domain swapping. Biochemistry 1998, 37, 10345–10353. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.D.; Olin, B.; Ridderstrom, M.; Mannervik, B.; Jones, T.A. Crystal structure of human glyoxalase I—Evidence for gene duplication and 3D domain swapping. EMBO J. 1997, 16, 3386–3395. [Google Scholar] [CrossRef] [PubMed]
- Clugston, S.L.; Barnard, J.F.; Kinach, R.; Miedema, D.; Ruman, R.; Daub, E.; Honek, J.F. Overproduction and characterization of a dimeric non-zinc glyoxalase I from Escherichia coli: Evidence for optimal activation by nickel ions. Biochemistry 1998, 37, 8754–8763. [Google Scholar] [CrossRef] [PubMed]
- Clugston, S.L.; Yajima, R.; Honek, J.F. Investigation of metal binding and activation of Escherichia coli glyoxalase I: Kinetic, thermodynamic and mutagenesis studies. Biochem. J. 2004, 377, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.; Clugston, S.L.; Honek, J.F.; Maroney, M.J. XAS investigation of the nickel active site structure in Escherichia coli glyoxalase I. Inorg. Chem. 2000, 39, 2962–2963. [Google Scholar] [CrossRef] [PubMed]
- He, M.M.; Clugston, S.L.; Honek, J.F.; Matthews, B.W. Determination of the structure of Escherichia coli glyoxalase I suggests a structural basis for differential metal activation. Biochemistry 2000, 39, 8719–8727. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.D.; Ridderstrom, M.; Olin, B.; Kavarana, M.J.; Creighton, D.J.; Mannervik, B. Reaction mechanism of glyoxalase I explored by an X-ray crystallographic analysis of the human enzyme in complex with a transition state analogue. Biochemistry 1999, 38, 13480–13490. [Google Scholar] [CrossRef]
- Sellin, S.; Eriksson, L.E.; Aronsson, A.C.; Mannervik, B. Octahedral metal coordination in the active site of glyoxalase I as evidenced by the properties of Co(II)-glyoxalase I. J. Biol. Chem. 1983, 258, 2091–2093. [Google Scholar]
- Himo, F.; Siegbahn, P.E. Catalytic mechanism of glyoxalase I: A theoretical study. J. Am. Chem. Soc. 2001, 123, 10280–10289. [Google Scholar] [CrossRef]
- Richter, U.; Krauss, M. Active site structure and mechanism of human glyoxalase I—An ab initio theoretical study. J. Am. Chem. Soc. 2001, 123, 6973–6982. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.; Clugston, S.L.; Honek, J.F.; Maroney, M.J. An XAS investigation of product and inhibitor complexes of Ni-containing GlxI from Escherichia coli: Mechanistic implications. Biochemistry 2001, 40, 4569–4582. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Suttisansanee, U.; Lau, K.; Lagishetty, S.; Rao, K.N.; Swaminathan, S.; Sauder, J.M.; Burley, S.K.; Honek, J.F. Structural variation in bacterial glyoxalase I enzymes: Investigation of the metalloenzyme glyoxalase I from Clostridium acetobutylicum. J. Biol. Chem. 2011, 286, 38367–38374. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M.; Sturm, N.; Mittler, S.; Harner, M.; Mack, H.; Becker, K. Allosteric coupling of two different functional active sites in monomeric Plasmodium falciparum glyoxalase I. J. Biol. Chem. 2007, 282, 28419–28430. [Google Scholar] [CrossRef] [PubMed]
- Frickel, E.M.; Jemth, P.; Widersten, M.; Mannervik, B. Yeast glyoxalase I is a monomeric enzyme with two active sites. J. Biol. Chem. 2001, 276, 1845–1849. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Maeta, K.; Nomura, W. Glyoxalase system in yeasts: Structure, function, and physiology. Semin. Cell Dev. Biol. 2011, 22, 278–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozef, R.; Rahlfs, S.; Chang, T.; Schirmer, H.; Becker, K. Glyoxalase I of the malarial parasite Plasmodium falciparum: Evidence for subunit fusion. FEBS Lett. 2003, 554, 284–288. [Google Scholar] [CrossRef]
- Turra, G.L.; Agostini, R.B.; Fauguel, C.M.; Presello, D.A.; Andreo, C.S.; Gonzalez, J.M.; Campos-Bermudez, V.A. Structure of the novel monomeric glyoxalase I from Zea mays. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 2009–2020. [Google Scholar] [CrossRef]
- Kaur, C.; Vishnoi, A.; Ariyadasa, T.U.; Bhattacharya, A.; Singla-Pareek, S.L.; Sopory, S.K. Episodes of horizontal gene-transfer and gene-fusion led to co-existence of different metal-ion specific glyoxalase I. Sci. Rep. 2013, 3, 3076. [Google Scholar] [CrossRef] [Green Version]
- Mustafiz, A.; Ghosh, A.; Tripathi, A.K.; Kaur, C.; Ganguly, A.K.; Bhavesh, N.S.; Tripathi, J.K.; Pareek, A.; Sopory, S.K.; Singla-Pareek, S.L. A unique Ni2+ -dependent and methylglyoxal-inducible rice glyoxalase I possesses a single active site and functions in abiotic stress response. Plant J. 2014, 78, 951–963. [Google Scholar] [CrossRef]
- Gonzalez, J.M.; Agostini, R.B.; Alvarez, C.E.; Klinke, S.; Andreo, C.S.; Campos-Bermudez, V.A. Deciphering the number and location of active sites in the monomeric glyoxalase I of Zea mays. FEBS J. 2019. [Google Scholar] [CrossRef]
- Su, Z.; Sukdeo, N.; Honek, J.F. 15N-1H HSQC NMR evidence for distinct specificity of two active sites in Escherichia coli glyoxalase I. Biochemistry 2008, 47, 13232–13241. [Google Scholar] [CrossRef]
- Bythell-Douglas, R.; Suttisansanee, U.; Flematti, G.R.; Challenor, M.; Lee, M.; Panjikar, S.; Honek, J.F.; Bond, C.S. The crystal structure of a homodimeric Pseudomonas glyoxalase I enzyme reveals asymmetric metallation commensurate with half-of-sites activity. Chemistry 2015, 21, 541–544. [Google Scholar] [CrossRef]
- Suttisansanee, U.; Ran, Y.; Mullings, K.Y.; Sukdeo, N.; Honek, J.F. Modulating glyoxalase I metal selectivity by deletional mutagenesis: Underlying structural factors contributing to nickel activation profiles. Metallomics 2015, 7, 605–612. [Google Scholar] [CrossRef]
- Hand, C.E.; Honek, J.F. Biological chemistry of naturally occurring thiols of microbial and marine origin. J. Nat. Prod. 2005, 68, 293–308. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, Q.; Liu, W. The versatile low-molecular-weight thiols: Beyond cell protection. Bioessays 2015, 37, 1262–1267. [Google Scholar] [CrossRef]
- Newton, G.L.; Arnold, K.; Price, M.S.; Sherrill, C.; Delcardayre, S.B.; Aharonowitz, Y.; Cohen, G.; Davies, J.; Fahey, R.C.; Davis, C. Distribution of thiols in microorganisms: Mycothiol is a major thiol in most actinomycetes. J. Bacteriol. 1996, 178, 1990–1995. [Google Scholar] [CrossRef]
- Gaballa, A.; Newton, G.L.; Antelmann, H.; Parsonage, D.; Upton, H.; Rawat, M.; Claiborne, A.; Fahey, R.C.; Helmann, J.D. Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in Bacilli. Proc. Natl. Acad. Sci. USA 2010, 107, 6482–6486. [Google Scholar] [CrossRef] [Green Version]
- Jothivasan, V.K.; Hamilton, C.J. Mycothiol: Synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 2008, 25, 1091–1117. [Google Scholar] [CrossRef]
- Vickers, T.J.; Greig, N.; Fairlamb, A.H. A trypanothione-dependent glyoxalase I with a prokaryotic ancestry in Leishmania major. Proc. Natl. Acad. Sci. USA 2004, 101, 13186–13191. [Google Scholar] [CrossRef]
- Oza, S.L.; Shaw, M.P.; Wyllie, S.; Fairlamb, A.H. Trypanothione biosynthesis in Leishmania major. Mol. Biochem. Parasitol. 2005, 139, 107–116. [Google Scholar] [CrossRef]
- Greig, N.; Wyllie, S.; Patterson, S.; Fairlamb, A.H. A comparative study of methylglyoxal metabolism in trypanosomatids. FEBS J. 2009, 276, 376–386. [Google Scholar] [CrossRef]
- Ariza, A.; Vickers, T.J.; Greig, N.; Armour, K.A.; Dixon, M.J.; Eggleston, I.M.; Fairlamb, A.H.; Bond, C.S. Specificity of the trypanothione-dependent Leishmania major glyoxalase I: Structure and biochemical comparison with the human enzyme. Mol. Microbiol. 2006, 59, 1239–1248. [Google Scholar] [CrossRef]
- Greig, N.; Wyllie, S.; Vickers, T.J.; Fairlamb, A.H. Trypanothione-dependent glyoxalase I in Trypanosoma cruzi. Biochem. J. 2006, 400, 217–223. [Google Scholar] [CrossRef]
- Fahey, R.C. Glutathione analogs in prokaryotes. Biochim. Biophys. Acta 2013, 1830, 3182–3198. [Google Scholar] [CrossRef]
- Sharma, S.V.; Arbach, M.; Roberts, A.A.; Macdonald, C.J.; Groom, M.; Hamilton, C.J. Biophysical features of bacillithiol, the glutathione surrogate of Bacillus subtilis and other firmicutes. ChemBioChem 2013, 14, 2160–2168. [Google Scholar] [CrossRef]
- Perera, V.R.; Newton, G.L.; Pogliano, K. Bacillithiol: A key protective thiol in Staphylococcus aureus. Expert Rev. Anti Infect. Ther. 2015, 13, 1089–1107. [Google Scholar] [CrossRef]
- Chandrangsu, P.; Loi, V.V.; Antelmann, H.; Helmann, J.D. The Role of Bacillithiol in Gram-Positive Firmicutes. Antioxid. Redox Signal. 2018, 28, 445–462. [Google Scholar] [CrossRef]
- Chandrangsu, P.; Dusi, R.; Hamilton, C.J.; Helmann, J.D. Methylglyoxal resistance in Bacillus subtilis: Contributions of bacillithiol-dependent and independent pathways. Mol. Microbiol. 2014, 91, 706–715. [Google Scholar] [CrossRef]
- Sao Emani, C.; Williams, M.J.; Wiid, I.J.; Baker, B. The functional interplay of low molecular weight thiols in Mycobacterium tuberculosis. J. Biomed. Sci. 2018, 25, 55. [Google Scholar] [CrossRef]
- Sharma, S.V.; Van Laer, K.; Messens, J.; Hamilton, C.J. Thiol Redox and pKa Properties of Mycothiol, the Predominant Low-Molecular-Weight Thiol Cofactor in the Actinomycetes. ChemBioChem 2016, 17, 1689–1692. [Google Scholar] [CrossRef]
- Liu, Y.B.; Long, M.X.; Yin, Y.J.; Si, M.R.; Zhang, L.; Lu, Z.Q.; Wang, Y.; Shen, X.H. Physiological roles of mycothiol in detoxification and tolerance to multiple poisonous chemicals in Corynebacterium glutamicum. Arch. Microbiol. 2013, 195, 419–429. [Google Scholar] [CrossRef]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef]
- Bentley, S.D.; Chater, K.F.; Cerdeno-Tarraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef]
- Park, J.H.; Cha, C.J.; Roe, J.H. Identification of genes for mycothiol biosynthesis in Streptomyces coelicolor A3(2). J. Microbiol. 2006, 44, 121–125. [Google Scholar]
- Kizil, G.; Wilks, K.; Wells, D.; Ala’Aldeen, D.A. Detection and characterisation of the genes encoding glyoxalase I and II from Neisseria meningitidis. J. Med. Microbiol. 2000, 49, 669–673. [Google Scholar] [CrossRef]
- Sukdeo, N.; Honek, J.F. Pseudomonas aeruginosa contains multiple glyoxalase I-encoding genes from both metal activation classes. Biochim. Biophys. Acta 2007, 1774, 756–763. [Google Scholar] [CrossRef]
- Hamilton, C.J.; Finlay, R.M.; Stewart, M.J.; Bonner, A. Mycothiol disulfide reductase: A continuous assay for slow time-dependent inhibitors. Anal. Biochem. 2009, 388, 91–96. [Google Scholar] [CrossRef]
- Unson, M.D.; Newton, G.L.; Davis, C.; Fahey, R.C. An immunoassay for the detection and quantitative determination of mycothiol. J. Immunol. Methods 1998, 214, 29–39. [Google Scholar] [CrossRef]
- Patel, M.P.; Blanchard, J.S. Synthesis of des-myo-inositol mycothiol and demonstration of a mycobacterial specific reductase activity. J. Am. Chem. Soc. 1998, 120, 11538–11539. [Google Scholar] [CrossRef]
- Vander Jagt, D.L.; Han, L.P.; Lehman, C.H. Kinetic evaluation of substrate specificity in the glyoxalase-I-catalyzed disproportionation of α-ketoaldehydes. Biochemistry 1972, 11, 3735–3740. [Google Scholar] [CrossRef]
- Vince, R.; Daluge, S.; Wadd, W.B. Studies on the inhibition of glyoxalase I by S-substituted glutathiones. J. Med. Chem. 1971, 14, 402–404. [Google Scholar] [CrossRef]
- Cliffe, E.E.; Waley, S.G. The mechanism of the glyoxalase I reaction, and the effect of ophthalmic acid as an inhibitor. Biochem. J. 1961, 79, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Creighton, D.J.; Migliorini, M.; Pourmotabbed, T.; Guha, M.K. Optimization of efficiency in the glyoxalase pathway. Biochemistry 1988, 27, 7376–7384. [Google Scholar] [CrossRef]
- Griffis, C.E.; Ong, L.H.; Buettner, L.; Creighton, D.J. Nonstereospecific substrate usage by glyoxalase I. Biochemistry 1983, 22, 2945–2951. [Google Scholar] [CrossRef]
- Vander Jagt, D.L.; Daub, E.; Krohn, J.A.; Han, L.P. Effects of pH and thiols on the kinetics of yeast glyoxalase I. An evaluation of the random pathway mechanism. Biochemistry 1975, 14, 3669–3675. [Google Scholar] [CrossRef]
- Mullings, K.Y.; Sukdeo, N.; Suttisansanee, U.; Ran, Y.; Honek, J.F. Ni2+-activated glyoxalase I from Escherichia coli: Substrate specificity, kinetic isotope effects and evolution within the betaalphabetabetabeta superfamily. J. Inorg. Biochem. 2012, 108, 133–140. [Google Scholar] [CrossRef]
- Rae, C.; O’Donoghue, S.I.; Bubb, W.A.; Kuchel, P.W. Stereospecificity of substrate usage by glyoxalase 1: Nuclear magnetic resonance studies of kinetics and hemithioacetal substrate conformation. Biochemistry 1994, 33, 3548–3559. [Google Scholar] [CrossRef]
- Akoachere, M.; Iozef, R.; Rahlfs, S.; Deponte, M.; Mannervik, B.; Creighton, D.J.; Schirmer, H.; Becker, K. Characterization of the glyoxalases of the malarial parasite Plasmodium falciparum and comparison with their human counterparts. Biol. Chem. 2005, 386, 41–52. [Google Scholar] [CrossRef]
- Mannervik, B.; Ridderstrom, M. Catalytic and molecular properties of glyoxalase I. Biochem. Soc. Trans. 1993, 21, 515–517. [Google Scholar] [CrossRef] [Green Version]
- Bergdoll, M.; Eltis, L.D.; Cameron, A.D.; Dumas, P.; Bolin, J.T. All in the family: Structural and evolutionary relationships among three modular proteins with diverse functions and variable assembly. Protein Sci. 1998, 7, 1661–1670. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.N. Mechanistic diversity in a metalloenzyme superfamily. Biochemistry 2000, 39, 13625–13632. [Google Scholar] [CrossRef]
- Honek, J.F. Nickel Glyuoxalase I. In The Biological Chemistry of Nickel; Kozlowski, H., Zamble, D., Rowinska-Zyrek, M., Eds.; Royal Societ of Chemistry: London, UK, 2017; Volume 10. [Google Scholar]
- McCarthy, A.A.; Baker, H.M.; Shewry, S.C.; Patchett, M.L.; Baker, E.N. Crystal structure of methylmalonyl-coenzyme A epimerase from P. shermanii: A novel enzymatic function on an ancient metal binding scaffold. Structure 2001, 9, 637–646. [Google Scholar] [CrossRef]
- Dumas, P.; Bergdoll, M.; Cagnon, C.; Masson, J.M. Crystal structure and site-directed mutagenesis of a bleomycin resistance protein and their significance for drug sequestering. EMBO J. 1994, 13, 2483–2492. [Google Scholar] [CrossRef]
- Martin, T.W.; Dauter, Z.; Devedjiev, Y.; Sheffield, P.; Jelen, F.; He, M.; Sherman, D.H.; Otlewski, J.; Derewenda, Z.S.; Derewenda, U. Molecular basis of mitomycin C resistance in streptomyces: Structure and function of the MRD protein. Structure 2002, 10, 933–942. [Google Scholar] [CrossRef]
- Thompson, M.K.; Keithly, M.E.; Harp, J.; Cook, P.D.; Jagessar, K.L.; Sulikowski, G.A.; Armstrong, R.N. Structural and chemical aspects of resistance to the antibiotic fosfomycin conferred by FosB from Bacillus cereus. Biochemistry 2013, 52, 7350–7362. [Google Scholar] [CrossRef]
- Gonzalez-Quinonez, N.; Corte-Rodriguez, M.; Alvarez-Fernandez-Garcia, R.; Rioseras, B.; Lopez-Garcia, M.T.; Fernandez-Garcia, G.; Montes-Bayon, M.; Manteca, A.; Yague, P. Cytosolic copper is a major modulator of germination, development and secondary metabolism in Streptomyces coelicolor. Sci. Rep. 2019, 9, 4214. [Google Scholar] [CrossRef]
- Aronsson, A.C.; Mannervik, B. Characterization of glyoxalase I purified from pig erythrocytes by affinity chromatography. Biochem. J. 1977, 165, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Mannervik, B.; Lindstrom, L.; Bartfai, T. Partial purification and characterization of glyoxalase I from porcine erythrocytes. Eur. J. Biochem. 1972, 29, 276–281. [Google Scholar] [CrossRef]
- Uotila, L.; Koivusalo, M. Purification and properties of glyoxalase I from sheep liver. Eur. J. Biochem. 1975, 52, 493–503. [Google Scholar] [CrossRef]
- Takatsume, Y.; Izawa, S.; Inoue, Y. Identification of thermostable glyoxalase I in the fission yeast Schizosaccharomyces pombe. Arch. Microbiol. 2004, 181, 371–377. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- O’Young, J.; Sukdeo, N.; Honek, J.F. Escherichia coli glyoxalase II is a binuclear zinc-dependent metalloenzyme. Arch. Biochem. Biophys. 2007, 459, 20–26. [Google Scholar] [CrossRef]
- Hunt, J.B.; Neece, S.H.; Ginsburg, A. The use of 4-(2-pyridylazo)resorcinol in studies of zinc release from Escherichia coli aspartate transcarbamoylase. Anal. Biochem. 1985, 146, 150–157. [Google Scholar] [CrossRef]
- McCall, K.A.; Fierke, C.A. Colorimetric and fluorimetric assays to quantitate micromolar concentrations of transition metals. Anal. Biochem. 2000, 284, 307–315. [Google Scholar] [CrossRef]
- Louis-Jeune, C.; Andrade-Navarro, M.A.; Perez-Iratxeta, C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 2011, 80, 374–381. [Google Scholar] [CrossRef]
Figure 1.
The overall reaction of the enzymes glyoxalase I (Glo1) and glyoxalase II (Glo2). The intracellular thiol utilized in many, but not all, organisms is the tripeptide glutathione.
Figure 1.
The overall reaction of the enzymes glyoxalase I (Glo1) and glyoxalase II (Glo2). The intracellular thiol utilized in many, but not all, organisms is the tripeptide glutathione.

Figure 2.
The ribbon structure of (A) the active Ni2+-bound Escherichia coli Glo1 (PDB ID: 1F9Z) forming an octahedral geometry with four metal-binding residues (His5, Glu56, His74 and Glu122) and two nearby water molecules around the metal center and (B) the inactive Zn2+-bound E. coli Glo1 (PDB ID: 1FA5) forming a trigonal bipyramidal metal coordination with the same four metal-binding residues but with only one water molecule [27]. The metal-binding residues are shown in sticks, and active site water molecules are shown in green. The divalent metals in the catalytic pocket are represented as red spheres. The 3D structures were generated by using the UCSF Chimera program (University of California, San Francisco, CA, USA) [33].
Figure 2.
The ribbon structure of (A) the active Ni2+-bound Escherichia coli Glo1 (PDB ID: 1F9Z) forming an octahedral geometry with four metal-binding residues (His5, Glu56, His74 and Glu122) and two nearby water molecules around the metal center and (B) the inactive Zn2+-bound E. coli Glo1 (PDB ID: 1FA5) forming a trigonal bipyramidal metal coordination with the same four metal-binding residues but with only one water molecule [27]. The metal-binding residues are shown in sticks, and active site water molecules are shown in green. The divalent metals in the catalytic pocket are represented as red spheres. The 3D structures were generated by using the UCSF Chimera program (University of California, San Francisco, CA, USA) [33].

Figure 3.
Several important intracellular thiols found in nature (trypanothione, bacillithiol, mycothiol). Des-myo-inositol mycothiol (tMSH) is an analog of mycothiol also employed in the current study.
Figure 3.
Several important intracellular thiols found in nature (trypanothione, bacillithiol, mycothiol). Des-myo-inositol mycothiol (tMSH) is an analog of mycothiol also employed in the current study.

Figure 4.
The overall reaction of a mycothiol-dependent glyoxalase I enzyme.

Figure 5.
The multiple sequence alignment of the putative dioxygenase (PDO, CAC42744) and the putative lyase (PLA, CAC37263) from Streptomyces coelicolor A3(2) with glyoxalase I from other organisms (organism name followed by National Center for Biotechnology Information (NCBI) accession number), including E. coli (NP_310387), Y. pestis (ZP_01887743.1), N. meningitides (CAA74673), P. aeruginosa GloA2 (ATE47122.1), L. major (AAT98624.1), P. aeruginosa GloA3 (AAG08496.1), H. sapiens (AAB49495), P. putida (AAN69360), B. subtilis bacillithiol-dependent MG resistance proposed Glo1 (P39586.1). The metal-binding residues are highlighted with asterisks. Three different Glo1 have been identified in P. aeruginosa PA01: GloA1 (Ni2+-activated), GloA2 (Ni2+-activated and GloA3 (Zn2+-activated) [68]. The alignment was created using CLC Main Workbench (version 8.1.2) with the accurate alignment algorithm (http://www.qiagenbioinformatics.com).
Figure 5.
The multiple sequence alignment of the putative dioxygenase (PDO, CAC42744) and the putative lyase (PLA, CAC37263) from Streptomyces coelicolor A3(2) with glyoxalase I from other organisms (organism name followed by National Center for Biotechnology Information (NCBI) accession number), including E. coli (NP_310387), Y. pestis (ZP_01887743.1), N. meningitides (CAA74673), P. aeruginosa GloA2 (ATE47122.1), L. major (AAT98624.1), P. aeruginosa GloA3 (AAG08496.1), H. sapiens (AAB49495), P. putida (AAN69360), B. subtilis bacillithiol-dependent MG resistance proposed Glo1 (P39586.1). The metal-binding residues are highlighted with asterisks. Three different Glo1 have been identified in P. aeruginosa PA01: GloA1 (Ni2+-activated), GloA2 (Ni2+-activated and GloA3 (Zn2+-activated) [68]. The alignment was created using CLC Main Workbench (version 8.1.2) with the accurate alignment algorithm (http://www.qiagenbioinformatics.com).

Figure 6.
(A) PDO metal activation profile after metal ion (5 equivalents) preincubation of the apo-form of the enzyme with metal chloride (conditions: PDO (3.1 µg in 200 µL) incubated with metal chlorides overnight at 4 °C); (B) % Relative specific enzyme activity of PDO versus metal ion titration with ( ![Inorganics 07 00099 i001]() ) Ni2+ and (
) Ni2+ and ( ![Inorganics 07 00099 i002]() ) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.
) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.
 ) Ni2+ and (
) Ni2+ and (  ) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.
) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.
Figure 6.
(A) PDO metal activation profile after metal ion (5 equivalents) preincubation of the apo-form of the enzyme with metal chloride (conditions: PDO (3.1 µg in 200 µL) incubated with metal chlorides overnight at 4 °C); (B) % Relative specific enzyme activity of PDO versus metal ion titration with ( ![Inorganics 07 00099 i001]() ) Ni2+ and (
) Ni2+ and ( ![Inorganics 07 00099 i002]() ) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.
) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.
) Ni2+ and ( ) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Kinetic data for the enzyme reaction catalyzed by Ni2+-reconstituted PDO.
| Thiol Cofactors | Vmax (μmol/min/mg) | Km (μM) | kcat (s−1) | kcat/Km (M−1s−1) |
|---|---|---|---|---|
| MSH | 11.5 ± 1.8 | 612 ± 56 | 6.4 | 10403 |
| tMSH | 5.8 ± 1.0 | 1247 ± 127 | 3.2 | 2545 |
Conditions: PDO (1.5–3.0 µg in 200 µL assay) using two different hemithioacetal substrates (0.08–1 mM, Kd = 3.3 mM, 30 min equilibrium time) that formed non-enzymatically between methylglyoxal and thiol cofactors, including isolated mycothiol (MSH) and synthesized truncated mycothiol (tMSH), in 50 mM KPB (pH 6.6) at 25 °C. The protein was prepared in its apo-form and incubated with five equivalents of NiCl2 overnight at 4 °C prior to performing the assay.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Suttisansanee, U.; Honek, J.F. Preliminary Characterization of a Ni2+-Activated and Mycothiol-Dependent Glyoxalase I Enzyme from Streptomyces coelicolor. Inorganics 2019, 7, 99. https://doi.org/10.3390/inorganics7080099
AMA Style
Suttisansanee U, Honek JF. Preliminary Characterization of a Ni2+-Activated and Mycothiol-Dependent Glyoxalase I Enzyme from Streptomyces coelicolor. Inorganics. 2019; 7(8):99. https://doi.org/10.3390/inorganics7080099
Chicago/Turabian StyleSuttisansanee, Uthaiwan, and John F. Honek. 2019. "Preliminary Characterization of a Ni2+-Activated and Mycothiol-Dependent Glyoxalase I Enzyme from Streptomyces coelicolor" Inorganics 7, no. 8: 99. https://doi.org/10.3390/inorganics7080099
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.