
Virus-Induced Epilepsy vs. Epilepsy Patients Acquiring Viral Infection: Unravelling the Complex Relationship for Precision Treatment


Abstract
:1. Introduction
2. Virus-Inducing Epilepsy
2.1. Mechanism Underlying Epileptogenesis and Neuroinflammation
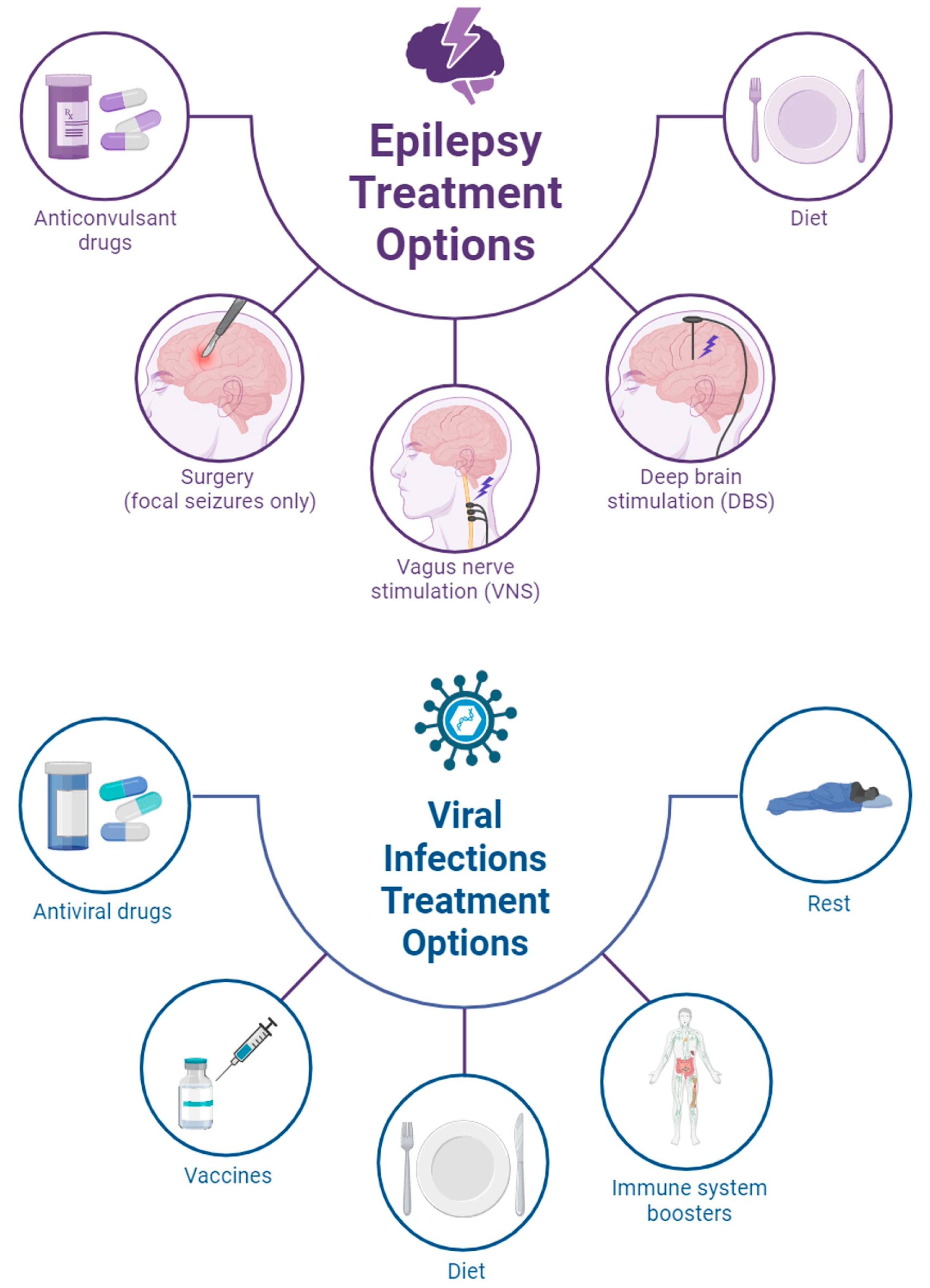
2.2. Strategies for Disease Management
3. Epilepsy Patients Acquiring Viral Infection
3.1. Mechanisms Underlying the Development of Epilepsy
3.2. Understanding the Susceptibility of Epilepsy Patients to Viral Infections
4. Pharmacokinetic Considerations in Epilepsy and Viral Infection
4.1. Drug Interactions
4.2. Treatment of Individuals with Epilepsy Who Acquired a Viral Infection Versus Those with Virus-Induced Epilepsy
4.3. Modeling of Pharmacokinetics
5. Interactions at the Neuroinflammatory Level
5.1. Molecular and Cellular Mechanisms of Neuroinflammation
5.2. Identification of Biomarkers
5.3. Development of Therapeutic Targets to Modulate Neuroinflammation
6. Clinical Implications
7. Future Directions and Precision Therapies
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Theodore, W.H. Epilepsy and Viral Infections. Epilepsy Curr. 2014, 14, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Bonello, M.; Michael, B.D.; Solomon, T. Infective Causes of Epilepsy. Semin. Neurol. 2015, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yang, Y.; Zou, J.; Yang, X.; Liu, Q.; Chen, Y. Seizures and Epilepsy Secondary to Viral Infection in the Central Nervous System. Acta Epileptol. 2020, 2, 12. [Google Scholar] [CrossRef]
- Singhi, P. Infectious Causes of Seizures and Epilepsy in the Developing World. Dev. Med. Child. Neurol. 2011, 53, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Parihar, J.; Tripathi, M.; Dhamija, R.K. Seizures and Epilepsy in Times of Corona Virus Disease 2019 Pandemic. J. Epilepsy Res. 2020, 10, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Basta-Kaim, A.; Budziszewska, B.; Lasoń, W. Effects of Antiepileptic Drugs on Immune System. Prz. Lek. 2008, 65, 799–802. [Google Scholar]
- Asadi-Pooya, A.A.; Emami, A.; Akbari, A.; Javanmardi, F. COVID-19 Presentations and Outcome in Patients with Epilepsy. Acta Neurol. Scand. 2021, 143, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Lang, B.; Aronica, E. Immunity and Inflammation in Epilepsy. Cold Spring Harb. Perspect. Med. 2016, 6, a022699. [Google Scholar] [CrossRef] [PubMed]
- Cherian, A.; Thomas, S. Status Epilepticus. Ann. Indian Acad. Neurol. 2009, 12, 140. [Google Scholar] [CrossRef]
- World Health Organiaztion. Epilepsy. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 7 December 2023).
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE Official Report: A Practical Clinical Definition of Epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.R.; Balcar, V.J.; Matsumoto, I.; Müller, M.; King, N.J.C. Viruses and the Immune System: Their Roles in Seizure Cascade Development. J. Neurochem. 2008, 104, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; Fujinami, R.S. Neurotropic Viral Infections Leading to Epilepsy: Focus on Theiler’s Murine Encephalomyelitis Virus. Future Virol. 2011, 6, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Nunez, N.V.; Gaudin, R. A Viral Journey to the Brain: Current Considerations and Future Developments. PLoS Pathog. 2020, 16, e1008434. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Michael, B.D.; Smith, P.E.; Sanderson, F.; Davies, N.W.S.; Hart, I.J.; Holland, M.; Easton, A.; Buckley, C.; Kneen, R.; et al. Management of Suspected Viral Encephalitis in Adults—Association of British Neurologists and British Infection Association National Guidelines. J. Infect. 2012, 64, 347–373. [Google Scholar] [CrossRef]
- Rocamonde, B.; Hasan, U.; Mathieu, C.; Dutartre, H. Viral-Induced Neuroinflammation: Different Mechanisms Converging to Similar Exacerbated Glial Responses. Front. Neurosci. 2023, 17, 1108212. [Google Scholar] [CrossRef] [PubMed]
- Kielian, T. Neuroinflammation: Good, Bad, or Indifferent? J. Neurochem. 2014, 130, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Fujinami, R.S.; White, H.S.; Preux, P.-M.; Blümcke, I.; Sander, J.W.; Löscher, W. Infections, Inflammation and Epilepsy. Acta Neuropathol. 2016, 131, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Swanson, P.A.; McGavern, D.B. Viral Diseases of the Central Nervous System. Curr. Opin. Virol. 2015, 11, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Ghaderi, S.; Olfati, M.; Ghaderi, M.; Hadizadeh, H.; Yazdanpanah, G.; Khodadadi, Z.; Karami, A.; Papi, Z.; Abdi, N.; Sharif Jalali, S.S.; et al. Neurological Manifestation in COVID-19 Disease with Neuroimaging Studies. Am. J. Neurodegener. Dis. 2023, 12, 42–84. [Google Scholar] [PubMed]
- Taquet, M.; Sillett, R.; Zhu, L.; Mendel, J.; Camplisson, I.; Dercon, Q.; Harrison, P.J. Neurological and Psychiatric Risk Trajectories after SARS-CoV-2 Infection: An Analysis of 2-Year Retrospective Cohort Studies Including 1 284 437 Patients. Lancet Psychiatry 2022, 9, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Taquet, M.; Geddes, J.R.; Husain, M.; Luciano, S.; Harrison, P.J. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: A retrospective cohort study using electronic health records. Lancet Psychiatry 2021, 8, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Nagib, M.M.; Yasmen, N.; Sluter, M.N.; Littlejohn, T.L.; Yu, Y.; Jiang, J. Neuroinflammatory Mediators in Acquired Epilepsy: An Update. Inflamm. Res. 2023, 72, 683–701. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.M.; Sun, M.; Crack, P.; O’Brien, T.J.; Shultz, S.R.; Semple, B.D. Inflammation in Epileptogenesis after Traumatic Brain Injury. J. Neuroinflamm. 2017, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Suleymanova, E.M. Behavioral Comorbidities of Epilepsy and Neuroinflammation: Evidence from Experimental and Clinical Studies. Epilepsy Behav. 2021, 117, 107869. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shan, W.; Yang, H.; Liu, R.; Wu, J.; Wang, Q. The Role of Neuroinflammation in Post-Traumatic Epilepsy. Front. Neurol. 2021, 12, 646152. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef] [PubMed]
- Bazhanova, E.D.; Kozlov, A.A.; Litovchenko, A.V. Mechanisms of Drug Resistance in the Pathogenesis of Epilepsy: Role of Neuroinflammation. A Literature Review. Brain Sci. 2021, 11, 663. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, G.; Giovannelli, F.; Giorgi, F.S.; Franco, V.; Gasparini, S.; Tacconi, F.M. Do Antiepileptic Drugs Increase the Risk of Infectious Diseases? A Meta-analysis of Placebo-controlled Studies. Br. J. Clin. Pharmacol. 2017, 83, 1873–1879. [Google Scholar] [CrossRef] [PubMed]
- Perucca, E. Clinically Relevant Drug Interactions with Antiepileptic Drugs. Br. J. Clin. Pharmacol. 2006, 61, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Howe, C.L. Molecular Mechanisms in the Genesis of Seizures and Epilepsy Associated with Viral Infection. Front. Mol. Neurosci. 2022, 15, 870868. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, M.D.; Lockhart, S.M.; Rathbun, R.C. Anticonvulsant and Antiretroviral Interactions. Ann. Pharmacother. 2004, 38, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, S.I.; Johannessen Landmark, C. Antiepileptic Drug Interactions—Principles and Clinical Implications. Curr. Neuropharmacol. 2010, 8, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Taburet, A.-M.; Singlas, E. Drug Interactions with Antiviral Drugs. Clin. Pharmacokinet. 1996, 30, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.J. Adverse Effects and Drug Interactions of Clinical Importance with Antiviral Drugs. Drug Saf. 1994, 10, 281–291. [Google Scholar] [CrossRef] [PubMed]
- DePaula-Silva, A.B.; Bell, L.A.; Wallis, G.J.; Wilcox, K.S. Inflammation Unleashed in Viral-Induced Epileptogenesis. Epilepsy Curr. 2021, 21, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Ghadimi, K.; Heidari, Z.; Kheradmand, M.; Najafi, M.A.; Chitsaz, A.; Khorvash, F.; Fahim, M.; Najafi, M.R. Prevalence, Clinical, Imaging, Electroencephalography and Laboratory Characteristics of Seizures in COVID-19. Am. J. Neurodegener. Dis. 2022, 11, 46–54. [Google Scholar] [PubMed]
- Ssentongo, P. Prevalence and Incidence of New-Onset Seizures and Epilepsy in Patients with Human Immunodeficiency Virus (HIV): Systematic Review and Meta-Analysis. Epilepsy Behav. 2019, 93, 49–55. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, W. Clinical Features and Surgical Treatment of Epilepsy after Viral Encephalitis. Brain Sci. Adv. 2019, 5, 41–50. [Google Scholar] [CrossRef]
- Huang, L.; Yu, D.; Luo, R.; Li, M.; Zhou, H.; Cai, X.-T.; Wang, Z.-L. Risk Factors and Prognosis of Secondary Epilepsy in Children with Viral Encephalitis. Sichuan Da Xue Xue Bao Yi Xue Ban 2017, 48, 257–262. [Google Scholar] [PubMed]
- Fiest, K.M.; Sauro, K.M.; Wiebe, S.; Patten, S.B.; Kwon, C.-S.; Dykeman, J.; Pringsheim, T.; Lorenzetti, D.L.; Jetté, N. Prevalence and Incidence of Epilepsy. Neurology 2017, 88, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Imai, Y.; Ishii, W.; Endo, A.; Arakawa, C.; Kohira, R.; Fuchigami, T.; Okubo, O.; Mugishima, H. Improvement of Intractable Childhood Epilepsy Following Acute Viral Infection. Brain Dev. 2011, 33, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Gifreu, A.; Falip, M.; Sala-Padró, J.; Mongay, N.; Morandeira, F.; Camins, Á.; Naval-Baudin, P.; Veciana, M.; Fernández, M.; Pedro, J.; et al. Risk of Developing Epilepsy after Autoimmune Encephalitis. Brain Sci. 2021, 11, 1182. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Schmidt, V.; Kaminski, M.; Stelzle, D.; De Meijere, R.; Bustos, J.; Sahu, P.S.; Garcia, H.H.; Bobić, B.; Cretu, C.; et al. Epidemiology and Surveillance of Human (Neuro)Cysticercosis in Europe: Is Enhanced Surveillance Required? Trop. Med. Int. Health 2020, 25, 566–578. [Google Scholar] [CrossRef]
- Steyn, T.J.S.; Awala, A.N.; de Lange, A.; Raimondo, J.V. What Causes Seizures in Neurocysticercosis? Epilepsy Curr. 2023, 23, 105–112. [Google Scholar] [CrossRef] [PubMed]
- De Lange, A.; Mahanty, S.; Raimondo, J.V. Model Systems for Investigating Disease Processes in Neurocysticercosis. Parasitology 2019, 146, 553–562. [Google Scholar] [CrossRef]
- Idro, R.; Marsh, K.; John, C.C.; Newton, C.R.J. Cerebral Malaria: Mechanisms of Brain Injury and Strategies for Improved Neurocognitive Outcome. Pediatr. Res. 2010, 68, 267–274. [Google Scholar] [CrossRef]
- Song, X.; Wei, W.; Cheng, W.; Zhu, H.; Wang, W.; Dong, H.; Li, J. Cerebral Malaria Induced by Plasmodium Falciparum: Clinical Features, Pathogenesis, Diagnosis, and Treatment. Front. Cell. Infect. Microbiol. 2022, 12, 939532. [Google Scholar] [CrossRef] [PubMed]
- Jaan, A.; Rajnik, M. TORCH Complex; StatPearls: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Barañano, K.; Burd, I. CNS Malformations in the Newborn. Matern. Health Neonatol. Perinatol. 2022, 8, 1. [Google Scholar] [CrossRef]
- Murthy, J.M.K.; Prabhakar, S. Bacterial Meningitis and Epilepsy. Epilepsia 2008, 49, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Yacubian, E.M.T.; Kakooza-Mwesige, A.; Singh, G.; Carpio, A.; de Figueiredo, N.V.; Lutzky Saute, R.; Marques de Haes, T. Common infectious and parasitic diseases as a cause of seizures: Geographic distribution and contribution to the burden of epilepsy. Epileptic Disord. 2022, 24, 994–1019. [Google Scholar] [CrossRef] [PubMed]
- Taquet, M.; Devinsky, O.; Cross, J.H.; Harrison, P.J.; Sen, A. Incidence of Epilepsy and Seizures over the First 6 Months after a COVID-19 Diagnosis. Neurology 2023, 100, e790–e799. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, S.; Yan, C.; Liu, X. Interaction between COVID-19 and Epilepsy during the Omicron Surge: A Cross-Sectional Survey Conducted in China Tertiary Hospital. Epilepsy Behav. Rep. 2023, 23, 100613. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, L.; Libbey, J.E.; Ravizza, T.; Fujinami, R.S.; Jacobson, S.; Gaillard, W.D. Viral Triggers and Inflammatory Mechanisms in Pediatric Epilepsy. Mol. Neurobiol. 2019, 56, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Lu, Y.-T.; Ho, C.-J.; Shih, F.-Y.; Tsai, M.-H. The Different Clinical Features between Autoimmune and Infectious Status Epilepticus. Front. Neurol. 2019, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, T.; Awaya, Y.; Matsuishi, T.; Nagai, T.; Yamamoto, K.; Kurihara, M.; Iyoda, K.; Maekawa, K. Survey of Vaccination and Viral Infections for Children with Severe Myoclonic Epilepsy in Infancy. No Hattatsu Brain Dev. 2004, 36, 318–323. [Google Scholar]
- Tro-Baumann, B.; von Spiczak, S.; Lotte, J.; Bast, T.; Haberlandt, E.; Sassen, R.; Freund, A.; Leiz, S.; Stephani, U.; Boor, R.; et al. A Retrospective Study of the Relation between Vaccination and Occurrence of Seizures in Dravet Syndrome. Epilepsia 2011, 52, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Auvin, S.; Jeljeli, M.; Desnous, B.; Soussi-Yanicostas, N.; Dournaud, P.; Sterkers, G. Altered Vaccine-induced Immunity in Children with Dravet Syndrome. Epilepsia 2018, 59, e45–e50. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, N.E.; van der Maas, N.A.T.; Sonsma, A.C.M.; Ippel, E.; Vermeer-de Bondt, P.E.; Hagebeuk, E.; Jansen, F.E.; Geesink, H.H.; Braun, K.P.; de Louw, A.; et al. Effect of Vaccinations on Seizure Risk and Disease Course in Dravet Syndrome. Neurology 2015, 85, 596–603. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, A.M.; McMahon, J.; Dibbens, L.M.; Iona, X.; Mulley, J.C.; Scheffer, I.E.; Berkovic, S.F. Effects of Vaccination on Onset and Outcome of Dravet Syndrome: A Retrospective Study. Lancet Neurol. 2010, 9, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; Hogue, I.B.; Enquist, L.W. Virus Infections in the Nervous System. Cell Host Microbe 2013, 13, 379–393. [Google Scholar] [CrossRef]
- Vezzani, A.; Aronica, E.; Mazarati, A.; Pittman, Q.J. Epilepsy and Brain Inflammation. Exp. Neurol. 2013, 244, 11–21. [Google Scholar] [CrossRef]
- Waltl, I.; Käufer, C.; Gerhauser, I.; Chhatbar, C.; Ghita, L.; Kalinke, U.; Löscher, W. Microglia Have a Protective Role in Viral Encephalitis-Induced Seizure Development and Hippocampal Damage. Brain Behav. Immun. 2018, 74, 186–204. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Patel, D.C.; Doty, D.J.; Fujinami, R.S. Infiltrating Macrophages Are Key to the Development of Seizures Following Virus Infection. J. Virol. 2013, 87, 1849–1860. [Google Scholar] [CrossRef]
- Aronica, E.; Ravizza, T.; Zurolo, E.; Vezzani, A. Astrocyte Immune Responses in Epilepsy. Glia 2012, 60, 1258–1268. [Google Scholar] [CrossRef]
- Vishwakarma, S.; Singh, S.; Singh, T.G. Pharmacological Modulation of Cytokines Correlating Neuroinflammatory Cascades in Epileptogenesis. Mol. Biol. Rep. 2022, 49, 1437–1452. [Google Scholar] [CrossRef]
- Wojtowicz, J.M. Potential consequences of altered neurogenesis on learning and memory in the epileptic brain. Epilepsia 2008, 49 (Suppl. S5), 42–49. [Google Scholar] [CrossRef] [PubMed]
- Dohgu, S.; Banks, W.A. Brain Pericytes Increase the Lipopolysaccharide-Enhanced Transcytosis of HIV-1 Free Virus across the in vitro Blood–Brain Barrier: Evidence for Cytokine-Mediated Pericyte-Endothelial Cell Crosstalk. Fluids Barriers CNS 2013, 10, 23. [Google Scholar] [CrossRef]
- Williams, R.; Dhillon, N.K.; Hegde, S.T.; Yao, H.; Peng, F.; Callen, S.; Chebloune, Y.; Davis, R.L.; Buch, S.J. Proinflammatory Cytokines and HIV-1 Synergistically Enhance CXCL10 Expression in Human Astrocytes. Glia 2009, 57, 734–743. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Michels, M.; Steckert, A.V.; Quevedo, J.; Barichello, T.; Dal-Pizzol, F. Mechanisms of Long-Term Cognitive Dysfunction of Sepsis: From Blood-Borne Leukocytes to Glial Cells. Intensive Care Med. Exp. 2015, 3, 30. [Google Scholar] [CrossRef]
- Scimemi, A.; Schorge, S.; Kullmann, D.M.; Walker, M.C. Epileptogenesis Is Associated with Enhanced Glutamatergic TransmissThus ion in the Perforant Path. J. Neurophysiol. 2006, 95, 1213–1220. [Google Scholar] [CrossRef]
- Meldrum, B.S. The Role of Glutamate in Epilepsy and Other CNS Disorders. Neurology 1994, 44, S14–S23. [Google Scholar]
- Barker-Haliski, M.; White, H.S. Glutamatergic Mechanisms Associated with Seizures and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022863. [Google Scholar] [CrossRef]
- Zhao, W.; Chuang, S.-C.; Young, S.R.; Bianchi, R.; Wong, R.K.S. Extracellular Glutamate Exposure Facilitates Group I MGluR-Mediated Epileptogenesis in the Hippocampus. J. Neurosci. 2015, 35, 308–315. [Google Scholar] [CrossRef]
- Wouk, J.; Rechenchoski, D.Z.; Rodrigues, B.C.D.; Ribelato, E.V.; Faccin-Galhardi, L.C. Viral Infections and Their Relationship to Neurological Disorders. Arch. Virol. 2021, 166, 733–753. [Google Scholar] [CrossRef]
- Kaul, M.; Zheng, J.; Okamoto, S.; Gendelman, H.E.; Lipton, S.A. HIV-1 Infection and AIDS: Consequences for the Central Nervous System. Cell Death Differ. 2005, 12, 878–892. [Google Scholar] [CrossRef]
- Everall, I.P.; Luthert, P.J.; Lantos, P.L. Neuronal Loss in the Frontal Cortex in HIV Infection. Lancet 1991, 337, 1119–1121. [Google Scholar] [CrossRef]
- Shirbhate, E.; Pandey, J.; Patel, V.K.; Kamal, M.; Jawaid, T.; Gorain, B.; Kesharwani, P.; Rajak, H. Understanding the Role of ACE-2 Receptor in Pathogenesis of COVID-19 Disease: A Potential Approach for Therapeutic Intervention. Pharmacol. Rep. 2021, 73, 1539–1550. [Google Scholar] [CrossRef]
- Desforges, M.; Le Coupanec, A.; Brison, É.; Meessen-Pinard, M.; Talbot, P.J. Neuroinvasive and Neurotropic Human Respiratory Coronaviruses: Potential Neurovirulent Agents in Humans. In Proceedings of the Infectious Diseases and Nanomedicine I: First International Conference (ICIDN–2012), Kathmandu, Nepal, 15–18 December 2012; Springer: New Delhi, India, 2014; pp. 75–96. [Google Scholar]
- Ohsawa, K.; Watanabe, Y.; Miyata, H.; Sato, H. Genetic Analysis of a Theiler-like Virus Isolated from Rats. Comp. Med. 2003, 53, 191–196. [Google Scholar]
- DePaula-Silva, A.B.; Hanak, T.J.; Libbey, J.E.; Fujinami, R.S. Theiler’s Murine Encephalomyelitis Virus Infection of SJL/J and C57BL/6J Mice: Models for Multiple Sclerosis and Epilepsy. J. Neuroimmunol. 2017, 308, 30–42. [Google Scholar] [CrossRef]
- Stewart, K.-A.A.; Wilcox, K.S.; Fujinami, R.S.; White, H.S. Development of Postinfection Epilepsy after Theiler’s Virus Infection of C57BL/6 Mice. J. Neuropathol. Exp. Neurol. 2010, 69, 1210–1219. [Google Scholar] [CrossRef]
- Wang, H.; Song, G.; Chuang, H.; Chiu, C.; Abdelmaksoud, A.; Ye, Y.; Zhao, L. Portrait of Glial Scar in Neurological Diseases. Int. J. Immunopathol. Pharmacol. 2018, 31, 205873841880140. [Google Scholar] [CrossRef]
- Appelbaum, L.G.; Shenasa, M.A.; Stolz, L.; Daskalakis, Z. Synaptic Plasticity and Mental Health: Methods, Challenges and Opportunities. Neuropsychopharmacology 2023, 48, 113–120. [Google Scholar] [CrossRef]
- Squire, L.R. The Legacy of Patient H.M. for Neuroscience. Neuron 2009, 61, 6–9. [Google Scholar] [CrossRef]
- Snyder, A. Explaining and Inducing Savant Skills: Privileged Access to Lower Level, Less-Processed Information. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 1399–1405. [Google Scholar] [CrossRef]
- Di Filippo, M.; Sarchielli, P.; Picconi, B.; Calabresi, P. Neuroinflammation and Synaptic Plasticity: Theoretical Basis for a Novel, Immune-Centred, Therapeutic Approach to Neurological Disorders. Trends Pharmacol. Sci. 2008, 29, 402–412. [Google Scholar] [CrossRef]
- Soares, B.P.; Provenzale, J.M. Imaging of Herpesvirus Infections of the CNS. Am. J. Roentgenol. 2016, 206, 39–48. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Schaefer, D.C.; Rose, J.W.; Stroop, W.G. Neuropathology of Herpes Simplex Virus Encephalitis in a Rat Seizure Model. J. Neuropathol. Exp. Neurol. 1993, 52, 241–252. [Google Scholar] [CrossRef]
- Cotter, C.R.; Kim, W.; Nguyen, M.L.; Yount, J.S.; López, C.B.; Blaho, J.A.; Moran, T.M. The Virion Host Shutoff Protein of Herpes Simplex Virus 1 Blocks the Replication-Independent Activation of NF-ΚB in Dendritic Cells in the Absence of Type I Interferon Signaling. J. Virol. 2011, 85, 12662–12672. [Google Scholar] [CrossRef]
- Lippi, G.; Cervellin, G. Updates on Rabies Virus Disease: Is Evolution toward “Zombie Virus” a Tangible Threat? Acta Biomed. 2021, 92, e2021045. [Google Scholar] [CrossRef]
- Lafon, M.; Mégret, F.; Meuth, S.G.; Simon, O.; Velandia Romero, M.L.; Lafage, M.; Chen, L.; Alexopoulou, L.; Flavell, R.A.; Prehaud, C.; et al. Detrimental Contribution of the Immuno-Inhibitor B7-H1 to Rabies Virus Encephalitis. J. Immunol. 2008, 180, 7506–7515. [Google Scholar] [CrossRef] [PubMed]
- Lafon, M.; Megret, F.; Lafage, M.; Prehaud, C. The Innate Immune Facet of Brain: Human Neurons Express TLR-3 and Sense Viral DsRNA. J. Mol. Neurosci. 2006, 29, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Baloul, L.; Camelo, S.; Lafon, M. Up-Regulation of Fas Ligand (FasL) in the Central Nervous System: A Mechanism of Immune Evasion by Rabies Virus. J. Neurovirol 2004, 10, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Fekete, R.; Cserép, C.; Lénárt, N.; Tóth, K.; Orsolits, B.; Martinecz, B.; Méhes, E.; Szabó, B.; Németh, V.; Gönci, B.; et al. Microglia Control the Spread of Neurotropic Virus Infection via P2Y12 Signalling and Recruit Monocytes through P2Y12-Independent Mechanisms. Acta Neuropathol. 2018, 136, 461–482. [Google Scholar] [CrossRef] [PubMed]
- Agostini, S.; Mancuso, R.; Baglio, F.; Cabinio, M.; Hernis, A.; Costa, A.S.; Calabrese, E.; Nemni, R.; Clerici, M. High Avidity HSV-1 Antibodies Correlate with Absence of Amnestic Mild Cognitive Impairment Conversion to Alzheimer’s Disease. Brain Behav. Immun. 2016, 58, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Butowt, R.; von Bartheld, C.S. The Route of SARS-CoV-2 to Brain Infection: Have We Been Barking up the Wrong Tree? Mol. Neurodegener. 2022, 17, 20. [Google Scholar] [CrossRef]
- Cavallieri, F.; Sellner, J.; Zedde, M.; Moro, E. Neurologic Complications of Coronavirus and Other Respiratory Viral Infections. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2022; pp. 331–358. [Google Scholar]
- Brola, W.; Wilski, M. Neurological Consequences of COVID-19. Pharmacol. Rep. 2022, 74, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.I.; Martynov, M.Y.; Boyko, A.N.; Voznyuk, I.A.; Latsh, N.Y.; Sivertseva, S.A.; Spirin, N.N.; Shamalov, N.A. The Novel Coronavirus Infection (COVID-19) and Nervous System Involvement: Mechanisms of Neurological Disorders, Clinical Manifestations, and the Organization of Neurological Care. Neurosci. Behav. Physiol. 2021, 51, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Dixit, A.B.; Banerjee, J.; Tripathi, M.; Sarat Chandra, P. Role of Inflammation and Its MiRNA Based Regulation in Epilepsy: Implications for Therapy. Clin. Chim. Acta 2016, 452, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Granata, T. Brain Inflammation in Epilepsy: Experimental and Clinical Evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Marchi, N.; Granata, T.; Freri, E.; Ciusani, E.; Ragona, F.; Puvenna, V.; Teng, Q.; Alexopolous, A.; Janigro, D. Efficacy of Anti-Inflammatory Therapy in a Model of Acute Seizures and in a Population of Pediatric Drug Resistant Epileptics. PLoS ONE 2011, 6, e18200. [Google Scholar] [CrossRef] [PubMed]
- Perucca, P.; Scheffer, I.E.; Kiley, M. The Management of Epilepsy in Children and Adults. Med. J. Aust. 2018, 208, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Von der Brelie, C.; Malter, M.P.; Niehusmann, P.; Elger, C.E.; von Lehe, M.; Schramm, J. Surgical Management and Long-term Seizure Outcome after Epilepsy Surgery for Different Types of Epilepsy Associated with Cerebral Cavernous Malformations. Epilepsia 2013, 54, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Beal, J.C.; Grinspan, Z.M. Automated Identification of Surgical Candidates and Estimation of Postoperative Seizure Freedom in Children—A Focused Review. Semin. Pediatr. Neurol. 2021, 39, 100914. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Liu, S.; Qin, H.; Li, B.; Zhang, X. Drug-Resistant Epilepsy and Surgery. Curr. Neuropharmacol. 2017, 16, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Anwar, H.; Khan, Q.U.; Nadeem, N.; Pervaiz, I.; Ali, M.; Cheema, F.F. Epileptic Seizures. Discoveries 2020, 8, e110. [Google Scholar] [CrossRef]
- Reid, C.A.; Berkovic, S.F.; Petrou, S. Mechanisms of Human Inherited Epilepsies. Prog. Neurobiol. 2009, 87, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Mulley, J.C.; Scheffer, I.E.; Petrou, S.; Berkovic, S.F. Channelopathies as a Genetic Cause of Epilepsy. Curr. Opin. Neurol. 2003, 16, 171–176. [Google Scholar] [CrossRef]
- Pandolfo, M. Genetics of Epilepsy. Semin. Neurol. 2011, 31, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, D.; Godet, B.; Druet-Cabanac, M.; Preux, P.-M. Etiologies of Epilepsy: A Comprehensive Review. Expert. Rev. Neurother. 2011, 11, 861–876. [Google Scholar] [CrossRef] [PubMed]
- Jóźwiak, S.; Kasprzyk-Obara, J.; Domańska-Pakieła, D. Phacomatoses: Structural Substare of Epilepsy. Neurol. Neurochir. Pol. 2000, 34 (Suppl. S1), 243–251. [Google Scholar] [PubMed]
- Shields, C.L.; Shields, J.A. Phakomatoses. In Retina; Elsevier: Amsterdam, The Netherlands, 2013; pp. 2170–2183. [Google Scholar]
- Stafstrom, C.E.; Staedtke, V.; Comi, A.M. Epilepsy Mechanisms in Neurocutaneous Disorders: Tuberous Sclerosis Complex, Neurofibromatosis Type 1, and Sturge–Weber Syndrome. Front. Neurol. 2017, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Northrup, H.; Aronow, M.E.; Bebin, E.M.; Bissler, J.; Darling, T.N.; de Vries, P.J.; Frost, M.D.; Fuchs, Z.; Gosnell, E.S.; Gupta, N.; et al. Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatr. Neurol. 2021, 123, 50–66. [Google Scholar] [CrossRef]
- Feliciano, D.M. The Neurodevelopmental Pathogenesis of Tuberous Sclerosis Complex (TSC). Front. Neuroanat. 2020, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M.D.; Tang, H.; Gallione, C.J.; Baugher, J.D.; Frelin, L.P.; Cohen, B.; North, P.E.; Marchuk, D.A.; Comi, A.M.; Pevsner, J. Sturge–Weber Syndrome and Port-Wine Stains Caused by Somatic Mutation in GNAQ. N. Engl. J. Med. 2013, 368, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Sudarsanam, A.; Ardern-Holmes, S.L. Sturge–Weber Syndrome: From the Past to the Present. Eur. J. Paediatr. Neurol. 2014, 18, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Pan, J.; Zhao, M.; Wang, X.; Zhang, C.; Li, T.; Wang, M.; Wang, J.; Zhou, J.; Liu, C.; et al. Characteristics, Surgical Outcomes, and Influential Factors of Epilepsy in Sturge-Weber Syndrome. Brain 2022, 145, 3431–3443. [Google Scholar] [CrossRef] [PubMed]
- Fortman, B.J.; Kuszyk, B.S.; Urban, B.A.; Fishman, E.K. Neurofibromatosis Type 1: A Diagnostic Mimicker at CT. RadioGraphics 2001, 21, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Yohay, K.H. The Genetic and Molecular Pathogenesis of NF1 and NF2. Semin. Pediatr. Neurol. 2006, 13, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, S.; Wójcik, M.; Harat, M.; Furtak, J. Influence of Epilepsy on the Quality of Life of Patients with Brain Tumors. Int. J. Environ. Res. Public Health 2021, 18, 6390. [Google Scholar] [CrossRef] [PubMed]
- Englot, D.J.; Chang, E.F.; Vecht, C.J. Epilepsy and Brain Tumors. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 267–285. [Google Scholar]
- Stafstrom, C.E.; Carmant, L. Seizures and Epilepsy: An Overview for Neuroscientists. Cold Spring Harb. Perspect. Med. 2015, 5, a022426. [Google Scholar] [CrossRef] [PubMed]
- Mody, I. Ion Channels in Epilepsy. Int. Rev. Neurobiol. 1998, 42, 199–226. [Google Scholar] [PubMed]
- Rana, A.; Musto, A.E. The Role of Inflammation in the Development of Epilepsy. J. Neuroinflamm. 2018, 15, 144. [Google Scholar] [CrossRef] [PubMed]
- Reschke, C.R.; Henshall, D.C. MicroRNA and Epilepsy. In microRNA: Medical Evidence. Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2015; pp. 41–70. [Google Scholar]
- Wang, J.; Zhao, J. MicroRNA Dysregulation in Epilepsy: From Pathogenetic Involvement to Diagnostic Biomarker and Therapeutic Agent Development. Front. Mol. Neurosci. 2021, 14, 650372. [Google Scholar] [CrossRef] [PubMed]
- Dadas, A.; Janigro, D. Breakdown of Blood Brain Barrier as a Mechanism of Post-Traumatic Epilepsy. Neurobiol. Dis. 2019, 123, 20–26. [Google Scholar] [CrossRef]
- Lopes, M.A.; Junges, L.; Woldman, W.; Goodfellow, M.; Terry, J.R. The Role of Excitability and Network Structure in the Emergence of Focal and Generalized Seizures. Front. Neurol. 2020, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.W.; Rutecki, P.A.; Sutula, T.P. The Effects of Seizures on the Brain. Curr. Opin. Neurol. 1996, 9, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.N.; Raffelt, D.; Curwood, E.; Tsai, M.; Tournier, J.; Connelly, A.; Jackson, G.D. Tract-specific Atrophy in Focal Epilepsy: Disease, Genetics, or Seizures? Ann. Neurol. 2017, 81, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Gloor, P.; Fariello, R.G. Generalized Epilepsy: Some of Its Cellular Mechanisms Differ from Those of Focal Epilepsy. Trends Neurosci. 1988, 11, 63–68. [Google Scholar] [CrossRef]
- Stafstrom, C.E. Dietary Approaches to Epilepsy Treatment: Old and New Options on the Menu. Epilepsy Curr. 2004, 4, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.M.; Singh, R.K.; Shellhaas, R.A. Advanced Treatments for Childhood Epilepsy. JAMA Pediatr. 2013, 167, 76. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Hernandes, M.S. Sepsis-Associated Encephalopathy and Blood-Brain Barrier Dysfunction. Inflammation 2021, 44, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E. Epidemiology, virology, and clinical features of severe acute respiratory syndrome -coronavirus-2 (SARS-CoV-2; Coronavirus Disease-19). Clin. Exp. Pediatr. 2020, 63, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Levite, M.; Goldberg, H. Autoimmune Epilepsy—Novel Multidisciplinary Analysis, Discoveries and Insights. Front. Immunol. 2022, 12, 762743. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E.; Shorvon, S. Antiepileptic drugs and the immune system. Epilepsia 2011, 52 (Suppl. S3), 40–44. [Google Scholar] [CrossRef] [PubMed]
- Patsalos, P.N.; Fröscher, W.; Pisani, F.; Van Rijn, C.M. The Importance of Drug Interactions in Epilepsy Therapy. Epilepsia 2002, 43, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Patsalos, P.N.; Perucca, E. Clinically Important Drug Interactions in Epilepsy: Interactions between Antiepileptic Drugs and Other Drugs. Lancet Neurol. 2003, 2, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Karaźniewicz-Łada, M.; Główka, A.K.; Mikulska, A.A.; Główka, F.K. Pharmacokinetic Drug-Drug Interactions among Antiepileptic Drugs, Including CBD, Drugs Used to Treat COVID-19 and Nutrients. Int. J. Mol. Sci. 2021, 22, 9582. [Google Scholar] [CrossRef] [PubMed]
- Wanounou, M.; Caraco, Y.; Levy, R.H.; Bialer, M.; Perucca, E. Clinically Relevant Interactions between Ritonavir-Boosted Nirmatrelvir and Concomitant Antiseizure Medications: Implications for the Management of COVID-19 in Patients with Epilepsy. Clin. Pharmacokinet. 2022, 61, 1219–1236. [Google Scholar] [CrossRef] [PubMed]
- Fırat, O.; Yalçın, N.; Demirkan, K. COVID-19 & Antiepileptic Drugs: Should We Pay Attention? Seizure 2020, 80, 240–241. [Google Scholar] [CrossRef] [PubMed]
- Kirmani, B.F.; Mungall-Robinson, D. Role of Anticonvulsants in the Management of AIDS Related Seizures. Front. Neurol. 2014, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Marvanova, M. Pharmacokinetic Characteristics of Antiepileptic Drugs (AEDs). Ment. Health Clin. 2016, 6, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Faught, E. Pharmacokinetic Considerations in Prescribing Antiepileptic Drugs. Epilepsia 2001, 42, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Stefan, H.; Feuerstein, T.J. Novel Anticonvulsant Drugs. Pharmacol. Ther. 2007, 113, 165–183. [Google Scholar] [CrossRef] [PubMed]
- French, J.A.; Gazzola, D.M. New Generation Antiepileptic Drugs: What Do They Offer in Terms of Improved Tolerability and Safety? Ther. Adv. Drug Saf. 2011, 2, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H. Antiviral Therapy (Non-HIV). In Goldman’s Cecil Medicine; Elsevier: Amsterdam, The Netherlands, 2012; pp. 2082–2089. [Google Scholar]
- Zaccara, G.; Perucca, E. Interactions between Antiepileptic Drugs, and between Antiepileptic Drugs and Other Drugs. Epileptic Disord. 2014, 16, 409–431. [Google Scholar] [CrossRef] [PubMed]
- Spina, E.; Pisani, F.; Perucca, E. Clinically Significant Pharmacokinetic Drug Interactions with Carbamazepine. Clin. Pharmacokinet. 1996, 31, 198–214. [Google Scholar] [CrossRef] [PubMed]
- Phenytoin/Rifampicin Interaction. React. Wkly. 2014, 1486, 25. [CrossRef]
- Grimaldi, R.; Lecchini, S.; Crema, F.; Perucca, E. In Vivo Plasma Protein Binding Interaction between Valproic Acid and Naproxen. Eur. J. Drug Metab. Pharmacokinet. 1984, 9, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Addison, R.S.; Parker-Scott, S.L.; Hooper, W.D.; Eadie, M.J.; Dickinson, R.G. Effect of Naproxen Co-administration on Valproate Disposition. Biopharm. Drug Dispos. 2000, 21, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Volk, A. Displacement of Valproic Acid and Carbamazepine from Protein Binding in Normal and Uremic Sera by Tolmetin, Ibuprofen, and Naproxen: Presence of Inhibitor in Uremic Serum That Blocks Valproic Acid-Naproxen Interactions. Ther. Drug Monit. 1996, 18, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Gulcebi, M.I.; Bartolini, E.; Lee, O.; Lisgaras, C.P.; Onat, F.; Mifsud, J.; Striano, P.; Vezzani, A.; Hildebrand, M.S.; Jimenez-Jimenez, D.; et al. Climate change and epilepsy: Insights from clinical and basic science studies. Epilepsy Behav. 2021, 116, 107791. [Google Scholar] [CrossRef] [PubMed]
- Akyüz, E.; Üner, A.K.; Köklü, B.; Arulsamy, A.; Shaikh, M.F. Cardiorespiratory Findings in Epilepsy: A Recent Review on Outcomes and Pathophysiology. J. Neurosci. Res. 2021, 99, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Fernández, I.; Goodkin, H.P.; Scott, R.C. Pathophysiology of Convulsive Status Epilepticus. Seizure 2019, 68, 16–21. [Google Scholar] [CrossRef]
- Siahaan, Y.M.T.; Ketaren, R.J.; Hartoyo, V.; Hariyanto, T.I. Epilepsy and the Risk of Severe Coronavirus Disease 2019 Outcomes: A Systematic Review, Meta-Analysis, and Meta-Regression. Epilepsy Behav. 2021, 125, 108437. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; Brigo, F.; Trinka, E. Acute Symptomatic Seizures Caused by Electrolyte Disturbances. J. Clin. Neurol. 2016, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-Z.; Liu, Y.-H.; Tseng, C.-M.; Tseng, Y.-H.; Chen, T.-H. Comparison of Clinical Characteristics between Febrile and Afebrile Seizures Associated with Acute Gastroenteritis in Childhood. Front. Pediatr. 2020, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D.; Dill, L.K.; O’Brien, T.J. Immune Challenges and Seizures: How Do Early Life Insults Influence Epileptogenesis? Front. Pharmacol. 2020, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Kocamaz, H.; Işıkay, S. Epilepsy and GI Disorders. In Epilepsy—Advances in Diagnosis and Therapy; IntechOpen: London, UK, 2019. [Google Scholar]
- Sanchez-Larsen, A.; Conde-Blanco, E.; Viloria-Alebesque, A.; Sánchez-Vizcaíno Buendía, C.; Espinosa Oltra, T.; Alvarez-Noval, A.; Aledo-Serrano, A.; Martin-Garcia, R.; Ramos-Araque, M.E.; Campos, D.; et al. COVID-19 Prevalence and Mortality in People with Epilepsy: A Nation-Wide Multicenter Study. Epilepsy Behav. 2021, 125, 108379. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.C.; Toovey, S.; Jick, S.S.; Meier, C.R. Previously Diagnosed Influenza Infections and the Risk of Developing Epilepsy. Epidemiol. Infect. 2015, 143, 2408–2415. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, G.; Turchi, G.; Mazzoli, M.; Vaudano, A.E.; Meletti, S. New Onset Status Epilepticus in Influenza Associated Encephalopathy: The Presenting Manifestation of Genetic Generalized Epilepsy. Epilepsy Behav. Rep. 2021, 16, 100413. [Google Scholar] [CrossRef] [PubMed]
- Galgani, A.; Palleria, C.; Iannone, L.F.; De Sarro, G.; Giorgi, F.S.; Maschio, M.; Russo, E. Pharmacokinetic Interactions of Clinical Interest between Direct Oral Anticoagulants and Antiepileptic Drugs. Front. Neurol. 2018, 9, 1067, Corrigendum in Front. Neurol. 2020, 10, 1381. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.Q.T.; Xu, L.H.R.; Moe, O.W. Drug-Induced Metabolic Acidosis. F1000Research 2015, 4, 1460. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E. Impact of Infectious and Inflammatory Disease on Cytochrome P450–Mediated Drug Metabolism and Pharmacokinetics. Clin. Pharmacol. Ther. 2009, 85, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.N.; Siddiqi, Z.A. Antiepileptic Drugs and Liver Disease. Seizure 2006, 15, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Asconapé, J.J. Use of Antiepileptic Drugs in Hepatic and Renal Disease. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 417–432. [Google Scholar]
- Khadka, B.; Lee, J.-Y.; Park, E.K.; Kim, K.-T.; Bae, J.-S. Impacts of Drug Interactions on Pharmacokinetics and the Brain Transporters: A Recent Review of Natural Compound-Drug Interactions in Brain Disorders. Int. J. Mol. Sci. 2021, 22, 1809. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Dohi, K.; Banks, W.A. Neuroinflammation: A Common Pathway in CNS Diseases as Mediated at the Blood-Brain Barrier. Neuroimmunomodulation 2012, 19, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Nwogu, J.N.; Ma, Q.; Babalola, C.P.; Adedeji, W.A.; Morse, G.D.; Taiwo, B. Pharmacokinetic, Pharmacogenetic, and Other Factors Influencing CNS Penetration of Antiretrovirals. AIDS Res. Treat. 2016, 2016, 2587094. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, B.C.; Lopes Barbosa, M.G.; Silva Olak, A.P.; Belebecha Terezo, N.; Nishi, L.; Watanabe, M.A.; Marinello, P.; Zendrini Rechenchoski, D.; Dejato Rocha, S.P.; Faccin-Galhardi, L.C. Antiviral Therapies: Advances and Perspectives. Fundam. Clin. Pharmacol. 2021, 35, 305–320. [Google Scholar] [CrossRef]
- Gao, B.; Wu, Y.; Yang, Y.-J.; Li, W.-Z.; Dong, K.; Zhou, J.; Yin, Y.-Y.; Huang, D.-K.; Wu, W.-N. Sinomenine Exerts Anticonvulsant Profile and Neuroprotective Activity in Pentylenetetrazole Kindled Rats: Involvement of Inhibition of NLRP1 Inflammasome. J. Neuroinflammation 2018, 15, 152. [Google Scholar] [CrossRef] [PubMed]
- Mahfoz, A.M.; Shahzad, N. Neuroinflammation Impact in Epileptogenesis and New Treatment Strategy. Behav. Pharmacol. 2019, 30, 660–674. [Google Scholar] [CrossRef]
- Ravizza, T.; Vezzani, A. Pharmacological Targeting of Brain Inflammation in Epilepsy: Therapeutic Perspectives from Experimental and Clinical Studies. Epilepsia Open 2018, 3, 133–142. [Google Scholar] [CrossRef]
- Sharma, R.; Leung, W.L.; Zamani, A.; O’Brien, T.J.; Casillas Espinosa, P.M.; Semple, B.D. Neuroinflammation in Post-Traumatic Epilepsy: Pathophysiology and Tractable Therapeutic Targets. Brain Sci. 2019, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Lazarini-Lopes, W.; Do Val-da Silva, R.A.; da Silva-Júnior, R.M.P.; Leite, J.P.; Garcia-Cairasco, N. The Anticonvulsant Effects of Cannabidiol in Experimental Models of Epileptic Seizures: From Behavior and Mechanisms to Clinical Insights. Neurosci. Biobehav. Rev. 2020, 111, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Natali, K.M.; Jimenez, H.R.; Slim, J. When Coadministration Cannot Be Avoided: Real World Experience of Direct Acting Antivirals for the Treatment of Hepatitis C Virus Infection in Patients on First Generation Anticonvulsants. J. Pharm. Pract. 2022, 35, 495–499. [Google Scholar] [CrossRef]
- Yin, J.; Hill, L.; Mansour, M.; Collier, S.; Barman, P. Successful Treatment of Hepatitis C Virus Infection in Patients on Anti-epileptics or Mood Stabilizers Using Pharmacokinetic Enhancers. Br. J. Clin. Pharmacol. 2023, 89, 1891–1895. [Google Scholar] [CrossRef] [PubMed]
- Blackmond, N.; Kanke, J.D.; Loh, T.; Weitzman, R. Challenges of Treating an Acute Hepatitis C Virus Infection with Concurrent Seizure Treatment in a Free Clinic. Cureus 2022, 14, e25530. [Google Scholar] [CrossRef] [PubMed]
- Birbeck, G.L.; French, J.A.; Perucca, E.; Simpson, D.M.; Fraimow, H.; George, J.M.; Okulicz, J.F.; Clifford, D.B.; Hachad, H.; Levy, R.H. Evidence-Based Guideline: Antiepileptic Drug Selection for People with HIV/AIDS: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Ad Hoc Task Force of the Commission on Therapeutic Strategies of the International League Against Epilepsy. Neurology 2012, 78, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Birbeck, G.L.; French, J.A.; Perucca, E.; Simpson, D.M.; Fraimow, H.; George, J.M.; Okulicz, J.F.; Clifford, D.B.; Hachad, H.; Levy, R.H. for the Quality Standards Subcommittee of the American Academy of Neurology and the ad hoc Task Force of the Commission on Therapeutic Strategies of the International League against Epilepsy. Antiepileptic Drug Selection for People with HIV/AIDS: Evidence-based Guidelines from the ILAE and AAN. Epilepsia 2012, 53, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Agbabiaka, T.B.; Savović, J.; Ernst, E. Methods for Causality Assessment of Adverse Drug Reactions. Drug Saf. 2008, 31, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Cao, P.; Wang, Y.; Xie, F.; Ma, J.; Yu, M.; Wang, J.; Xu, Y.; Zhang, Y.; Wan, J. A Review of Approaches for Predicting Drug–Drug Interactions Based on Machine Learning. Front. Pharmacol. 2022, 12, 814858. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Lu, R.-M.; Li, M.-C.; Huang, J.-L.; Hsu, F.-F.; Ko, S.-H.; Ke, F.-Y.; Su, S.-C.; Liang, K.-H.; Yuan, J.P.-Y.; et al. A Critical Overview of Current Progress for COVID-19: Development of Vaccines, Antiviral Drugs, and Therapeutic Antibodies. J. Biomed. Sci. 2022, 29, 68. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, N. Epilepsy and COVID-19: Updated Evidence and Narrative Review. Epilepsy Behav. 2021, 116, 107785. [Google Scholar] [CrossRef] [PubMed]
- Hensel, A.; Bauer, R.; Heinrich, M.; Spiegler, V.; Kayser, O.; Hempel, G.; Kraft, K. Challenges at the Time of COVID-19: Opportunities and Innovations in Antivirals from Nature. Planta Med. 2020, 86, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Pazgan-Simon, M.; Jaroszewicz, J.; Simon, K.; Lorenc, B.; Sitko, M.; Zarębska-Michaluk, D.; Dybowska, D.; Tudrujek-Zdunek, M.; Berak, H.; Mazur, W.; et al. Real-World Effectiveness and Safety of Direct-Acting Antivirals in Patients with Chronic Hepatitis C and Epilepsy: An Epi-Ter-2 Study in Poland. J. Pers. Med. 2023, 13, 1111. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Mantilla, J.D.; Huang, F.; Peters, S.A. Can Mechanistic Static Models for Drug-Drug Interactions Support Regulatory Filing for Study Waivers and Label Recommendations? Clin. Pharmacokinet. 2023, 62, 457–480. [Google Scholar] [CrossRef] [PubMed]
- Taskar, K.S.; Pilla Reddy, V.; Burt, H.; Posada, M.M.; Varma, M.; Zheng, M.; Ullah, M.; Emami Riedmaier, A.; Umehara, K.; Snoeys, J.; et al. Physiologically-Based Pharmacokinetic Models for Evaluating Membrane Transporter Mediated Drug–Drug Interactions: Current Capabilities, Case Studies, Future Opportunities, and Recommendations. Clin. Pharmacol. Ther. 2020, 107, 1082–1115. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, Y.; Wang, L.; Xu, S.; Fang, G.; Guo, X.; Chen, Z.; Gu, Z. PBPK Modeling on Organs-on-Chips: An Overview of Recent Advancements. Front. Bioeng. Biotechnol. 2022, 10, 900481. [Google Scholar] [CrossRef]
- Zhuang, X.; Lu, C. PBPK Modeling and Simulation in Drug Research and Development. Acta Pharm. Sin. B 2016, 6, 430–440. [Google Scholar] [CrossRef]
- Stader, F.; Battegay, M.; Sendi, P.; Marzolini, C. Physiologically Based Pharmacokinetic Modelling to Investigate the Impact of the Cytokine Storm on CYP3A Drug Pharmacokinetics in COVID-19 Patients. Clin. Pharmacol. Ther. 2022, 111, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Grimstein, M.; Yang, Y.; Zhang, X.; Grillo, J.; Huang, S.-M.; Zineh, I.; Wang, Y. Physiologically Based Pharmacokinetic Modeling in Regulatory Science: An Update From the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J. Pharm. Sci. 2019, 108, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Taskar, K.S.; Harada, I.; Alluri, R.V. Physiologically-Based Pharmacokinetic (PBPK) Modelling of Transporter Mediated Drug Absorption, Clearance and Drug-Drug Interactions. Curr. Drug Metab. 2021, 22, 523–531. [Google Scholar] [CrossRef]
- Fan, J.; Yang, Y.; Grimstein, M.; Zhang, X.; Kitabi, E.; Earp, J.C.; Arya, V.; Reynolds, K.S.; Zhu, H.; Wang, Y. Whole Body PBPK Modeling of Remdesivir and Its Metabolites to Aid in Estimating Active Metabolite Exposure in the Lung and Liver in Patients with Organ Dysfunction. Clin. Pharmacol. Ther. 2022, 111, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Humeniuk, R.; Mathias, A.; Kirby, B.J.; Lutz, J.D.; Cao, H.; Osinusi, A.; Babusis, D.; Porter, D.; Wei, X.; Ling, J.; et al. Pharmacokinetic, Pharmacodynamic, and Drug-Interaction Profile of Remdesivir, a SARS-CoV-2 Replication Inhibitor. Clin. Pharmacokinet. 2021, 60, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.M.; Coulter, D.A. Mechanisms of Epileptogenesis: A Convergence on Neural Circuit Dysfunction. Nat. Rev. Neurosci. 2013, 14, 337–349. [Google Scholar] [CrossRef]
- Pracucci, E.; Pillai, V.; Lamers, D.; Parra, R.; Landi, S. Neuroinflammation: A Signature or a Cause of Epilepsy? Int. J. Mol. Sci. 2021, 22, 6981. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, H.; Bryceson, Y.T. Neuroinflammation Associated with Inborn Errors of Immunity. Front. Immunol. 2022, 12, 827815. [Google Scholar] [CrossRef] [PubMed]
- Jesus, A.A.; Osman, M.; Silva, C.A.; Kim, P.W.; Pham, T.-H.; Gadina, M.; Yang, B.; Bertola, D.R.; Carneiro-Sampaio, M.; Ferguson, P.J.; et al. A Novel Mutation of IL1RN in the Deficiency of Interleukin-1 Receptor Antagonist Syndrome: Description of Two Unrelated Cases from Brazil. Arthritis Rheum. 2011, 63, 4007–4017. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Peri, A.M.; Cagliani, R.; Forni, D.; Riva, S.; Biasin, M.; Clerici, M.; Gori, A. TLR3 Mutations in Adult Patients with Herpes Simplex Virus and Varicella-Zoster Virus Encephalitis. J. Infect. Dis. 2017, 215, 1430–1434. [Google Scholar] [CrossRef]
- Mosaffa, F.; Kalalinia, F.; Lage, H.; Afshari, J.T.; Behravan, J. Pro-Inflammatory Cytokines Interleukin-1 Beta, Interleukin 6, and Tumor Necrosis Factor-Alpha Alter the Expression and Function of ABCG2 in Cervix and Gastric Cancer Cells. Mol. Cell. Biochem. 2012, 363, 385–393. [Google Scholar] [CrossRef]
- Scatton, B. The NMDA Receptor Complex. Fundam. Clin. Pharmacol. 1993, 7, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Koda, S.; Hu, J.; Ju, X.; Sun, G.; Shao, S.; Tang, R.-X.; Zheng, K.-Y.; Yan, J. The Role of Glutamate Receptors in the Regulation of the Tumor Microenvironment. Front. Immunol. 2023, 14, 1123841. [Google Scholar] [CrossRef] [PubMed]
- Mottahedin, A.; Ardalan, M.; Chumak, T.; Riebe, I.; Ek, J.; Mallard, C. Effect of Neuroinflammation on Synaptic Organization and Function in the Developing Brain: Implications for Neurodevelopmental and Neurodegenerative Disorders. Front. Cell. Neurosci. 2017, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, E. Brain-to-Brain Communication: The Possible Role of Brain Electromagnetic Fields (As a Potential Hypothesis). Heliyon 2021, 7, e06363. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory Pathways as Treatment Targets and Biomarkers in Epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Politsky, J.M. Brain Tumor-Related Epilepsy: A Current Review of the Etiologic Basis and Diagnostic and Treatment Approaches. Curr. Neurol. Neurosci. Rep. 2017, 17, 70. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.K. Neurocysticercosis and Epilepsy in Developing Countries. J. Neurol. Neurosurg. Psychiatry 2000, 68, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.S.; Wadhwa, A.; Garg, J.; Mahajan, R.K. HIV-Associated Neurocysticercosis. J. Int. Assoc. Provid. AIDS Care (JIAPAC) 2015, 14, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A. Herpes Simplex Virus Encephalitis: Toll-Free Access to the Brain. Cell Host Microbe 2012, 12, 731–732. [Google Scholar] [CrossRef] [PubMed]
- Ellison, P.H.; Hanson, P.A. Herpes Simplex: A Possible Cause of Brain-Stem Encephalitis. Pediatrics 1977, 59, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Kuwajima, M.; Kido, H. Pathologic Mechanisms of Influenza Encephalitis with an Abnormal Expression of Inflammatory Cytokines and Accumulation of Mini-Plasmin. J. Med. Investig. 2003, 50, 1–8. [Google Scholar]
- Studahl, M. Influenza Virus and CNS Manifestations. J. Clin. Virol. 2003, 28, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Türkoğlu, R.; Tüzün, E. Brainstem Encephalitis Following Influenza Vaccination: Favorable Response to Steroid Treatment. Vaccine 2009, 27, 7253–7256. [Google Scholar] [CrossRef] [PubMed]
- Meijer, W.J.; Linn, F.H.H.; Wensing, A.M.J.; Leavis, H.L.; van Riel, D.; GeurtsvanKessel, C.H.; Wattjes, M.P.; Murk, J.-L. Acute Influenza Virus-Associated Encephalitis and Encephalopathy in Adults: A Challenging Diagnosis. JMM Case Rep. 2016, 3, e005076. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Rothan, H.A.; Natekar, J.P.; Stone, S.; Pathak, H.; Strate, P.G.; Arora, K.; Brinton, M.A.; Kumar, M. Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-HACE2 Mice. Viruses 2021, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Moench, T.R.; Griffin, D.E.; Obriecht, C.R.; Vaisberg, A.J.; Johnson, R.T. Acute Measles in Patients with and without Neurological Involvement: Distribution of Measles Virus Antigen and RNA. J. Infect. Dis. 1988, 158, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Norrby, E.; Kristensson, K. Measles Virus in the Brain. Brain Res. Bull. 1997, 44, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Fuchigami, T.; Ishii, W.; Imai, Y.; Tanabe, S.; Kuwabara, R.; Fujita, Y.; Mugishima, H.; Aoki, K. Mumps-Virus-Associated Clinically Mild Encephalopathy with a Reversible Splenial Lesion. Int. J. Clin. Pediatr. 2012, 1, 124–128. [Google Scholar] [CrossRef]
- Löve, A.; Rydbeck, R.; Kristensson, K.; Orvell, C.; Norrby, E. Hemagglutinin-Neuraminidase Glycoprotein as a Determinant of Pathogenicity in Mumps Virus Hamster Encephalitis: Analysis of Mutants Selected with Monoclonal Antibodies. J. Virol. 1985, 53, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, D.; Uchiyama, T.; Sugiyama, T.; Sato, K.; Shimizu, T.; Ohashi, T. An Adult Case of Mumps-Associated Encephalitis/Encephalopathy Successfully Treated with Steroid Pulse Therapy. Rinsho Shinkeigaku 2013, 53, 839–842. [Google Scholar] [CrossRef]
- Dore, G.J.; Law, M.G.; Brew, B.J. Prospective Analysis of Seizures Occurring in Human Immunodeficiency Virus Type-1 Infection. J. Neuro-AIDS 1997, 1, 59–69. [Google Scholar] [CrossRef]
- Wong, M.C.; Suite, N.D.A.; Labar, D.R. Seizures in Human Immunodeficiency Virus Infection. Arch. Neurol. 1990, 47, 640–642. [Google Scholar] [CrossRef]
- Ene, L. Human Immunodeficiency Virus in the Brain—Culprit or Facilitator? Infect. Dis. Res. Treat. 2018, 11, 117863371775268. [Google Scholar] [CrossRef]
- Jones, J.F.; Straus, S.E. Chronic Epstein-Barr Virus Infection. Annu. Rev. Med. 1987, 38, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Satishchandra, P.; Sinha, S. Seizures in HIV-seropositive individuals: NIMHANS experience and review. Epilepsia 2008, 49 (Suppl. S6), 33–41. [Google Scholar] [CrossRef] [PubMed]
- Krstanović, F.; Britt, W.J.; Jonjić, S.; Brizić, I. Cytomegalovirus Infection and Inflammation in Developing Brain. Viruses 2021, 13, 1078. [Google Scholar] [CrossRef] [PubMed]
- Kamakura, T.; Hiraki, A.; Kikuchi, M. Transient Seizure-related MRI Abnormalities in a Child with Primary Epstein–Barr Virus Infection. Pediatr. Int. 2016, 58, 525–527. [Google Scholar] [CrossRef]
- Pascual-Sedano, B.; Iranzo, A.; Marti-Fàbregas, J.; Domingo, P.; Escartin, A.; Fuster, M.; Barrio, J.L.; Sambeat, M.A. Prospective study of new-onset seizures in patients with human immunodeficiency virus infection: Etiologic and clinical aspects. Arch. Neurol. 1999, 56, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.G.; Unterholzner, L. Viral Evasion and Subversion of Pattern-Recognition Receptor Signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Li, H.; Luo, X.; Liu, C.; Zhang, Y.; Guo, S.; Xu, J.; Bao, C.; Dong, W.; Yu, Y. Expression and Mechanisms of Interferon-Stimulated Genes in Viral Infection of the Central Nervous System (CNS) and Neurological Diseases. Front. Immunol. 2022, 13, 1008072. [Google Scholar] [CrossRef] [PubMed]
- Könnecke, H.; Bechmann, I. The Role of Microglia and Matrix Metalloproteinases Involvement in Neuroinflammation and Gliomas. Clin. Dev. Immunol. 2013, 2013, 914104. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Restrepo-Jiménez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.-M. Molecular Mimicry and Autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [CrossRef]
- Neațu, M.; Jugurt, A.; Covaliu, A.; Davidescu, E.I.; Popescu, B.O. Autoimmune Encephalitis—A Multifaceted Pathology. Biomedicines 2023, 11, 2176. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Friedman, A. Brain Inflammation as a Biomarker in Epilepsy. Biomark. Med. 2011, 5, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Bronisz, E.; Cudna, A.; Wierzbicka, A.; Kurkowska-Jastrzębska, I. Blood-Brain Barrier-Associated Proteins Are Elevated in Serum of Epilepsy Patients. Cells 2023, 12, 368. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Gao, Y.; Zhang, S.-J.; Zhe, X.; Meng, F.-L.; Qian, H.; Zhang, B.; Li, Y.-J. Alteration of Plasma Cytokines in Patients with Active Epilepsy. Acta Neurol. Scand. 2017, 135, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Gershen, L.D.; Zanotti-Fregonara, P.; Dustin, I.H.; Liow, J.-S.; Hirvonen, J.; Kreisl, W.C.; Jenko, K.J.; Inati, S.K.; Fujita, M.; Morse, C.L.; et al. Neuroinflammation in Temporal Lobe Epilepsy Measured Using Positron Emission Tomographic Imaging of Translocator Protein. JAMA Neurol. 2015, 72, 882. [Google Scholar] [CrossRef] [PubMed]
- Dickstein, L.P.; Liow, J.; Austermuehle, A.; Zoghbi, S.; Inati, S.K.; Zaghloul, K.; Zanotti-Fregonara, P.; Theodore, W.H. Neuroinflammation in Neocortical Epilepsy Measured by PET Imaging of Translocator Protein. Epilepsia 2019, 60, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 3161. [Google Scholar] [CrossRef] [PubMed]
- Ching, A.S.C.; Kuhnast, B.; Damont, A.; Roeda, D.; Tavitian, B.; Dollé, F. Current Paradigm of the 18-KDa Translocator Protein (TSPO) as a Molecular Target for PET Imaging in Neuroinflammation and Neurodegenerative Diseases. Insights Imaging 2012, 3, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, Y.; Caleo, M. Epilepsy, Seizures, and Inflammation: Role of the C-C Motif Ligand 2 Chemokine. DNA Cell Biol. 2016, 35, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Kothur, K.; Bandodkar, S.; Wienholt, L.; Chu, S.; Pope, A.; Gill, D.; Dale, R.C. Etiology Is the Key Determinant of Neuroinflammation in Epilepsy: Elevation of Cerebrospinal Fluid Cytokines and Chemokines in Febrile Infection-related Epilepsy Syndrome and Febrile Status Epilepticus. Epilepsia 2019, 60, 1678–1688. [Google Scholar] [CrossRef] [PubMed]
- Noe, F.M.; Polascheck, N.; Frigerio, F.; Bankstahl, M.; Ravizza, T.; Marchini, S.; Beltrame, L.; Reschke Banderó, C.; Löscher, W.; Vezzani, A. Pharmacological blockade of IL-1β/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol. Dis. 2013, 59, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Cole-Edwards, K.K.; Bazan, N.G. Lipid Signaling in Experimental Epilepsy. Neurochem. Res. 2005, 30, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Terrone, G.; Salamone, A.; Vezzani, A. Inflammation and Epilepsy: Preclinical Findings and Potential Clinical Translation. Curr. Pharm. Des. 2017, 23, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Kamali, A.N.; Zian, Z.; Bautista, J.M.; Hamedifar, H.; Hossein-Khannazer, N.; Hosseinzadeh, R.; Yazdani, R.; Azizi, G. The Potential Role of Pro-Inflammatory and Anti-Inflammatory Cytokines in Epilepsy Pathogenesis. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.E.; Smesny, S.; Amminger, G.P. Bioactive Lipids in Schizophrenia. Int. Rev. Psychiatry 2006, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.T. Epilepsy after Brain Insult: Targeting Epileptogenesis. Neurology 2002, 59, S21–S26. [Google Scholar] [CrossRef]
- Van Dijkman, S.C.; Voskuyl, R.A.; de Lange, E.C. Biomarkers in Epilepsy–A Modelling Perspective. Eur. J. Pharm. Sci. 2017, 109, S47–S52. [Google Scholar] [CrossRef] [PubMed]
- Kamnaksh, A.; Puhakka, N.; Ali, I.; Smith, G.; Aniceto, R.; McCullough, J.; Das Gupta, S.; Ndode-Ekane, X.E.; Brady, R.; Casillas-Espinosa, P.; et al. Harmonization of Pipeline for Preclinical Multicenter Plasma Protein and MiRNA Biomarker Discovery in a Rat Model of Post-Traumatic Epileptogenesis. Epilepsy Res. 2019, 149, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Simonato, M.; Agoston, D.V.; Brooks-Kayal, A.; Dulla, C.; Fureman, B.; Henshall, D.C.; Pitkänen, A.; Theodore, W.H.; Twyman, R.E.; Kobeissy, F.H.; et al. Identification of Clinically Relevant Biomarkers of Epileptogenesis—A Strategic Roadmap. Nat. Rev. Neurol. 2021, 17, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Engel, J. Biomarkers in Epilepsy: Introduction. Biomark. Med. 2011, 5, 537–544. [Google Scholar] [CrossRef]
- Van Vliet, E.A.; Puhakka, N.; Mills, J.D.; Srivastava, P.K.; Johnson, M.R.; Roncon, P.; Das Gupta, S.; Karttunen, J.; Simonato, M.; Lukasiuk, K.; et al. Standardization procedure for plasma biomarker analysis in rat models of epileptogenesis: Focus on circulating microRNAs. Epilepsia 2017, 58, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Banote, R.K.; Akel, S.; Zelano, J. Blood Biomarkers in Epilepsy. Acta Neurol. Scand. 2022, 146, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Li, J.; Shao, Q.; Chen, H.; Lin, Z.; Sun, Z.S.; Wu, J. EpilepsyGene: A Genetic Resource for Genes and Mutations Related to Epilepsy. Nucleic Acids Res. 2015, 43, D893–D899. [Google Scholar] [CrossRef] [PubMed]
- Epi4K Consortium. Epi4K: Gene Discovery in 4000 Genomes. Epilepsia 2012, 53, 1457–1467. [Google Scholar] [CrossRef]
- Liu, Y.; Elsworth, B.; Erola, P.; Haberland, V.; Hemani, G.; Lyon, M.; Zheng, J.; Lloyd, O.; Vabistsevits, M.; Gaunt, T.R. EpiGraphDB: A Database and Data Mining Platform for Health Data Science. Bioinformatics 2021, 37, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Kobylarek, D.; Iwanowski, P.; Lewandowska, Z.; Limphaibool, N.; Szafranek, S.; Labrzycka, A.; Kozubski, W. Advances in the Potential Biomarkers of Epilepsy. Front. Neurol. 2019, 10, 685. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Wendtner, M.-H.; Korting, H.C. Penciclovir Cream—Improved Topical Treatment for Herpes Simplex Infections. Skin. Pharmacol. Physiol. 2004, 17, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Mi, X.; Gao, B.; Gu, J.; Wang, W.; Zhang, Y.; Wang, X. Minocycline Inhibits Brain Inflammation and Attenuates Spontaneous Recurrent Seizures Following Pilocarpine-Induced Status Epilepticus. Neuroscience 2015, 287, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Miller, D.B.; O’Callaghan, J.P. Minocycline Attenuates Microglial Activation but Fails to Mitigate Striatal Dopaminergic Neurotoxicity: Role of Tumor Necrosis Factor-α. J. Neurochem. 2006, 96, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Rolland, W.B.; Lekic, T.; Krafft, P.R.; Hasegawa, Y.; Altay, O.; Hartman, R.; Ostrowski, R.; Manaenko, A.; Tang, J.; Zhang, J.H. Fingolimod Reduces Cerebral Lymphocyte Infiltration in Experimental Models of Rodent Intracerebral Hemorrhage. Exp. Neurol. 2013, 241, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shi, D.; Cao, K.; Wu, F.; Zhu, X.; Wen, S.; You, Q.; Zhang, K.; Liu, L.; Zhou, H. Fingolimod Targets Cerebral Endothelial Activation to Block Leukocyte Recruitment in the Central Nervous System. J. Leukoc. Biol. 2018, 103, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Hartung, H.-P. Mechanism of Action of Oral Fingolimod (FTY720) in Multiple Sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Pfender, N.; Jelcic, I.; Linnebank, M.; Schwarz, U.; Martin, R. Reactivation of Herpesvirus under Fingolimod: A Case of Severe Herpes Simplex Encephalitis. Neurology 2015, 84, 2377–2378. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.A.; Aronica, E.; Vezzani, A.; Ravizza, T. Review: Neuroinflammatory Pathways as Treatment Targets and Biomarker Candidates in Epilepsy: Emerging Evidence from Preclinical and Clinical Studies. Neuropathol. Appl. Neurobiol. 2018, 44, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Iori, V.; Iyer, A.M.; Ravizza, T.; Beltrame, L.; Paracchini, L.; Marchini, S.; Cerovic, M.; Hill, C.; Ferrari, M.; Zucchetti, M.; et al. Blockade of the IL-1R1/TLR4 Pathway Mediates Disease-Modification Therapeutic Effects in a Model of Acquired Epilepsy. Neurobiol. Dis. 2017, 99, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Kang, X.; Qiu, J.; Du, Y.; Jiang, J. Anti-Inflammatory Small Molecules to Treat Seizures and Epilepsy: From Bench to Bedside. Trends Pharmacol. Sci. 2016, 37, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Hanamsagar, R.; Hanke, M.L.; Kielian, T. Toll-like Receptor (TLR) and Inflammasome Actions in the Central Nervous System. Trends Immunol. 2012, 33, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Vitaliti, G.; Pavone, P.; Mahmood, F.; Nunnari, G.; Falsaperla, R. Targeting Inflammation as a Therapeutic Strategy for Drug-Resistant Epilepsies. Hum. Vaccin. Immunother. 2014, 10, 868–875. [Google Scholar] [CrossRef]
- Engel, T.; Smith, J.; Alves, M. Targeting Neuroinflammation via Purinergic P2 Receptors for Disease Modification in Drug-Refractory Epilepsy. J. Inflamm. Res. 2021, 14, 3367–3392. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.A.; Petrovski, S.; Berkovic, S.F. Epilepsy Genetics: Clinical Impacts and Biological Insights. Lancet Neurol. 2020, 19, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Epstein, C.M. Epilepsy; Butterworth-Heinemann: Oxford, UK, 1990; ISBN 040990077X. [Google Scholar]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The Consequences of Refractory Epilepsy and Its Treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef]
- Toledano, R.; Villanueva, V.; Toledo, M.; Sabaniego, J.; Pérez-Domper, P. Clinical and Economic Implications of Epilepsy Management across Treatment Lines in Spain: A Real-Life Database Analysis. J. Neurol. 2023, 270, 5945–5957. [Google Scholar] [CrossRef] [PubMed]
- Landmark, C.J.; Johannessen, S.I.; Tomson, T. Dosing Strategies for Antiepileptic Drugs: From a Standard Dose for All to Individualised Treatment by Implementation of Therapeutic Drug Monitoring. Epileptic Disord. 2016, 18, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, S.; Mei, D.; Sisodiya, S.M.; Guerrini, R. Steps to Improve Precision Medicine in Epilepsy. Mol. Diagn. Ther. 2023, 27, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Striano, P.; Minassian, B.A. From Genetic Testing to Precision Medicine in Epilepsy. Neurotherapeutics 2020, 17, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-T.; Hong, S.-Y.; Lin, W.-D.; Lin, C.-H.; Lin, S.-S.; Tsai, F.-J.; Chou, I.-C. Genetic Testing in Children with Developmental and Epileptic Encephalopathies: A Review of Advances in Epilepsy Genomics. Children 2023, 10, 556. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.K.; Helbig, I.; Metcalf, C.S.; Lubbers, L.S.; Isom, L.L.; Demarest, S.; Goldberg, E.M.; George, A.L.; Lerche, H.; Weckhuysen, S.; et al. Precision Medicine for Genetic Epilepsy on the Horizon: Recent Advances, Present Challenges, and Suggestions for Continued Progress. Epilepsia 2022, 63, 2461–2475. [Google Scholar] [CrossRef] [PubMed]
- Vawter-Lee, M.; Franz, D.N.; Fuller, C.E.; Greiner, H.M. Clinical Letter: A Case Report of Targeted Therapy with Sirolimus for NPRL3 Epilepsy. Seizure 2019, 73, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Matin, N.; Tabatabaie, O.; Falsaperla, R.; Lubrano, R.; Pavone, P.; Mahmood, F.; Gullotta, M.; Serra, A.; Di Mauro, P.; Cocuzza, S.; et al. Epilepsy and innate immune system: A possible immunogenic predisposition and related therapeutic implications. Hum. Vaccines Immunother. 2015, 11, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Arya, K.; Willis, E.; Samanta, D. Immunotherapy in Autoimmune and Neuroinflammation-Related Epilepsies. J. Pediatr. Epilepsy 2018, 7, 69–75. [Google Scholar] [CrossRef]
- Zimmern, V.; Minassian, B.; Korff, C. A Review of Targeted Therapies for Monogenic Epilepsy Syndromes. Front. Neurol. 2022, 13, 829116. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.-S.; Ripa, V.; Al-Awar, O.; Panov, F.; Ghatan, S.; Jetté, N. Epilepsy and Neuromodulation—Randomized Controlled Trials. Brain Sci. 2018, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Dalkilic, E.B. Neurostimulation Devices Used in Treatment of Epilepsy. Curr. Treat. Options Neurol. 2017, 19, 7. [Google Scholar] [CrossRef] [PubMed]
- Abouelleil, M.; Deshpande, N.; Ali, R. Emerging Trends in Neuromodulation for Treatment of Drug-Resistant Epilepsy. Front. Pain Res. 2022, 3, 839463. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Considerations | Epilepsy Patients Acquiring Viral Infection | Individuals Developing Epilepsy after Viral Infection |
|---|---|---|
| Type and severity of the viral infection | Epilepsy patients acquiring viral infections may exhibit diverse types and severities, influenced by their brain and immune system susceptibility. Examples include severe or chronic infections like human immunodeficiency virus (HIV), cytomegalovirus, or Epstein-Barr virus, leading to persistent brain infection, chronic neuroinflammation, and seizures. | Individuals developing epilepsy after viral infection may experience less severe or acute infections, such as herpes simplex virus, influenza virus, or severe acute respiratory syndrome -coronavirus-2 (SARS-CoV-2). These infections may cause transient brain infection, acute neuroinflammation, and seizures. |
| Time and duration of the infection | Epilepsy patients acquiring viral infections may have varying infection timelines and durations, depending on the pre-existing epilepsy status. Viral infections may exacerbate or modify existing epilepsy. | Individuals developing epilepsy after viral infection may lack a history of epilepsy before the viral event. The infection may serve as an initiator or trigger, influencing the onset and progression of epilepsy. |
| Individual susceptibility and response to the infection | Epilepsy patients acquiring viral infections may display distinct susceptibilities and responses, influenced by genetic, environmental, and immunological factors. Genetic factors, like polymorphisms or mutations in inflammatory genes, may modulate the response and epilepsy susceptibility. | Individuals developing epilepsy after viral infection may be influenced by environmental factors (age, sex, comorbidities, or medication) that modulate the inflammatory response and susceptibility to epilepsy. Immunological factors, including the immune response type and magnitude, balance between pathways, and resolution, may also play a role. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, B.; Vale, N. Virus-Induced Epilepsy vs. Epilepsy Patients Acquiring Viral Infection: Unravelling the Complex Relationship for Precision Treatment. Int. J. Mol. Sci. 2024, 25, 3730. https://doi.org/10.3390/ijms25073730
Costa B, Vale N. Virus-Induced Epilepsy vs. Epilepsy Patients Acquiring Viral Infection: Unravelling the Complex Relationship for Precision Treatment. International Journal of Molecular Sciences. 2024; 25(7):3730. https://doi.org/10.3390/ijms25073730
Chicago/Turabian StyleCosta, Bárbara, and Nuno Vale. 2024. "Virus-Induced Epilepsy vs. Epilepsy Patients Acquiring Viral Infection: Unravelling the Complex Relationship for Precision Treatment" International Journal of Molecular Sciences 25, no. 7: 3730. https://doi.org/10.3390/ijms25073730