
A miRNA-Disease Association Identification Method Based on Reliable Negative Sample Selection and Improved Single-Hidden Layer Feedforward Neural Network

Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Disease Semantic Similarity
2.2.1. Disease Semantic Similarity Model 1
2.2.2. Disease Semantic Similarity Model 2
2.3. miRNA Functional Similarity
2.4. Gaussian Interaction Profile Kernel Similarity for Diseases
2.5. Gaussian Interaction Profile Kernel Similarity for miRNAs
2.6. Integrated Similarity for miRNAs and Diseases
2.7. RNSSLFN Prediction Model
- (i)
- when to add nodes
- (ii)
- how to update the learnable parameters of new nodes
- (iii)
- when to terminate node self-growth
3. Results
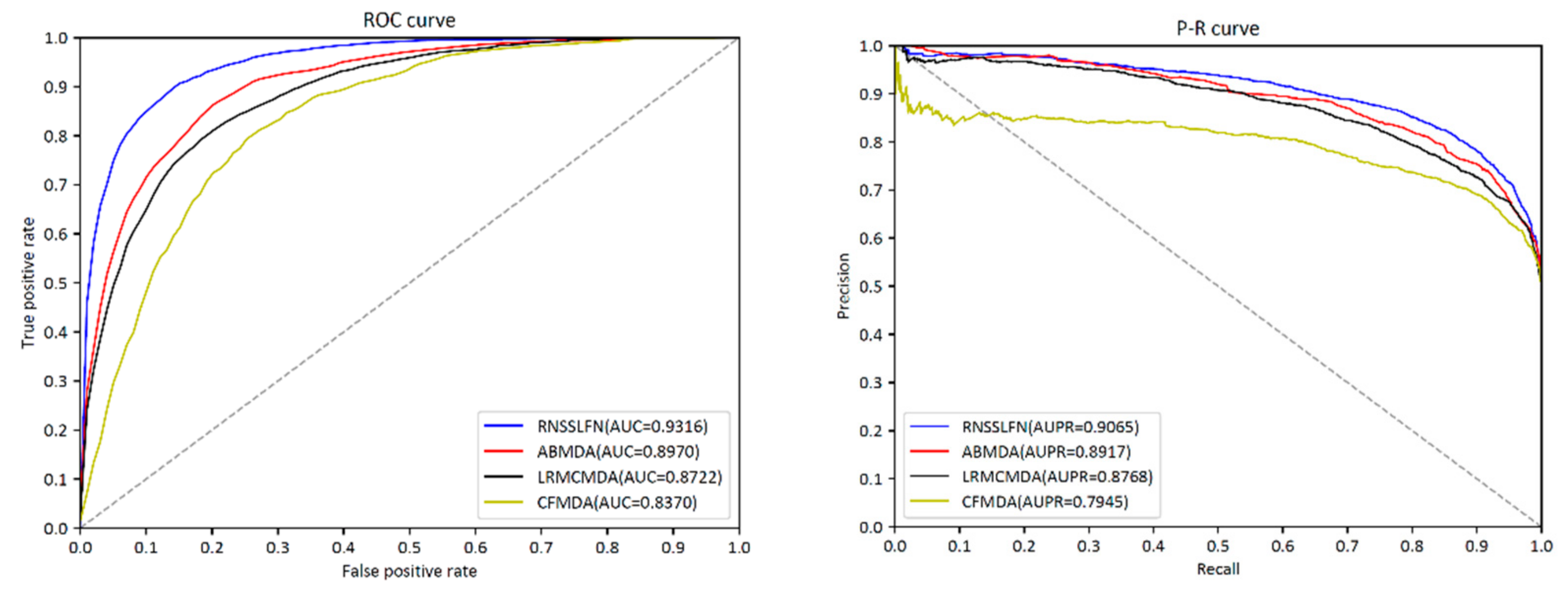
3.1. Performance Evaluation
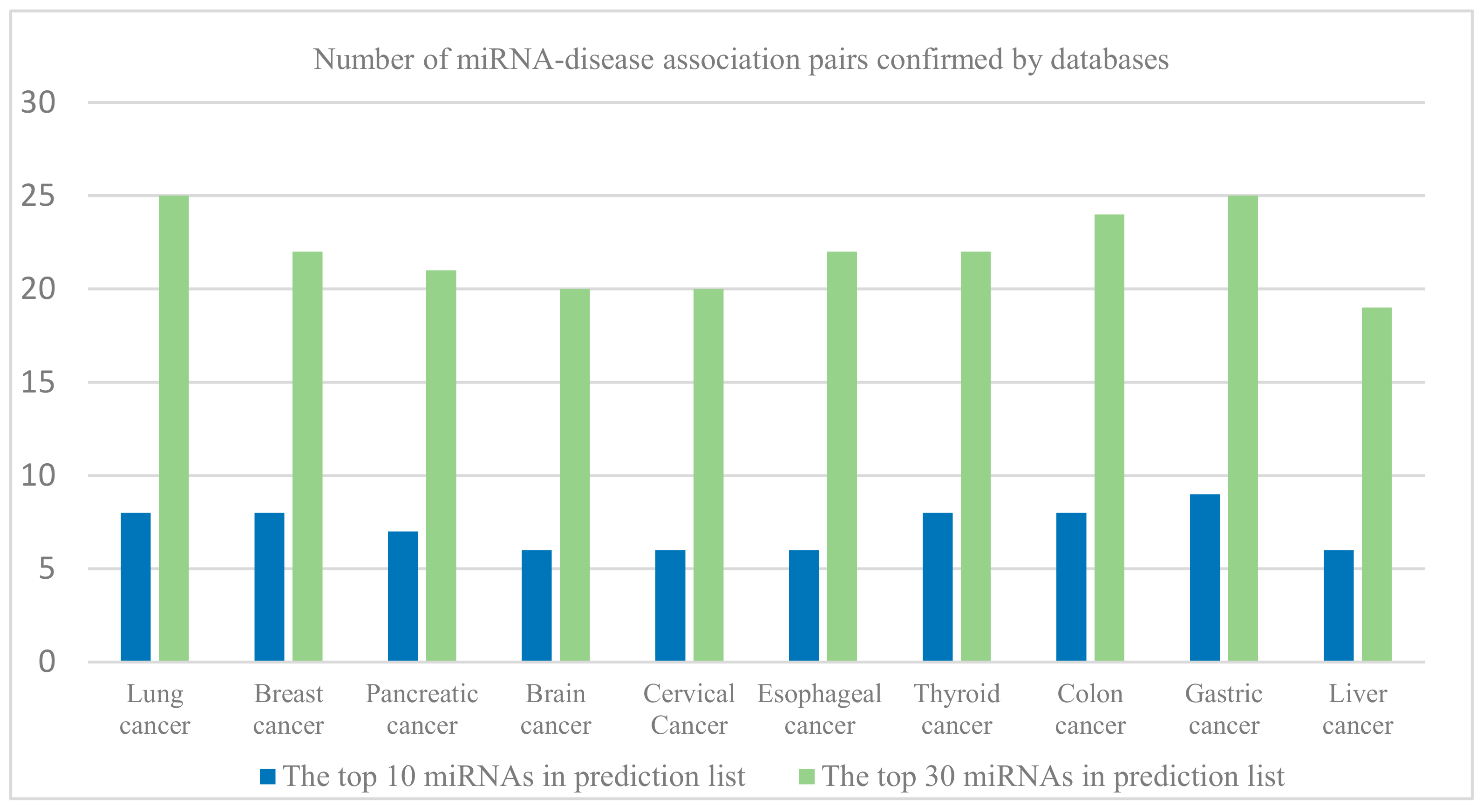
3.2. Case Studies
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Siomi, H.; Siomi, M.C. Posttranscriptional Regulation of MicroRNA Biogenesis in Animals. Mol. Cell 2010, 38, 323–332. [Google Scholar] [CrossRef]
- Sayed, D.; Abdellatif, M. MicroRNAs in Development and Disease. Physiol. Rev. 2011, 91, 827–887. [Google Scholar] [CrossRef]
- Chang, T.-C.; Mendell, J.T. microRNAs in Vertebrate Physiology and Human Disease. Annu. Rev. Genom. Hum. Genet. 2007, 8, 215–239. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Cai, L.; Liao, B.; Zhu, W.; Wang, P.; Meng, Y.; Lang, J.; Tian, G.; Yang, J. Identifying Potential miRNAs–Disease Associations With Probability Matrix Factorization. Front. Genet. 2019, 10, 1234. [Google Scholar] [CrossRef]
- Chen, X.; Yin, J.; Qu, J.; Huang, L.; Wang, E. MDHGI: Matrix Decomposition and Heterogeneous Graph Inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018, 8, e1006418. [Google Scholar] [CrossRef]
- Ha, J.; Park, C.; Park, C.; Park, S. Improved Prediction of miRNA-Disease Associations Based on Matrix Completion with Network Regularization. Cells 2020, 3, 881. [Google Scholar] [CrossRef] [Green Version]
- Ha, J.; Park, C. MLMD: Metric Learning for predicting miRNA-Disease associations. IEEE Access 2021, 5, 78847–78858. [Google Scholar] [CrossRef]
- Li, J.-Q.; Rong, Z.-H.; Chen, X.; Yan, G.-Y.; You, Z.-H. MCMDA: Matrix completion for miRNA-disease association prediction. Oncotarget 2017, 28, 21187–21199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.; Park, C.; Park, C.; Park, S. IMIPMF: Inferring miRNA-disease interactions using probabilistic matrix factorization. J Biomed Inform. 2020, 102, 103358. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Park, S.J. Big-ECG: Cardiographic Predictive Cyber-Physical System for Stroke Management. IEEE Access 2021, 9, 123146–123164. [Google Scholar] [CrossRef]
- Hussain, I.; Park, S.J. HealthSOS: Real-Time Health Monitoring System for Stroke Prognostics. IEEE Access 2020, 8, 213574–213586. [Google Scholar] [CrossRef]
- Perez-Iratxeta, C.; Wjst, M.; Bork, P.; Andrade, M.A. G2D: A tool for mining genes associated with disease. BMC Genet. 2005, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Ji, B.-Y.; You, Z.-H.; Cheng, L.; Zhou, J.-R.; Alghazzawi, D.; Li, L.-P. Predicting miRNA-disease association from heterogeneous information network with GraRep embedding model. Sci. Rep. 2020, 10, 6658. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhu, W.; Cai, L.; Liao, B.; Meng, Y.; Xiang, J.; Yuan, D.; Tian, G.; Yang, J. LRMCMDA: Predicting miRNA-Disease Association by Integrating Low-Rank Matrix Completion With miRNA and Disease Similarity Information. IEEE Access 2020, 8, 80728–80738. [Google Scholar] [CrossRef]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.-H.; Liu, H. BNPMDA: Bipartite Network Projection for MiRNA–Disease Association prediction. Bioinformatics 2018, 34, 3178–3186. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, B.; Yan, C. CFMDA: Collaborative filtering-based miRNA-disease association prediction. Multimed. Tools Appl. 2019, 78, 605–618. [Google Scholar] [CrossRef]
- Peng, L.-H.; Sun, C.-N.; Guan, N.-N.; Li, J.-Q.; Chen, X. HNMDA: Heterogeneous network-based miRNA–disease association prediction. Mol. Genet. Genom. 2018, 293, 983–995. [Google Scholar] [CrossRef]
- Luo, J.; Long, Y. NTSHMDA: Prediction of Human Microbe-Disease Association Based on Random Walk by Integrating Network Topological Similarity. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2018, 17, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Badillo, S.; Banfai, B.; Birzele, F.; Davydov, I.I.; Hutchinson, L.; Kam-Thong, T.; Siebourg-Polster, J.; Steiert, B.; Zhang, J.D. An Introduction to Machine Learning. Clin. Pharmacol. Ther. 2020, 107, 871–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yin, J.; Zhang, X. A Semi-Supervised Learning Algorithm for Predicting Four Types miRNA-Disease Associations by Mutual Information in a Heterogeneous Network. Genes 2018, 9, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, S.-L.; Zhang, J.-F.; Zhang, W.; Li, W. A neural collaborative filtering method for identifying miRNA-disease associations. Neurocomputing 2021, 422, 176–185. [Google Scholar] [CrossRef]
- Xuan; Zhang; Zhang; Li; Zhao Predicting miRNA-Disease Associations by Incorporating Projections in Low-Dimensional Space and Local Topological Information. Genes 2019, 10, 685. [CrossRef] [Green Version]
- Che, K.; Guo, M.; Wang, C.; Liu, X.; Chen, X. Predicting miRNA-Disease Association by Latent Feature Extraction with Positive Samples. Genes 2019, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, X.; Yin, J. Adaptive boosting-based computational model for predicting potential miRNA-disease associations. Bioinformatics 2019, 35, 4730–4738. [Google Scholar] [CrossRef]
- Meng, Y.; Jin, M.; Tang, X.; Xu, J. Degree-Based Similarity Indexes for Identifying Potential miRNA-Disease Associations. IEEE Access 2020, 8, 133170–133179. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.-F.; Yan, G.-Y. RKNNMDA: Ranking-based KNN for miRNA-Disease Association prediction. RNA Biol. 2017, 14, 952–962. [Google Scholar] [CrossRef] [Green Version]
- Xuan, P.; Han, K.; Guo, Y.; Li, J.; Li, X.; Zhong, Y.; Zhang, Z.; Ding, J. Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics 2015, 31, 1805–1815. [Google Scholar] [CrossRef]
- Chen, X.; Yan, G.-Y. Semi-supervised learning for potential human microRNA-disease associations inference. Sci. Rep. 2015, 4, 5501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Liu, B.; Li, Z.; Zhu, X.; Liang, Z.; An, J. Predicting miRNA-disease associations by multiple meta-paths fusion graph embedding model. BMC Bioinform. 2020, 21, 470. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014, 42, D1070–D1074. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Ding, Q.; Han, H.; Wu, D. miRCancer: A microRNA-cancer association database constructed by text mining on literature. Bioinformatics 2013, 29, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wu, L.; Wang, A.; Tang, W.; Zhao, Y.; Zhao, H.; Teschendorff, A.E. dbDEMC 2.0: Updated database of differentially expressed miRNAs in human cancers. Nucleic Acids Res. 2017, 45, D812–D818. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An Analysis of Human MicroRNA and Disease Associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA–disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Yue, X.; Xiong, Z.; Yu, Z.; Liu, S.; Zhang, W. Tensor decomposition with relational constraints for predicting multiple types of microRNA-disease associations. Brief. Bioinform. 2021, 22, bbaa140. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Wang, S.; Wu, Q.; Azim, R.; Li, W. Predicting potential miRNA-disease associations by combining gradient boosting decision tree with logistic regression. Comput. Biol. Chem. 2020, 85, 107200. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Han, K.; Guo, M.; Guo, Y.; Li, J.; Ding, J.; Liu, Y.; Dai, Q.; Li, J.; Teng, Z.; et al. Prediction of microRNAs Associated with Human Diseases Based on Weighted k Most Similar Neighbors. PLoS ONE 2013, 8, e70204. [Google Scholar] [CrossRef] [PubMed]
- van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, S.; Wan, Y.; Zhao, Y.; Shi, J.; Zhou, Y.; Cui, Q. MISIM v2.0: A web server for inferring microRNA functional similarity based on microRNA-disease associations. Nucleic Acids Res. 2019, 47, W536–W541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Zhang, M.; Li, Z.; Cao, Y.; He, B.; Feng, W. A Classfication Algorithm based on Self-organizing Neural Network Using Growing-Combination Structure. In Proceedings of the 2020 39th Chinese Control Conference (CCC), Shenyang, China, 27–29 July 2020. [Google Scholar]
- Sondermann, A.; Andreghetto, F.M.; Moulatlet, A.C.B.; da Silva Victor, E.; de Castro, M.G.; Nunes, F.D.; Brandão, L.G.; Severino, P. MiR-9 and miR-21 as prognostic biomarkers for recurrence in papillary thyroid cancer. Clin. Exp. Metastasis 2015, 32, 521–530. [Google Scholar] [CrossRef]
- Wang, P.; Meng, X.; Huang, Y.; Lv, Z.; Liu, J.; Wang, G.; Meng, W.; Xue, S.; Zhang, Q.; Zhang, P.; et al. MicroRNA-497 inhibits thyroid cancer tumor growth and invasion by suppressing BDNF. Oncotarget 2017, 8, 2825–2834. [Google Scholar] [CrossRef] [Green Version]
- Coulouarn, C.; Factor, V.M.; Andersen, J.B.; Durkin, M.E.; Thorgeirsson, S.S. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene 2009, 28, 3526–3536. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Wong, C.-M.; Ying, Q.; Fan, D.N.-Y.; Huang, S.; Ding, J.; Yao, J.; Yan, M.; Li, J.; Yao, M.; et al. MicroRNA-125b suppressesed human liver cancer cell proliferation and metastasis by directly targeting oncogene LIN28B2. Hepatology 2010, 52, 1731–1740. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| AUC | AUPR | Accuracy | Precision | Recall | F1-Score | |
|---|---|---|---|---|---|---|
| RNSSLFN | 0.9316 | 0.9065 | 0.8319 | 0.8238 | 0.8481 | 0.8358 |
| ABMDA | 0.8970 | 0.8917 | 0.8287 | 0.8160 | 0.7832 | 0.7993 |
| LRMCMDA | 0.8722 | 0.8768 | 0.8156 | 0.8105 | 0.8273 | 0.8219 |
| CFMDA | 0.8370 | 0.8014 | 0.7978 | 0.7443 | 0.7421 | 0.7434 |
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-137 | I, II | hsa-let-497 | I |
| hsa-mir-17 | I | hsa-mir-210 | unconfirmed |
| hsa-mir-18a | I | hsa-mir-214 | I, II |
| hsa-mir-16 | II | hsa-mir-223 | I |
| hsa-mir-150 | I | hsa-mir-29a | unconfirmed |
| hsa-mir-143 | II | hsa-mir-29c | I |
| hsa-mir-145 | unconfirmed | hsa-let-7a | II |
| hsa-mir-106b | unconfirmed | hsa-mir-15a | I, II |
| hsa-mir-19b | I | hsa-let-21 | I |
| hsa-mir-20a | I | hsa-mir-30a | I |
| hsa-let-7c | unconfirmed | hsa-let-9 | I, II |
| hsa-mir-142 | I, II | hsa-mir-101 | II |
| hsa-mir-29b | I | hsa-mir-34c | unconfirmed |
| hsa-mir-34a | unconfirmed | hsa-mir-195 | II |
| hsa-mir-199a | unconfirmed | hsa-mir-203 | I |
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-155 | I, II | hsa-mir-142 | unconfirmed |
| hsa-mir-145 | II | hsa-mir-15b | I |
| hsa-mir-19a | unconfirmed | hsa-mir-194 | I, II |
| hsa-mir-222 | I | hsa-mir-141 | unconfirmed |
| hsa-mir-16 | unconfirmed | hsa-mir-203 | II |
| hsa-mir-146a | unconfirmed | hsa-mir-101 | I, II |
| hsa-mir-126 | II | hsa-mir-107 | I |
| hsa-mir-17 | unconfirmed | hsa-mir-125b | I, II |
| hsa-mir-221 | I | hsa-mir-106a | unconfirmed |
| hsa-mir-200b | I | hsa-mir-132 | II |
| hsa-mir-20a | unconfirmed | hsa-mir-100 | unconfirmed |
| hsa-mir-122 | I, II | hsa-mir-375 | I, II |
| hsa-mir-1 | unconfirmed | hsa-mir-22 | II |
| hsa-mir-181a | I | hsa-mir-9 | unconfirmed |
| hsa-let-7c | I | hsa-mir-181b | I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Q.; Zhou, S.; Wu, Q. A miRNA-Disease Association Identification Method Based on Reliable Negative Sample Selection and Improved Single-Hidden Layer Feedforward Neural Network. Information 2022, 13, 108. https://doi.org/10.3390/info13030108
Tian Q, Zhou S, Wu Q. A miRNA-Disease Association Identification Method Based on Reliable Negative Sample Selection and Improved Single-Hidden Layer Feedforward Neural Network. Information. 2022; 13(3):108. https://doi.org/10.3390/info13030108
Chicago/Turabian StyleTian, Qinglong, Su Zhou, and Qi Wu. 2022. "A miRNA-Disease Association Identification Method Based on Reliable Negative Sample Selection and Improved Single-Hidden Layer Feedforward Neural Network" Information 13, no. 3: 108. https://doi.org/10.3390/info13030108